
Immune-Mediated Pathogenesis in Dengue Virus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Involvement of T Cells in the Pathogenesis of DENV Infection
3. Antibody-Dependent Enhancement-Mediated Immunopathogenesis
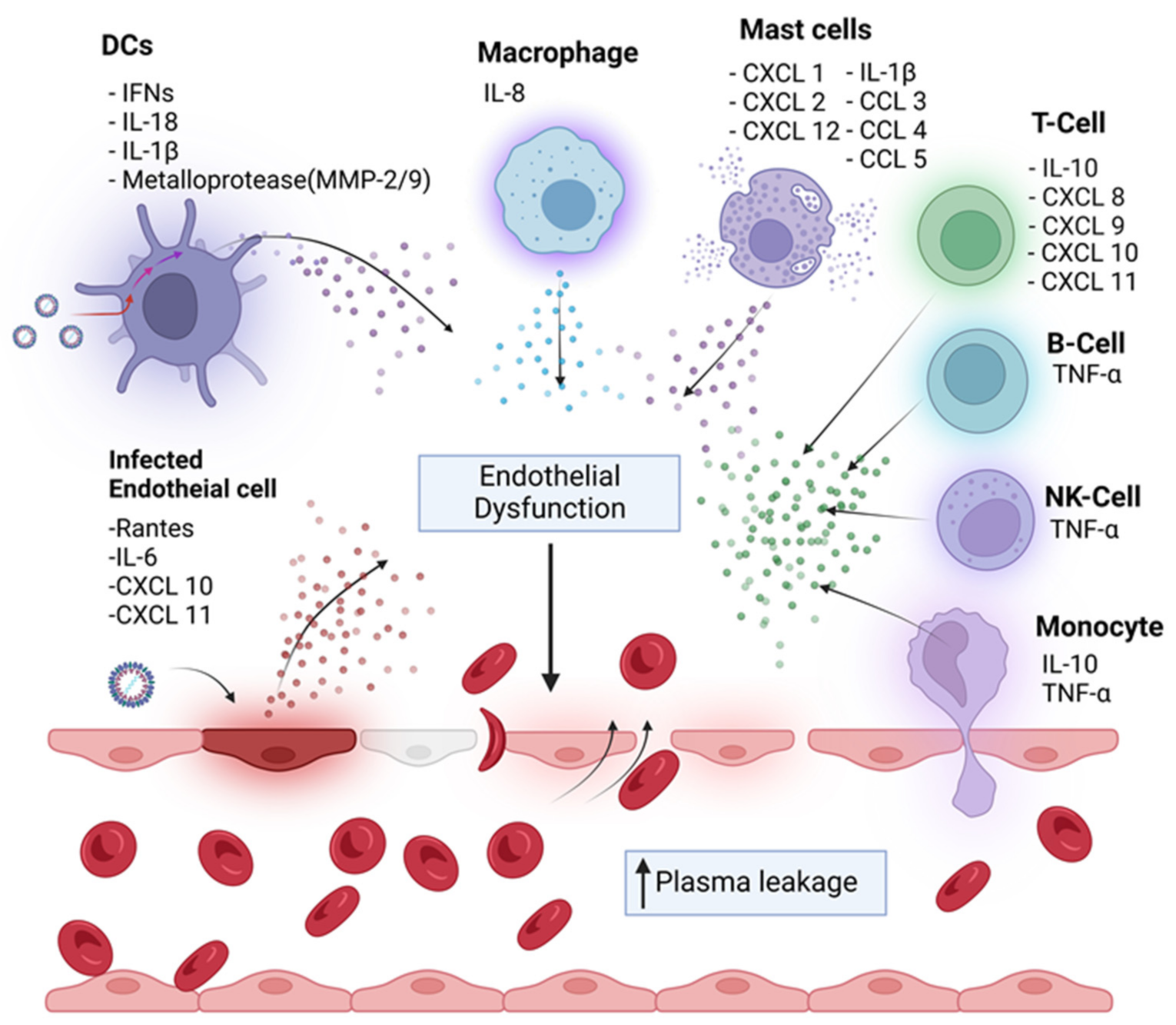
4. Cytokine Storm-Mediated Immunopathogenesis
5. Infection of Immune Cells with DENV Contribute to the Pathogenesis
6. Contribution of Exosomes during Dengue Infection
7. Role of Interferon-Stimulated Genes during Dengue Infection
8. Immune Escape Mechanism of DENV Contributes to Disease Severity
9. Dengue Serotypes Contribute to Disease Severity
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Alvarez, M.; Halstead, S.B. Secondary infection as a risk factor for dengue hemorrhagic fever/dengue shock syndrome: An historical perspective and role of antibody-dependent enhancement of infection. Arch. Virol. 2013, 158, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-W.; Tseng, H.-C.; Lee, C.-H.; Chuang, H.-Y.; Lin, S.-H. Clinical significance of skin rash in dengue fever: A focus on discomfort, complications, and disease outcome. Asian Pac. J. Trop. Med. 2016, 9, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, P.; Sabeena, S.P.; Varma, M.; Arunkumar, G. Current Understanding of the Pathogenesis of Dengue Virus Infection. Curr. Microbiol. 2021, 78, 17–32. [Google Scholar] [CrossRef]
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, C.M.D.; Basavannacharya, C.; Chan, K.W.K.; Chan, S.A.; Singh, D.; Wei, N.; Phoo, W.W.; Luo, D.; Lescar, J.; Vasudevan, S.G. NS3 helicase from dengue virus specifically recognizes viral RNA sequence to ensure optimal replication. Nucleic Acids Res. 2017, 45, 12904–12920. [Google Scholar] [CrossRef] [Green Version]
- Acharya, B.; Gyeltshen, S.; Chaijaroenkul, W.; Na-Bangchang, K. Significance of Autophagy in Dengue Virus Infection: A Brief Review. Am. J. Trop. Med. Hyg. 2019, 100, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Umareddy, I.; Chao, A.; Sampath, A.; Gu, F.; Vasudevan, S.G. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 2006, 87 Pt 9, 2605–2614. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Sreekanth, G.P.; Murali-Krishna, K.; Chandele, A.; Sitaraman, R. Dengue Virus Non-Structural Protein 5 as a Versatile, Multi-Functional Effector in Host-Pathogen Interactions. Front. Cell. Infect. Microbiol. 2021, 11, 574067. [Google Scholar] [CrossRef]
- Wijeratne, D.T.; Fernando, S.; Gomes, L.; Jeewandara, C.; Jayarathna, G.; Perera, Y.; Wickramanayake, S.; Wijewickrama, A.; Ogg, G.S.; Malavige, G.N. Association of dengue virus-specific polyfunctional T-cell responses with clinical disease severity in acute dengue infection. Immun. Inflamm. Dis. 2019, 7, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef] [Green Version]
- Nasirudeen, A.M.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.P.; Liu, D.X. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef]
- Wang, J.P.; Liu, P.; Latz, E.; Golenbock, D.T.; Finberg, R.W.; Libraty, D.H. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J. Immunol. 2006, 177, 7114–7121. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3ε regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial Interactome: A Focus on Antiviral Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Małkowska, P.; Niedźwiedzka-Rystwej, P. Factors affecting RIG-I-Like receptors activation-New research direction for viral hemorrhagic fevers. Front. Immunol. 2022, 13, 1010635. [Google Scholar] [CrossRef]
- Angleró-Rodríguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IκB kinase epsilon interaction. Clin. Vaccine Immunol. CVI 2014, 21, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Yung, C.F.; Lee, K.S.; Thein, T.L.; Tan, L.K.; Gan, V.C.; Wong, J.G.X.; Lye, D.C.; Ng, L.C.; Leo, Y.S. Dengue serotype-specific differences in clinical manifestation, laboratory parameters and risk of severe disease in adults, singapore. Am. J. Trop. Med. Hyg. 2015, 92, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Najioullah, F.; Besnier, F.; Valentino, R.; Césaire, R.; Rosine, J.; Cabié, A. Clinical presentation of dengue by serotype and year of epidemic in Martinique. Am. J. Trop. Med. Hyg. 2014, 91, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A. Dengue vaccine development by the year 2020: Challenges and prospects. Curr. Opin. Virol. 2020, 43, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Paz-Bailey, G.; Adams, L.; Wong, J.M.; Poehling, K.A.; Chen, W.H.; McNally, V.; Atmar, R.L.; Waterman, S.H. Dengue Vaccine: Recommendations of the Advisory Committee on Immunization Practices, United States, 2021. MMWR Recomm. Rep. 2021, 70, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Singla, M.; Kar, M.; Sethi, T.; Kabra, S.K.; Lodha, R.; Chandele, A.; Medigeshi, G.R. Immune Response to Dengue Virus Infection in Pediatric Patients in New Delhi, India—Association of Viremia, Inflammatory Mediators and Monocytes with Disease Severity. PLOS Negl. Trop. Dis. 2016, 10, e0004497. [Google Scholar]
- Narayan, R.; Tripathi, S. Intrinsic ADE: The Dark Side of Antibody Dependent Enhancement During Dengue Infection. Front. Cell. Infect. Microbiol. 2020, 10, 580096. [Google Scholar] [CrossRef]
- Kulkarni, R. Antibody-Dependent Enhancement of Viral Infections. Dyn. Immune Act. Viral Dis. 2019, 9–14. [Google Scholar]
- Keeler, S.P.; Fox, J.M. Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses 2021, 16, 1037. [Google Scholar] [CrossRef]
- John, A.L.; Rathore, A.P.S. Adaptive immune responses to primary and secondary dengue virus infections. Nat. Rev. Immunol. 2019, 19, 218–230. [Google Scholar] [CrossRef]
- Weiskopf, D.; Angelo, M.A.; de Azeredo, E.L.; Sidney, J.; Greenbaum, J.A.; Fernando, A.N.; Broadwater, A.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2046-53. [Google Scholar] [CrossRef] [Green Version]
- Rivino, L.; Kumaran, E.A.; Jovanovic, V.; Nadua, K.; Teo, E.W.; Pang, S.W.; Teo, G.H.; Gan, V.C.; Lye, D.C.; Leo, Y.S.; et al. Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. J. Virol. 2013, 87, 2693–2706. [Google Scholar] [CrossRef] [Green Version]
- Rivino, L.; Lim, M.Q. CD4(+) and CD8(+) T-cell immunity to Dengue-lessons for the study of Zika virus. Immunology 2017, 150, 146–154. [Google Scholar] [CrossRef]
- Tian, Y.; Grifoni, A.; Sette, A.; Weiskopf, D. Human T Cell Response to Dengue Virus Infection. Front. Immunol. 2019, 10, 2125. [Google Scholar] [CrossRef] [Green Version]
- Townsley, E.; Woda, M.; Thomas, S.J.; Kalayanarooj, S.; Gibbons, R.V.; Nisalak, A.; Srikiatkhachorn, A.; Green, S.; Stephens, H.A.; Rothman, A.L.; et al. Distinct activation phenotype of a highly conserved novel HLA-B57-restricted epitope during dengue virus infection. Immunology 2014, 141, 27–38. [Google Scholar] [CrossRef]
- Chandele, A.; Sewatanon, J.; Gunisetty, S.; Singla, M.; Onlamoon, N.; Akondy, R.S.; Kissick, H.T.; Nayak, K.; Reddy, E.S.; Kalam, H.; et al. Characterization of Human CD8 T Cell Responses in Dengue Virus-Infected Patients from India. J. Virol. 2016, 90, 11259–11278. [Google Scholar] [CrossRef] [Green Version]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol. 2017, 39, 563–574. [Google Scholar] [CrossRef]
- Patro, A.R.K.; Mohanty, S.; Prusty, B.K.; Singh, D.K.; Gaikwad, S.; Saswat, T.; Chattopadhyay, S.; Das, B.K.; Tripathy, R.; Ravindran, B. Cytokine Signature Associated with Disease Severity in Dengue. Viruses 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.; Valero, N.; Mosquera, J.; Montiel, M.; Reyes, E.; Larreal, Y.; Alvarez-Mon, M. Increased expression of cytokines, soluble cytokine receptors, soluble apoptosis ligand and apoptosis in dengue. Virology 2014, 452–453, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Hatch, S.; Endy, T.P.; Thomas, S.; Mathew, A.; Potts, J.; Pazoles, P.; Libraty, D.H.; Gibbons, R.; Rothman, A.L. Intracellular cytokine production by dengue virus-specific T cells correlates with subclinical secondary infection. J. Infect. Dis. 2011, 203, 1282–1291. [Google Scholar] [CrossRef]
- Graham, N.; Eisenhauer, P.; Diehl, S.A.; Pierce, K.K.; Whitehead, S.S.; Durbin, A.P.; Kirkpatrick, B.D.; Sette, A.; Weiskopf, D.; Boyson, J.E.; et al. Rapid Induction and Maintenance of Virus-Specific CD8(+) T(EMRA) and CD4(+) T(EM) Cells Following Protective Vaccination Against Dengue Virus Challenge in Humans. Front. Immunol. 2020, 11, 479. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Babor, M.; Lane, J.; Seumois, G.; Liang, S.; Goonawardhana, N.D.S.; De Silva, A.D.; Phillips, E.J.; Mallal, S.A.; da Silva Antunes, R.; et al. Dengue-specific CD8+ T cell subsets display specialized transcriptomic and TCR profiles. J. Clin. Investig. 2019, 129, 1727–1741. [Google Scholar] [CrossRef] [Green Version]
- Silveira, G.F.; Wowk, P.F.; Cataneo, A.H.D.; Dos Santos, P.F.; Delgobo, M.; Stimamiglio, M.A.; Lo Sarzi, M.; Thomazelli, A.; Conchon-Costa, I.; Pavanelli, W.R.; et al. Human T Lymphocytes Are Permissive for Dengue Virus Replication. J. Virol. 2018, 92, e02181-17. [Google Scholar] [CrossRef]
- Martins Sde, T.; Silveira, G.F.; Alves, L.R.; Duarte dos Santos, C.N.; Bordignon, J. Dendritic cell apoptosis and the pathogenesis of dengue. Viruses 2012, 4, 2736–2753. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.A.; Urcuqui-Inchima, S. Mechanisms of monocyte cell death triggered by dengue virus infection. Apoptosis Int. J. Program. Cell Death 2018, 23, 576–586. [Google Scholar] [CrossRef]
- Matsuda, T.; Almasan, A.; Tomita, M.; Tamaki, K.; Saito, M.; Tadano, M.; Yagita, H.; Ohta, T.; Mori, N. Dengue virus-induced apoptosis in hepatic cells is partly mediated by Apo2 ligand/tumour necrosis factor-related apoptosis-inducing ligand. J. Gen. Virol. 2005, 86 Pt 4, 1055–1065. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A. Plasma leakage in dengue haemorrhagic fever. Thromb. Haemost. 2009, 102, 1042–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahala, W.M.; Huang, C.; Butrapet, S.; White, L.J.; de Silva, A.M. Recombinant dengue type 2 viruses with altered e protein domain III epitopes are efficiently neutralized by human immune sera. J. Virol. 2012, 86, 4019–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, M.G.; Vazquez, S. The complexity of antibody-dependent enhancement of dengue virus infection. Viruses 2010, 2, 2649–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Sun, P.; Williams, M.; Nagabhushana, N.; Jani, V.; Defang, G.; Morrison, B.J. NK Cells Activated through Antibody-Dependent Cell Cytotoxicity and Armed with Degranulation/IFN-γ Production Suppress Antibody-dependent Enhancement of Dengue Viral Infection. Sci. Rep. 2019, 9, 1109. [Google Scholar] [CrossRef] [Green Version]
- Nanaware, N.; Banerjee, A.; Mullick Bagchi, S.; Bagchi, P.; Mukherjee, A. Dengue Virus Infection: A Tale of Viral Exploitations and Host Responses. Viruses 2021, 13, 1967. [Google Scholar] [CrossRef]
- Modhiran, N.; Kalayanarooj, S.; Ubol, S. Subversion of innate defenses by the interplay between DENV and pre-existing enhancing antibodies: TLRs signaling collapse. PLoS Negl. Trop. Dis. 2010, 4, e924. [Google Scholar] [CrossRef] [Green Version]
- Flipse, J.; Diosa-Toro, M.A.; Hoornweg, T.E.; van de Pol, D.P.; Urcuqui-Inchima, S.; Smit, J.M. Antibody-Dependent Enhancement of Dengue Virus Infection in Primary Human Macrophages; Balancing Higher Fusion against Antiviral Responses. Sci. Rep. 2016, 6, 29201. [Google Scholar] [CrossRef] [Green Version]
- Ubol, S.; Halstead, S.B. How innate immune mechanisms contribute to antibody-enhanced viral infections. Clin. Vaccine Immunol. CVI 2010, 17, 1829–1835. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.; Li, G.; Shen, M.; Yu, Z.; Ge, W.; Lao, Z.; Fan, Y.; Chen, K.; Ding, Z.; Wang, W.; et al. DENV NS1 and MMP-9 cooperate to induce vascular leakage by altering endothelial cell adhesion and tight junction. PLoS Pathog. 2021, 17, e1008603. [Google Scholar] [CrossRef]
- Luplertlop, N.; Missé, D.; Bray, D.; Deleuze, V.; Gonzalez, J.P.; Leardkamolkarn, V.; Yssel, H.; Veas, F. Dengue-virus-infected dendritic cells trigger vascular leakage through metalloproteinase overproduction. EMBO Rep. 2006, 7, 1176–1181. [Google Scholar] [CrossRef]
- Kelley, J.F.; Kaufusi, P.H.; Volper, E.M.; Nerurkar, V.R. Maturation of dengue virus nonstructural protein 4B in monocytes enhances production of dengue hemorrhagic fever-associated chemokines and cytokines. Virology 2011, 418, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Azizan, A.; Sweat, J.; Espino, C.; Gemmer, J.; Stark, L.; Kazanis, D. Differential proinflammatory and angiogenesis-specific cytokine production in human pulmonary endothelial cells, HPMEC-ST1.6R infected with dengue-2 and dengue-3 virus. J. Virol. Methods 2006, 138, 211–217. [Google Scholar] [CrossRef]
- Cruz Hernández, S.I.; Puerta-Guardo, H.N.; Flores Aguilar, H.; González Mateos, S.; López Martinez, I.; Ortiz-Navarrete, V.; Ludert, J.E.; Angel, R.M. Primary dengue virus infections induce differential cytokine production in Mexican patients. Mem. Do Inst. Oswaldo Cruz 2016, 111, 161–167. [Google Scholar] [CrossRef]
- Sierra, B.; Pérez, A.B.; Alvarez, M.; García, G.; Vogt, K.; Aguirre, E.; Schmolke, K.; Volk, H.D.; Guzmán, M.G. Variation in inflammatory/regulatory cytokines in secondary, tertiary, and quaternary challenges with dengue virus. Am. J. Trop. Med. Hyg. 2012, 87, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Ogg, G.S. Pathogenesis of vascular leak in dengue virus infection. Immunology 2017, 151, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Tedgui, A. Apoptosis in the vasculature: Mechanisms and functional importance. Br. J. Pharmacol. 2000, 130, 947–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, M.; Kamath, K.R.; Iyngkaran, N.; Sinniah, M. Dengue haemorrhagic fever and dengue shock syndrome: Are they tumour necrosis factor-mediated disorders? FEMS Microbiol. Immunol. 1991, 4, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Masood, K.I.; Jamil, B.; Rahim, M.; Islam, M.; Farhan, M.; Hasan, Z. Role of TNF α, IL-6 and CXCL10 in Dengue disease severity. Iran. J. Microbiol. 2018, 10, 202–207. [Google Scholar]
- Züst, R.; Toh, Y.X.; Valdés, I.; Cerny, D.; Heinrich, J.; Hermida, L.; Marcos, E.; Guillén, G.; Kalinke, U.; Shi, P.Y.; et al. Type I interferon signals in macrophages and dendritic cells control dengue virus infection: Implications for a new mouse model to test dengue vaccines. J. Virol. 2014, 88, 7276–7285. [Google Scholar] [CrossRef] [Green Version]
- Puc, I.; Ho, T.C.; Yen, K.L.; Vats, A.; Tsai, J.J.; Chen, P.L.; Chien, Y.W.; Lo, Y.C.; Perng, G.C. Cytokine Signature of Dengue Patients at Different Severity of the Disease. Int. J. Mol. Sci. 2021, 22, 2879. [Google Scholar] [CrossRef]
- Chaturvedi, U.C.; Shrivastava, R.; Tripathi, R.K.; Nagar, R. Dengue virus-specific suppressor T cells: Current perspectives. FEMS Immunol. Med. Microbiol. 2007, 50, 285–299. [Google Scholar] [CrossRef]
- Begum, F.; Das, S.; Mukherjee, D.; Mal, S.; Ray, U. Insight into the Tropism of Dengue Virus in Humans. Viruses 2019, 11, 1136. [Google Scholar] [CrossRef] [Green Version]
- Limonta, D.; Falcón, V.; Torres, G.; Capó, V.; Menéndez, I.; Rosario, D.; Castellanos, Y.; Alvarez, M.; Rodríguez-Roche, R.; de la Rosa, M.C.; et al. Dengue virus identification by transmission electron microscopy and molecular methods in fatal dengue hemorrhagic fever. Infection 2012, 40, 689–694. [Google Scholar] [CrossRef]
- Jessie, K.; Fong, M.Y.; Devi, S.; Lam, S.K.; Wong, K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004, 189, 1411–1418. [Google Scholar] [CrossRef]
- Póvoa, T.F.; Alves, A.M.; Oliveira, C.A.; Nuovo, G.J.; Chagas, V.L.; Paes, M.V. The pathology of severe dengue in multiple organs of human fatal cases: Histopathology, ultrastructure and virus replication. PLoS ONE 2014, 9, e83386. [Google Scholar] [CrossRef] [Green Version]
- Samanta, J.; Sharma, V. Dengue and its effects on liver. World J. Clin. Cases 2015, 3, 125–131. [Google Scholar] [CrossRef]
- Tomo, S.; Sindhujadevi, M.; Kumar, V.; Sevathy, S.; Daisy, M.S.; Agieshkumar, B.P.; Soundravally, R. Differential platelet receptor expression for viral capture (DC-SIGN) and plasma leakage in patients with dengue infection. J. Clin. Virol. Plus 2021, 1, 100039. [Google Scholar] [CrossRef]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Sung, P.S.; Hsieh, S.L. CLEC2 and CLEC5A: Pathogenic Host Factors in Acute Viral Infections. Front. Immunol. 2019, 10, 2867. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.L.; de Wet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Zhang, H.; Liang, Z.; Yang, X.; Cao, R. AXL, an Important Host Factor for DENV and ZIKV Replication. Front. Cell. Infect. Microbiol. 2021, 11, 575346. [Google Scholar] [CrossRef]
- Troupin, A.; Shirley, D.; Londono-Renteria, B.; Watson, A.M.; McHale, C.; Hall, A.; Hartstone-Rose, A.; Klimstra, W.B.; Gomez, G.; Colpitts, T.M. A Role for Human Skin Mast Cells in Dengue Virus Infection and Systemic Spread. J. Immunol. 2016, 197, 4382–4391. [Google Scholar] [CrossRef] [Green Version]
- Syenina, A.; Jagaraj, C.J.; Aman, S.A.B.; Sridharan, A.; St John, A.L. Dengue vascular leakage is augmented by mast cell degranulation mediated by immunoglobulin Fcγ receptors. eLife 2015, 4, e05291. [Google Scholar] [CrossRef] [PubMed]
- Begum, F.; Das, S.; Mukherjee, D.; Ray, U. Hijacking the Host Immune Cells by Dengue Virus: Molecular Interplay of Receptors and Dengue Virus Envelope. Microorganisms 2019, 7, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, J.; Rico-Hesse, R. Dengue virus tropism in humanized mice recapitulates human dengue fever. PLoS ONE 2011, 6, e20762. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Ruiz, C.; Gutiérrez-Barbosa, H.; Medina-Moreno, S.; Velandia-Romero, M.L.; Chua, J.V.; Castellanos, J.E.; Zapata, J.C. Humanized Mice in Dengue Research: A Comparison with Other Mouse Models. Vaccines 2020, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upasani, V.; Vo, H.T.M.; Auerswald, H.; Laurent, D.; Heng, S.; Duong, V.; Rodenhuis-Zybert, I.A.; Dussart, P.; Cantaert, T. Direct Infection of B Cells by Dengue Virus Modulates B Cell Responses in a Cambodian Pediatric Cohort. Front. Immunol. 2020, 11, 594813. [Google Scholar] [CrossRef]
- Kwissa, M.; Nakaya, H.I.; Onlamoon, N.; Wrammert, J.; Villinger, F.; Perng, G.C.; Yoksan, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Ahmed, R.; et al. Dengue virus infection induces expansion of a CD14(+)CD16(+) monocyte population that stimulates plasmablast differentiation. Cell Host Microbe 2014, 16, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Lata, S.; Ali, A.; Banerjea, A.C. Dengue haemorrhagic fever: A job done via exosomes? Emerg. Microbes Infect. 2019, 8, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Ruiz, J.M.; Osuna-Ramos, J.F.; De Jesús-González, L.A.; Hurtado-Monzón, A.M.; Farfan-Morales, C.N.; Cervantes-Salazar, M.; Bolaños, J.; Cigarroa-Mayorga, O.E.; Martín-Martínez, E.S.; Medina, F.; et al. Isolation and characterization of exosomes released from mosquito cells infected with dengue virus. Virus Res. 2019, 266, 1–14. [Google Scholar] [CrossRef]
- Vora, A.; Zhou, W.; Londono-Renteria, B.; Woodson, M.; Sherman, M.B.; Colpitts, T.M.; Neelakanta, G.; Sultana, H. Arthropod EVs mediate dengue virus transmission through interaction with a tetraspanin domain containing glycoprotein Tsp29Fb. Proc. Natl. Acad. Sci. USA 2018, 115, E6604–E6613. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Mettling, C.; Wu, S.R.; Yu, C.Y.; Perng, G.C.; Lin, Y.S.; Lin, Y.L. Autophagy-associated dengue vesicles promote viral transmission avoiding antibody neutralization. Sci. Rep. 2016, 6, 32243. [Google Scholar] [CrossRef]
- Sung, P.-S.; Huang, T.-F.; Hsieh, S.-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [Green Version]
- Martins, S.T.; Kuczera, D.; Lötvall, J.; Bordignon, J.; Alves, L.R. Characterization of Dendritic Cell-Derived Extracellular Vesicles During Dengue Virus Infection. Front. Microbiol. 2018, 9, 1792. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Hong, P.; Wang, Z.; Tang, Z.; Li, K. MicroRNAs in Transforming Growth Factor-Beta Signaling Pathway Associated With Fibrosis Involving Different Systems of the Human Body. Front. Mol. Biosci. 2021, 8, 707461. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef] [Green Version]
- Velandia-Romero, M.L.; Calderón-Peláez, M.A.; Balbás-Tepedino, A.; Márquez-Ortiz, R.A.; Madroñero, L.J.; Barreto Prieto, A.; Castellanos, J.E. Extracellular vesicles of U937 macrophage cell line infected with DENV-2 induce activation in endothelial cells EA.hy926. PLoS ONE 2020, 15, e0227030. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Wang, K.; Zou, C.; Wang, X.; Huang, C.; Feng, T.; Pan, W.; Wu, Q.; Wang, P.; Dai, J. Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLoS Pathog. 2018, 14, e1007287. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Pan, W.; Wang, P. ISG15 facilitates cellular antiviral response to dengue and west nile virus infection in vitro. Virol. J. 2011, 8, 468. [Google Scholar] [CrossRef] [Green Version]
- Castillo Ramirez, J.A.; Urcuqui-Inchima, S. Dengue Virus Control of Type I IFN Responses: A History of Manipulation and Control. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2015, 35, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Li, Y.; Xie, J.; Wu, S.; Xia, J.; Zhang, P.; Liu, C.; Zhang, P.; Huang, X. Protein kinase regulated by dsRNA downregulates the interferon production in dengue virus- and dsRNA-stimulated human lung epithelial cells. PLoS ONE 2013, 8, e55108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Roberts, T.G.; Edgil, D.; Lu, B.; Ernst, J.; Harris, E. Modulation of Dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 2000, 74, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Duan, X.; Yao, M.; Kang, L.; Li, Y.; Li, S.; Li, B.; Chen, L. USP18 Mediates Interferon Resistance of Dengue Virus Infection. Front. Microbiol. 2021, 12, 682380. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, S.; Li, Y.; He, L.; Wu, M.; Jiang, L.; Feng, L.; Zhang, P.; Huang, X. Activation of Toll-like receptor 3 impairs the dengue virus serotype 2 replication through induction of IFN-β in cultured hepatoma cells. PLoS ONE 2011, 6, e23346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.T.; Chang, S.Y.; Lee, C.N.; Kao, C.L. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Rodriguez-Madoz, J.R.; Bernal-Rubio, D.; Kaminski, D.; Boyd, K.; Fernandez-Sesma, A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 2010, 84, 4845–4850. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.; Aguirre, S.; Fernandez-Sesma, A. Innate immunity evasion by Dengue virus. Viruses 2012, 4, 397–413. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.C.; Hoang, L.T.; Mohamed Naim, A.N.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.Y. Evasion of early innate immune response by 2′-O-methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef]
- Waickman, A.T.; Lu, J.Q.; Fang, H.; Waldran, M.J.; Gebo, C.; Currier, J.R.; Ware, L.; Van Wesenbeeck, L.; Verpoorten, N.; Lenz, O.; et al. Evolution of inflammation and immunity in a dengue virus 1 human infection model. Sci. Transl. Med. 2022, 14, eabo5019. [Google Scholar] [CrossRef]
- Barnes, W.J.; Rosen, L. Fatal hemorrhagic disease and shock associated with primary dengue infection on a Pacific island. Am. J. Trop. Med. Hyg. 1974, 23, 495–506. [Google Scholar] [CrossRef]
- OhAinle, M.; Balmaseda, A.; Macalalad, A.R.; Tellez, Y.; Zody, M.C.; Saborío, S.; Nuñez, A.; Lennon, N.J.; Birren, B.W.; Gordon, A.; et al. Dynamics of dengue disease severity determined by the interplay between viral genetics and serotype-specific immunity. Sci. Transl. Med. 2011, 3, 114ra128. [Google Scholar] [CrossRef]
- Balmaseda, A.; Hammond, S.N.; Pérez, L.; Tellez, Y.; Saborío, S.I.; Mercado, J.C.; Cuadra, R.; Rocha, J.; Pérez, M.A.; Silva, S.; et al. Serotype-specific differences in clinical manifestations of dengue. Am. J. Trop. Med. Hyg. 2006, 74, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.R.; Herbinger, K.H.; Fröschl, G.; Malta Romano, C.; de Souza Areias Cabidelle, A.; Cerutti Junior, C. Serotype influences on dengue severity: A cross-sectional study on 485 confirmed dengue cases in Vitória, Brazil. BMC Infect. Dis. 2016, 16, 320. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.; Verlaeten, O.; Cabié, A.; Kaidomar, S.; Moravie, V.; Martial, J.; Najioullah, F.; Plumelle, Y.; Fonteau, C.; Dussart, P.; et al. Influence of the dengue serotype, previous dengue infection, and plasma viral load on clinical presentation and outcome during a dengue-2 and dengue-4 co-epidemic. Am. J. Trop. Med. Hyg. 2008, 78, 990–998. [Google Scholar] [CrossRef]
- Fried, J.R.; Gibbons, R.V.; Kalayanarooj, S.; Thomas, S.J.; Srikiatkhachorn, A.; Yoon, I.K.; Jarman, R.G.; Green, S.; Rothman, A.L.; Cummings, D.A. Serotype-specific differences in the risk of dengue hemorrhagic fever: An analysis of data collected in Bangkok, Thailand from 1994 to 2006. PLoS Negl. Trop. Dis. 2010, 4, e617. [Google Scholar] [CrossRef] [Green Version]
- Huy, N.T.; Van Giang, T.; Thuy, D.H.; Kikuchi, M.; Hien, T.T.; Zamora, J.; Hirayama, K. Factors associated with dengue shock syndrome: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2013, 7, e2412. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, U.C.; Nagar, R. Nitric oxide in dengue and dengue haemorrhagic fever: Necessity or nuisance? FEMS Immunol. Med. Microbiol. 2009, 56, 9–24. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, L.; Zhang, Z.; Hong, W.; Wang, J.; Qiu, S.; Yang, H.; Gan, M.; Sun, J.; Zhao, J.; et al. Genetic and pathogenicity diversity of dengue virus type 2 strains circulating in Guangdong, China. Biosaf. Health 2021, 3, 333–342. [Google Scholar] [CrossRef]
- Vaughn, D.W.; Green, S.; Kalayanarooj, S.; Innis, B.L.; Nimmannitya, S.; Suntayakorn, S.; Endy, T.P.; Raengsakulrach, B.; Rothman, A.L.; Ennis, F.A.; et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 2000, 181, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, N.L.; Quyen, N.T.H.; Tien, N.T.H.; Tuan, N.M.; Kien, D.T.H.; Lam, P.K.; Tam, D.T.H.; Van Ngoc, T.; Yacoub, S.; Jaenisch, T.; et al. Higher Plasma Viremia in the Febrile Phase Is Associated with Adverse Dengue Outcomes Irrespective of Infecting Serotype or Host Immune Status: An Analysis of 5642 Vietnamese Cases. Clin. Infect. Dis. 2021, 72, e1074–e1083. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanam, A.; Gutiérrez-Barbosa, H.; Lyke, K.E.; Chua, J.V. Immune-Mediated Pathogenesis in Dengue Virus Infection. Viruses 2022, 14, 2575. https://doi.org/10.3390/v14112575
Khanam A, Gutiérrez-Barbosa H, Lyke KE, Chua JV. Immune-Mediated Pathogenesis in Dengue Virus Infection. Viruses. 2022; 14(11):2575. https://doi.org/10.3390/v14112575
Chicago/Turabian StyleKhanam, Arshi, Hector Gutiérrez-Barbosa, Kirsten E. Lyke, and Joel V. Chua. 2022. "Immune-Mediated Pathogenesis in Dengue Virus Infection" Viruses 14, no. 11: 2575. https://doi.org/10.3390/v14112575