
Short ‘1.2× Genome’ Infectious Clone Initiates Kolmiovirid Replication in Boa constrictor Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
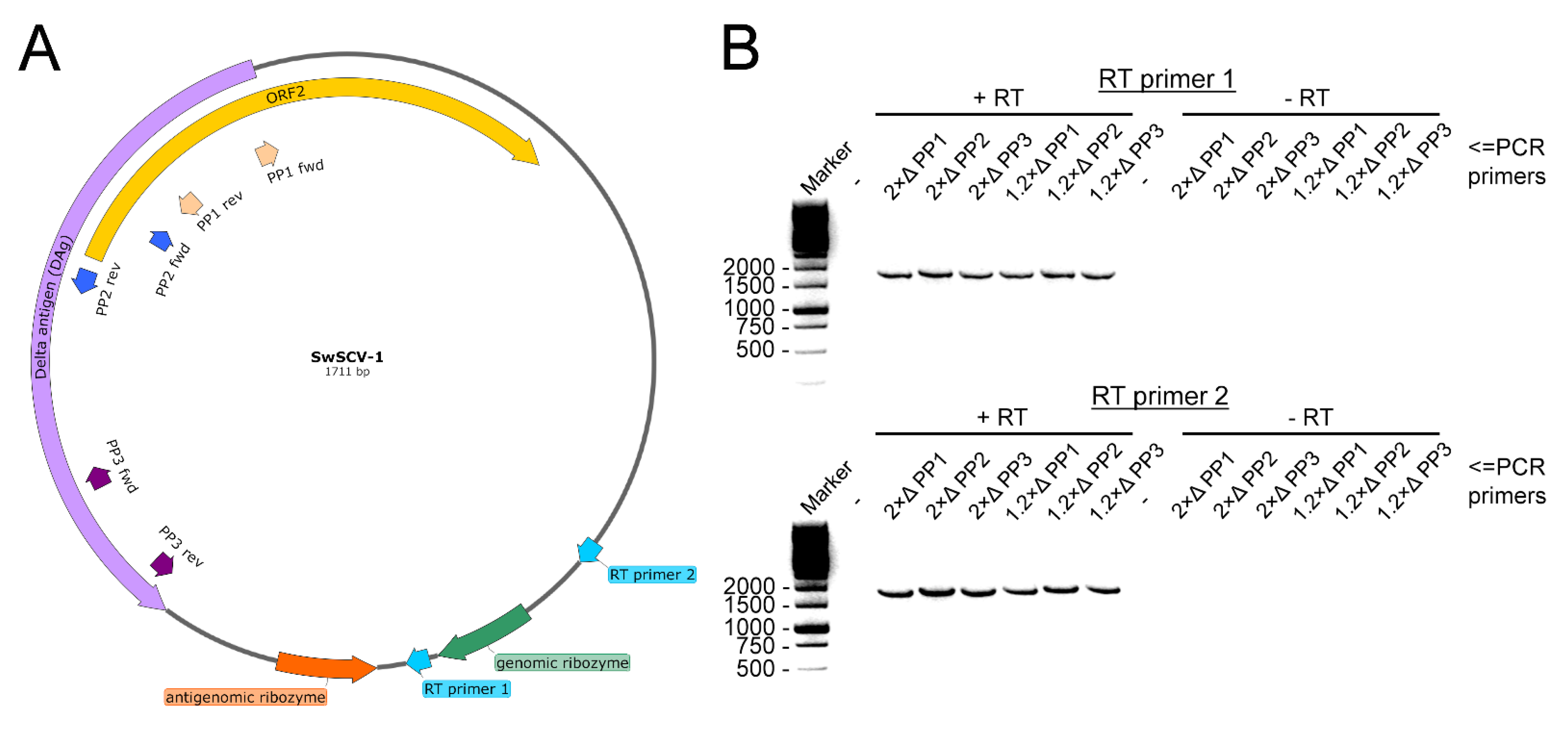{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Superinfection
2.2. Plasmids and Cloning
2.3. Transfection
2.4. Western Blot (WB)
2.5. Immunofluorescence Staining
2.6. Detection of Circular RNA Genome
2.7. Quantitative Reverse Transcription PCR (qRT-PCR)
2.8. Near-Infrared Fluorescent Northern Blot
2.9. SwSCV-1 Infection Dynamics in Naïve I/1Ki Cells
3. Results
3.1. Transfection with 1.2× Genome Construct Initiates Replication of Kolmiovirids
3.2. The 1.2× and 2× SwSCV-1 Genome Infectious Clones Induce Similar Infection as Judged by Antigen Expression and Replication
3.3. Superinfection of Cells Transfected with 1.2× SwSCV-1 FWD Construct Induces Infectious Particle Formation
3.4. Transfection of Cells with the 1.2× SwSCV-1 Construct Results in Persistent Infection
3.5. Inoculation of Naïve I/1Ki Cells with SwSCV-1 Results in Productive
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizzetto, M.; Canese, M.G.; Aricò, S.; Crivelli, O.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 1977, 18, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Rizzetto, M.; Canese, M.G.; Gerin, J.L.; London, W.T.; Sly, D.L.; Purcell, R.H. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J. Infect. Dis. 1980, 141, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Tavanez, J.P.; Gudima, S. Hepatitis delta virus: A fascinating and neglected pathogen. World J. Virol. 2015, 4, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.Y.; Chao, M.; Coates, L.; Taylor, J. Hepatitis delta virus genome replication: A polyadenylated mRNA for delta antigen. J. Virol. 1990, 64, 3192–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.; Hsieh, S.Y.; Taylor, J. Role of two forms of hepatitis delta virus antigen: Evidence for a mechanism of self-limiting genome replication. J. Virol. 1990, 64, 5066–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef] [Green Version]
- Macnaughton, T.B.; Shi, S.T.; Modahl, L.E.; Lai, M.M. Rolling circle replication of hepatitis delta virus RNA is carried out by two different cellular RNA polymerases. J. Virol. 2002, 76, 3920–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Been, M.D.; Wickham, G.S. Self-cleaving ribozymes of hepatitis delta virus RNA. Eur. J. Biochem. 1997, 247, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnius, L.; Taylor, J.; Mason, W.S.; Sureau, C.; Deny, P.; Norder, H.; Ictv Report, C. ICTV Virus Taxonomy Profile: Deltavirus. J. Gen. Virol. 2018, 99, 1565–1566. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Netter, H.J.; Littlejohn, M.; Yuen, L.; Shi, M.; Eden, J.S.; Klaassen, M.; Holmes, E.C.; Hurt, A.C. A Divergent Hepatitis D-Like Agent in Birds. Viruses 2018, 10, 720. [Google Scholar] [CrossRef] [Green Version]
- Hetzel, U.; Szirovicza, L.; Smura, T.; Prahauser, B.; Vapalahti, O.; Kipar, A.; Hepojoki, J. Identification of a Novel Deltavirus in Boa Constrictors. mBio 2019, 10, e00014-19. [Google Scholar] [CrossRef] [Green Version]
- Perez-Vargas, J.; Amirache, F.; Boson, B.; Mialon, C.; Freitas, N.; Sureau, C.; Fusil, F.; Cosset, F.L. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 2019, 10, 2098. [Google Scholar] [CrossRef] [Green Version]
- Szirovicza, L.; Hetzel, U.; Kipar, A.; Martinez-Sobrido, L.; Vapalahti, O.; Hepojoki, J. Snake Deltavirus Utilizes Envelope Proteins of Different Viruses To Generate Infectious Particles. mBio 2020, 11, e03250-19. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.S.; Pettersson, J.H.; Le Lay, C.; Shi, M.; Lo, N.; Wille, M.; Eden, J.S.; Holmes, E.C. Novel hepatitis D-like agents in vertebrates and invertebrates. Virus Evol. 2019, 5, vez021. [Google Scholar] [CrossRef]
- Paraskevopoulou, S.; Pirzer, F.; Goldmann, N.; Schmid, J.; Corman, V.M.; Gottula, L.T.; Schroeder, S.; Rasche, A.; Muth, D.; Drexler, J.F.; et al. Mammalian deltavirus without hepadnavirus coinfection in the neotropical rodent Proechimys semispinosus. Proc. Natl. Acad. Sci. USA 2020, 117, 17977–17983. [Google Scholar] [CrossRef]
- Bergner, L.M.; Orton, R.J.; Broos, A.; Tello, C.; Becker, D.J.; Carrera, J.E.; Patel, A.H.; Biek, R.; Streicker, D.G. Diversification of mammalian deltaviruses by host shifting. Proc. Natl. Acad. Sci. USA 2021, 118, e2019907118. [Google Scholar] [CrossRef]
- Iwamoto, M.; Shibata, Y.; Kawasaki, J.; Kojima, S.; Li, Y.T.; Iwami, S.; Muramatsu, M.; Wu, H.L.; Wada, K.; Tomonaga, K.; et al. Identification of novel avian and mammalian deltaviruses provides new insights into deltavirus evolution. Virus Evol. 2021, 7, veab003. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Basem, A.; et al. Petabase-scale sequence alignment catalyses viral discovery. BioRxiv 2021. [Google Scholar] [CrossRef]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and Evolution of Hepatitis B Virus and Hepatitis D Virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef]
- Perez-Vargas, J.; de Oliveira, R.P.; Jacquet, S.; Pontier, D.; Cosset, F.L.; Freitas, N. HDV-Like Viruses. Viruses 2021, 13, 1207. [Google Scholar] [CrossRef] [PubMed]
- Netter, H.J.; Barrios, M.H.; Littlejohn, M.; Yuen, L.K.W. Hepatitis Delta Virus (HDV) and Delta-Like Agents: Insights Into Their Origin. Front. Microbiol. 2021, 12, 652962. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Hepojoki, J.; Hetzel, U.; Paraskevopoulou, S.; Drosten, C.; Harrach, B.; Zerbini, M.; Koonin, E.V.; Krupovic, M.; Dolja, V.; Kuhn, J.H. Create One New Realm (Ribozyviria) Including One New Family (Kolmioviridae) Including Genus Deltavirus and Seven New Genera for a Total of 15 Species. Available online: https://ictv.global/ictv/proposals/2020.012D.R.Ribozyviria.zip (accessed on 16 August 2021).
- Tai, F.P.; Chen, P.J.; Chang, F.J.; Chen, D.S. Hepatitis delta virus cDNA monomer can be used in transfection experiments to initiate viral RNA replication. Virology 1993, 197, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, T.B.; Lai, M.M. Hepatitis delta virus RNA transfection for the cell culture model. Methods Mol. Med. 2004, 96, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Bergmann, K.F.; Brown, T.L.; Gerin, J.L. Structural requirements for RNA editing in hepatitis delta virus: Evidence for a uridine-to-cytidine editing mechanism. Proc. Natl. Acad. Sci. USA 1992, 89, 7149–7153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazinski, D.W.; Taylor, J.M. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 1993, 67, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Lieber, A.; Sandig, V.; Strauss, M. A mutant T7 phage promoter is specifically transcribed by T7-RNA polymerase in mammalian cells. Eur. J. Biochem. 1993, 217, 387–394. [Google Scholar] [CrossRef]
- Sandig, V.; Lieber, A.; Bähring, S.; Strauss, M. A phage T7 class-III promoter functions as a polymerase II promoter in mammalian cells. Gene 1993, 131, 255–259. [Google Scholar] [CrossRef]
- Hetzel, U.; Sironen, T.; Laurinmaki, P.; Liljeroos, L.; Patjas, A.; Henttonen, H.; Vaheri, A.; Artelt, A.; Kipar, A.; Butcher, S.J.; et al. Isolation, identification, and characterization of novel arenaviruses, the etiological agents of boid inclusion body disease. J. Virol. 2013, 87, 10918–10935. [Google Scholar] [CrossRef] [Green Version]
- Hepojoki, J.; Hepojoki, S.; Smura, T.; Szirovicza, L.; Dervas, E.; Prahauser, B.; Nufer, L.; Schraner, E.M.; Vapalahti, O.; Kipar, A.; et al. Characterization of Haartman Institute snake virus-1 (HISV-1) and HISV-like viruses-The representatives of genus Hartmanivirus, family Arenaviridae. PLoS Pathog. 2018, 14, e1007415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef]
- Korzyukov, Y.; Iheozor-Ejiofor, R.; Levanov, L.; Smura, T.; Hetzel, U.; Szirovicza, L.; de la Torre, J.C.; Martinez-Sobrido, L.; Kipar, A.; Vapalahti, O.; et al. Differences in Tissue and Species Tropism of Reptarenavirus Species Studied by Vesicular Stomatitis Virus Pseudotypes. Viruses 2020, 12, 395. [Google Scholar] [CrossRef] [Green Version]
- Korzyukov, Y.; Hetzel, U.; Kipar, A.; Vapalahti, O.; Hepojoki, J. Generation of Anti-Boa Immunoglobulin Antibodies for Serodiagnostic Applications, and Their Use to Detect Anti-Reptarenavirus Antibodies in Boa Constrictor. PLoS ONE 2016, 11, e0158417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Hetzel, U.; Sironen, T.; Korzyukov, Y.; Vapalahti, O.; Kipar, A.; Hepojoki, J. Co-infecting Reptarenaviruses Can Be Vertically Transmitted in Boa Constrictor. PLoS Pathog. 2017, 13, e1006179. [Google Scholar] [CrossRef] [PubMed]
- Argenta, F.F.; Hepojoki, J.; Smura, T.; Szirovicza, L.; Hammerschmitt, M.E.; Driemeier, D.; Kipar, A.; Hetzel, U. Identification of Reptarenaviruses, Hartmaniviruses, and a Novel Chuvirus in Captive Native Brazilian Boa Constrictors with Boid Inclusion Body Disease. J. Virol. 2020, 94, e00001-20. [Google Scholar] [CrossRef] [PubMed]
- Windbichler, K.; Michalopoulou, E.; Palamides, P.; Pesch, T.; Jelinek, C.; Vapalahti, O.; Kipar, A.; Hetzel, U.; Hepojoki, J. Antibody response in snakes with boid inclusion body disease. PLoS ONE 2019, 14, e0221863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, F.H.; Pestov, D.G. Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal. Biochem. 2013, 441, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.R.; Wei, T.; Fields, C.J.; Sheng, P.; Xie, M. Near-infrared fluorescent northern blot. RNA 2018, 24, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Chen, P.J.; Kuo, M.Y.; Lee, S.D.; Chen, D.S.; Ting, L.P. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. J. Virol. 1991, 65, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.M. Hepatitis D Virus Replication. Cold Spring Harb. Perspect. Med. 2015, 5, a021568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.; Grubb, D.; Elleuch, A.; Nohales, M.A.; Delgado, S.; Gago, S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: Variations on a theme. RNA Biol. 2011, 8, 200–206. [Google Scholar] [CrossRef] [Green Version]
- De la Pena, M.; Ceprian, R.; Casey, J.L.; Cervera, A. Hepatitis delta virus-like circular RNAs from diverse metazoans encode conserved hammerhead ribozymes. Virus Evol. 2021, 7, veab016. [Google Scholar] [CrossRef]
- Kuo, M.Y.; Sharmeen, L.; Dinter-Gottlieb, G.; Taylor, J. Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J. Virol. 1988, 62, 4439–4444. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.J.; Kuo, M.Y.; Chen, M.L.; Tu, S.J.; Chiu, M.N.; Wu, H.L.; Hsu, H.C.; Chen, D.S. Continuous expression and replication of the hepatitis delta virus genome in Hep G2 hepatoblastoma cells transfected with cloned viral DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5253–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, K.S.; Daniel, A.; Lai, M.M. A pseudoknot ribozyme structure is active in vivo and required for hepatitis delta virus RNA replication. J. Virol. 1996, 70, 2403–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Amaran, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olague, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-beta induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, T.B.; Beard, M.R.; Chao, M.; Gowans, E.J.; Lai, M.M. Endogenous promoters can direct the transcription of hepatitis delta virus RNA from a recircularized cDNA template. Virology 1993, 196, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Canese, M.G.; Rizzetto, M.; Arico, S.; Crivelli, O.; Zanetti, A.R.; Macchiorlatti, E.; Ponzetto, A.; Leone, L.; Mollo, F.; Verme, G. An ultrastructural and immunohistochemical study on the delta antigen associated with the hepatitis B virus. J. Pathol. 1979, 128, 169–175. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szirovicza, L.; Hetzel, U.; Kipar, A.; Hepojoki, J. Short ‘1.2× Genome’ Infectious Clone Initiates Kolmiovirid Replication in Boa constrictor Cells. Viruses 2022, 14, 107. https://doi.org/10.3390/v14010107
Szirovicza L, Hetzel U, Kipar A, Hepojoki J. Short ‘1.2× Genome’ Infectious Clone Initiates Kolmiovirid Replication in Boa constrictor Cells. Viruses. 2022; 14(1):107. https://doi.org/10.3390/v14010107
Chicago/Turabian StyleSzirovicza, Leonora, Udo Hetzel, Anja Kipar, and Jussi Hepojoki. 2022. "Short ‘1.2× Genome’ Infectious Clone Initiates Kolmiovirid Replication in Boa constrictor Cells" Viruses 14, no. 1: 107. https://doi.org/10.3390/v14010107