
Inhibitors of Venezuelan Equine Encephalitis Virus Identified Based on Host Interaction Partners of Viral Non-Structural Protein 3

 ,
,  ,
, {kind=link}
{kind=link}
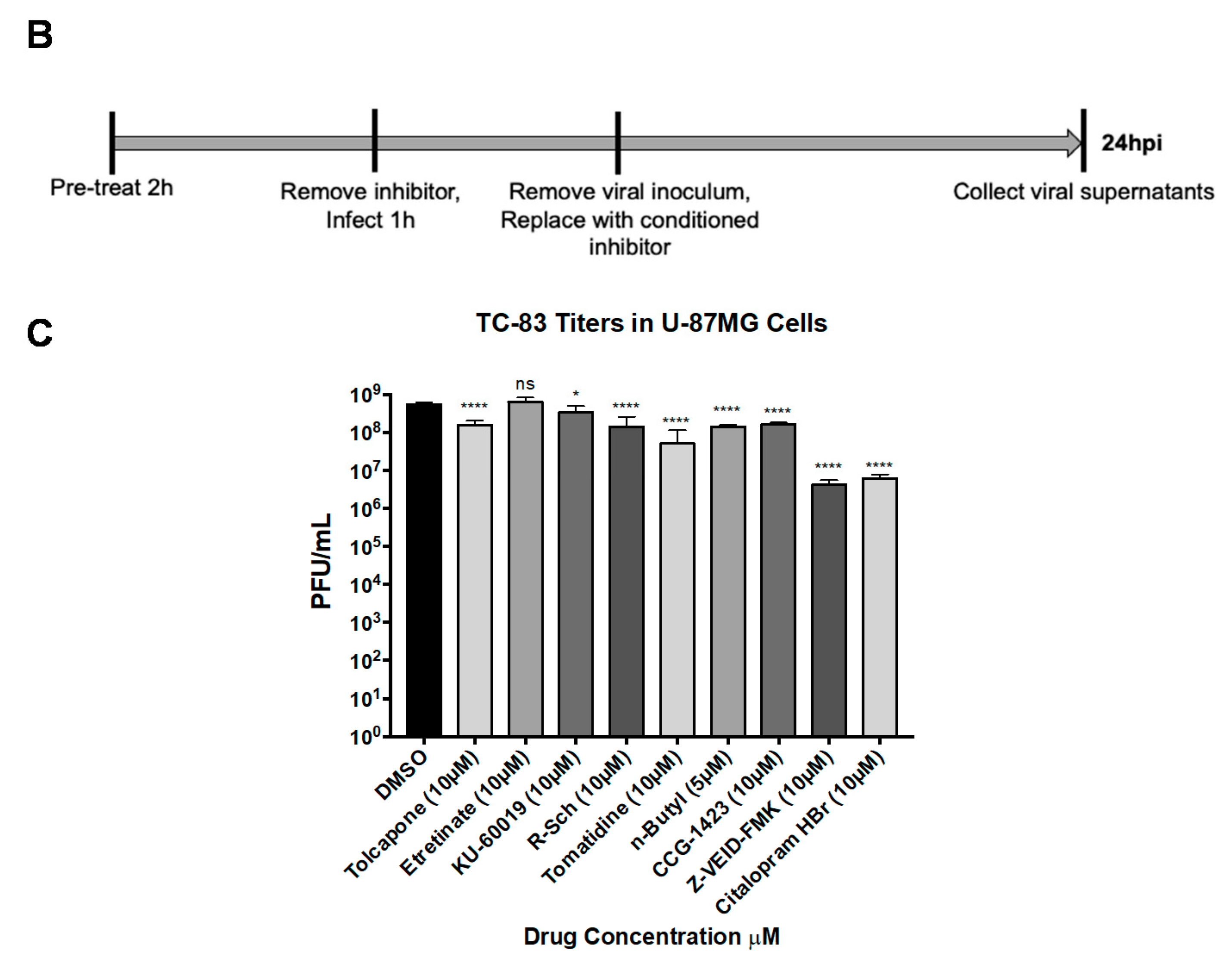{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Transfection and Preparation of Cellular Lysates
2.3. Immunoprecipitation
2.4. Western Blot
2.5. Liquid Chromatography-Mass Spectrometry
2.6. Ingenuity Pathway Analysis
2.7. Inhibitors
2.8. Toxicity Screens
2.9. Infections
2.10. Plaque Assay
2.11. Luciferase and Bradford Protein Assay
2.12. RNA Extraction
2.13. Primer/Probes and cDNA
2.14. RT-PCR
2.15. Statistical Analysis and Curve Fitting
3. Results
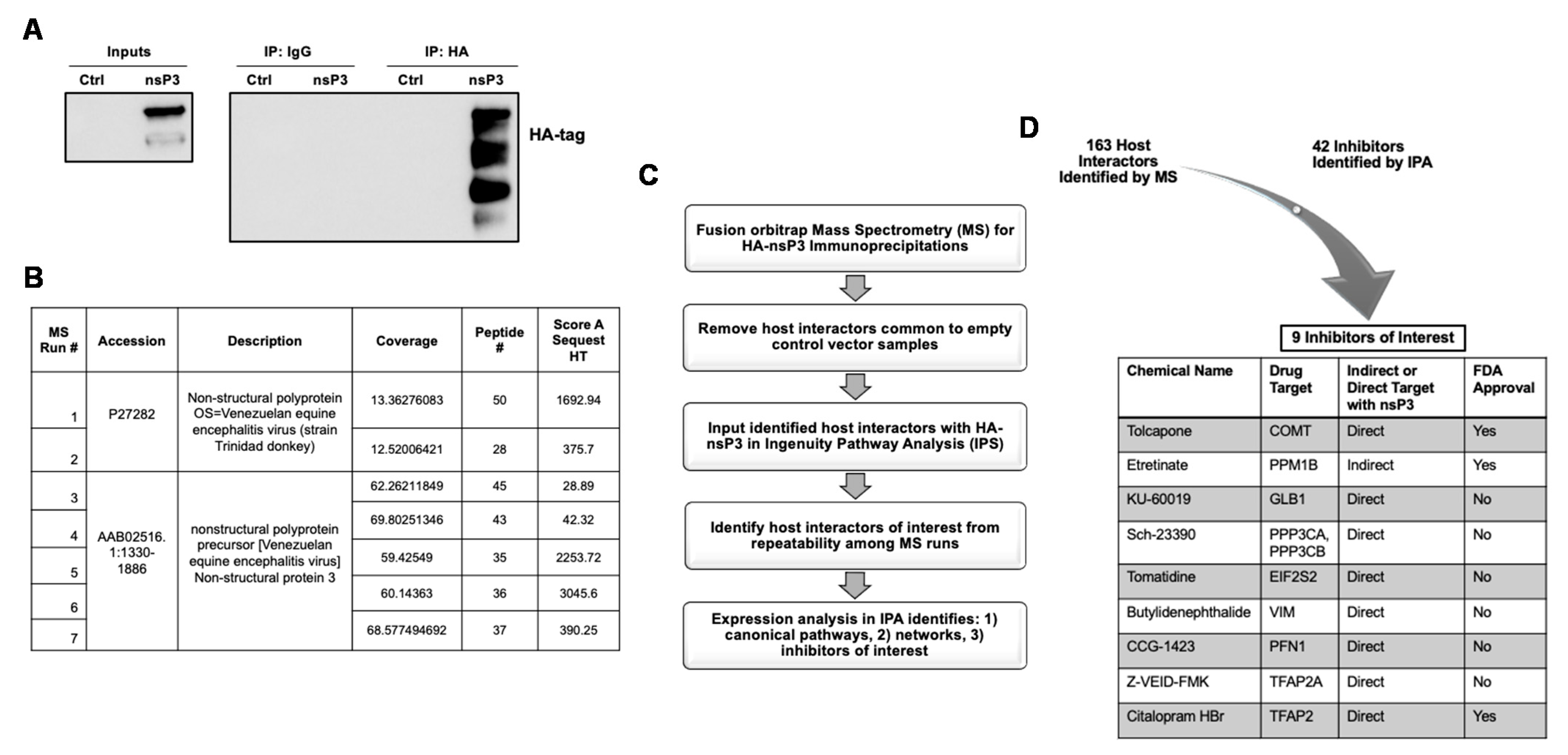
3.1. Mass Spectrometry Identifies VEEV nsP3 Host Interaction Partners as Part of the nsP3 Interactome
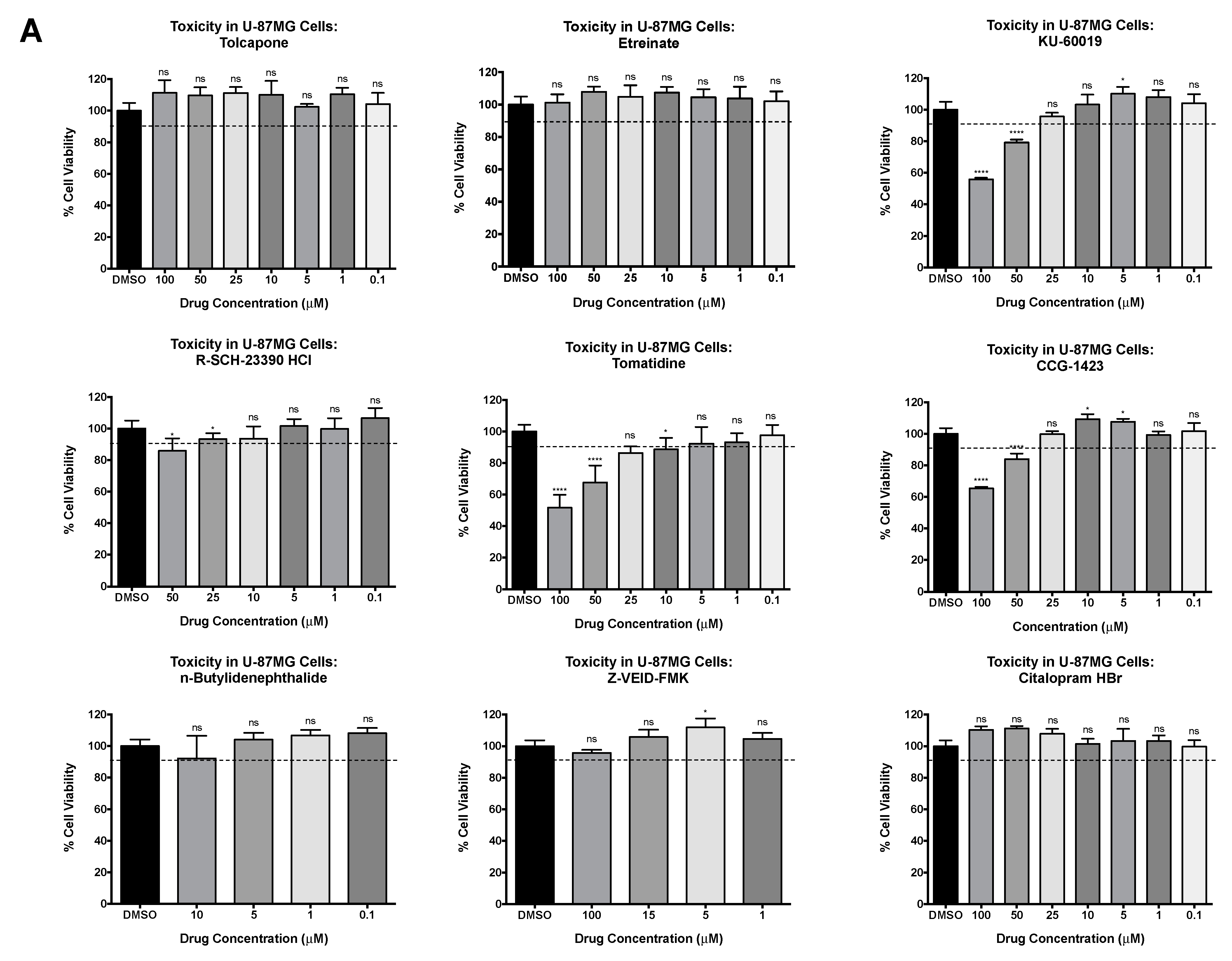
3.2. Host-Based Inhibitors Display Strong Antiviral Activity against VEEV TC-83
3.3. Tomatidine, Citalopram HBr, and Z-VEID-FMK Display Efficacy against VEEV TC-83 Replication in Multiple Cell-Types
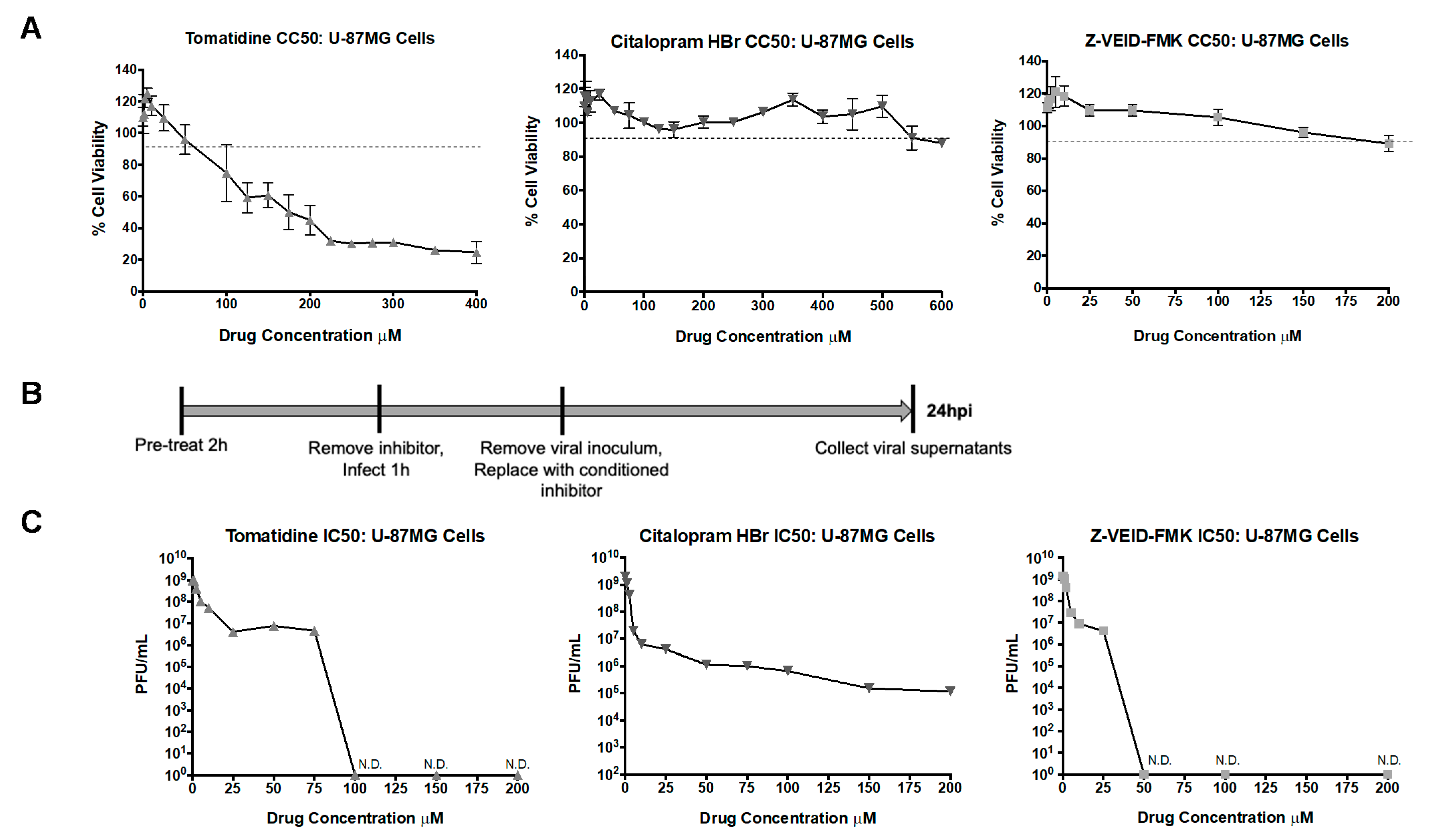
3.4. In Vitro Selectivity Indexes (SI) of Tomatidine, Citalopram HBr, and Z-VEID-FMK
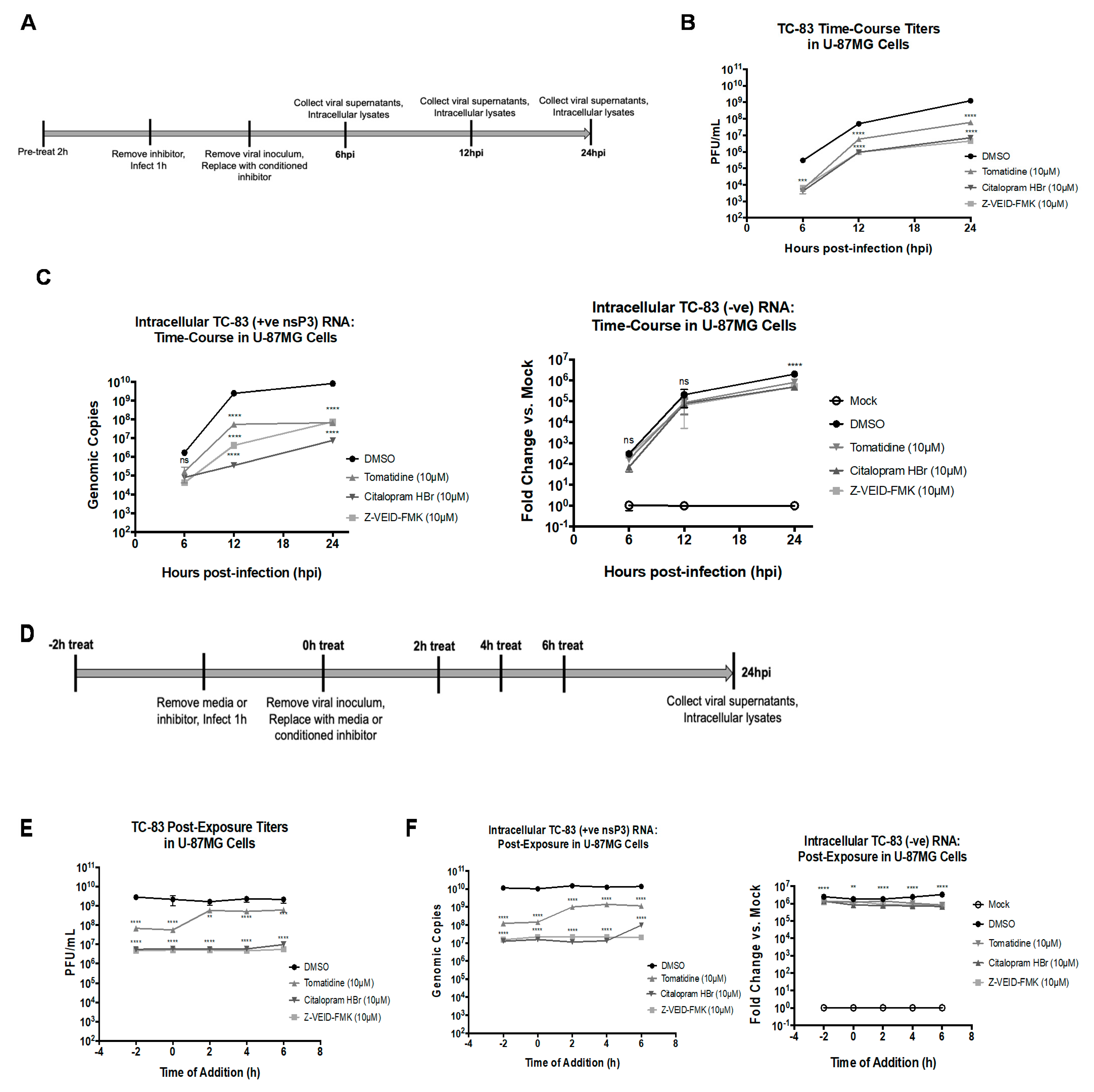
3.5. Tomatidine, Citalopram HBr, and Z-VEID-FMK Inhibit Positive-Sense, but Not Negative-Sense Viral RNA Synthesis, and Post-Infection Treatment with Citalopram HBr, and Z-VEID-FMK Reduces Viral Replication
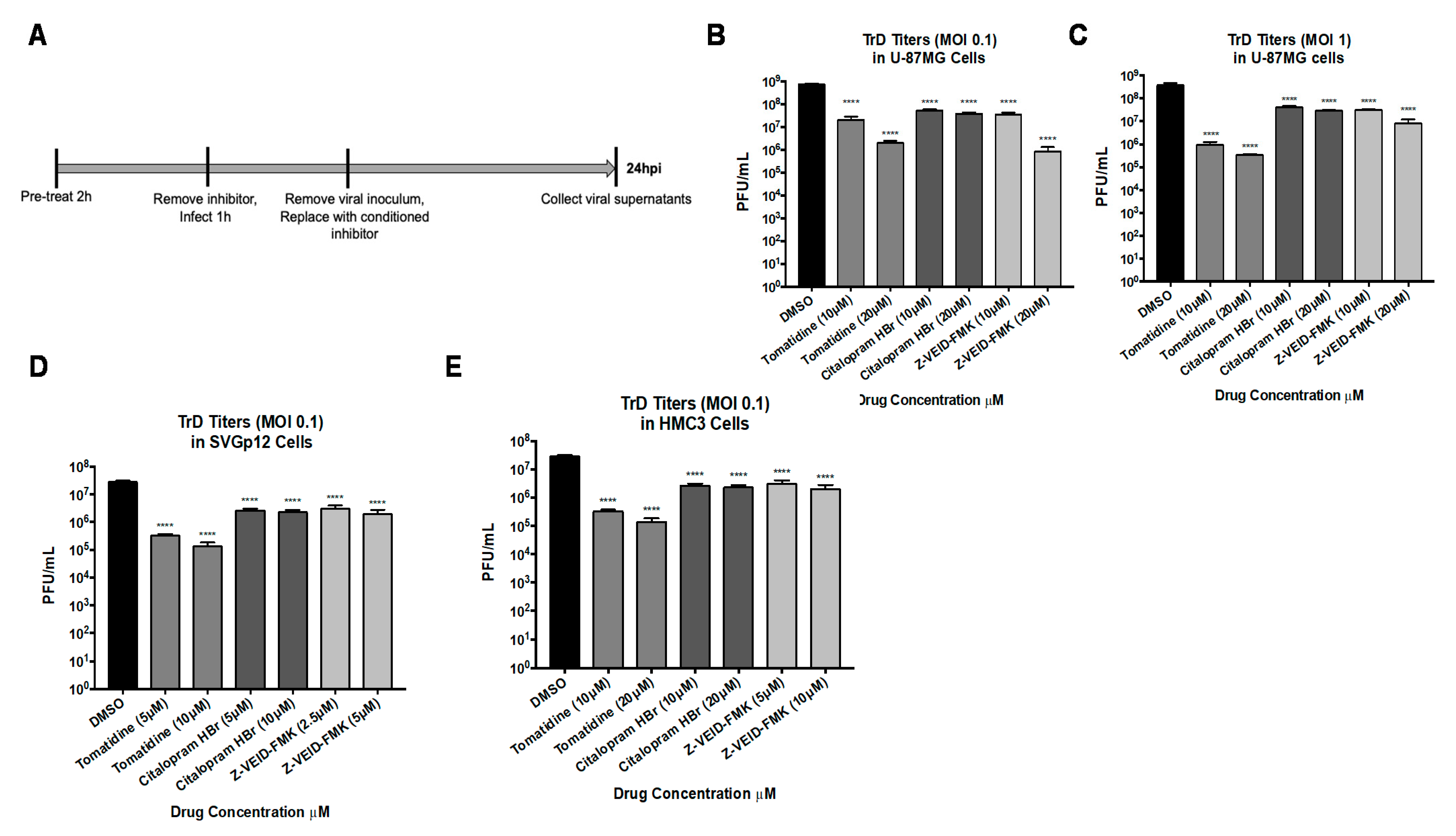
3.6. Tomatidine, Citalopram HBr, and Z-VEID-FMK Are Efficacious against the Virulent VEEV TrD Strain
3.7. Tomatidine, Citalopram HBr, and Z-VEID-FMK Can Inhibit EEEV Replication
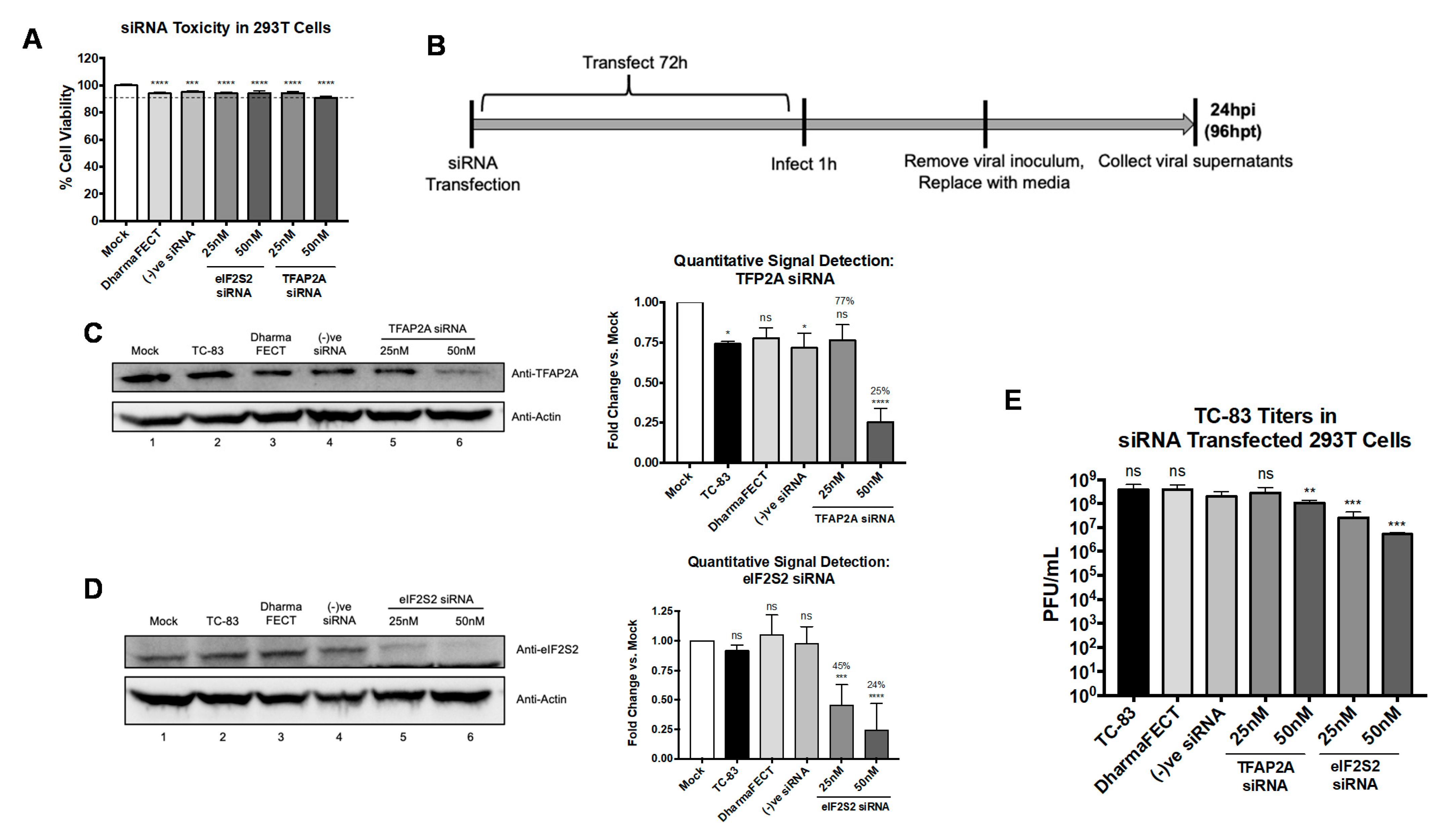
3.8. eIF2S2 Supports Efficient TC-83 Replication and Genomic RNA Translation, and VEEV nsP3 Colocalizes with TFAP2A
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bennett, J.E.; Dolin, R.; Blaser, M.J. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2015; p. 2. [Google Scholar]
- Aguilar, P.; Estrada-Franco, J.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.; Weaver, S. Endemic Venezuelan Equine Encephalitis in the Americas: Hidden under the Dengue Umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.C.; Barrett, A.D.T. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004, 2, 789–801. [Google Scholar] [CrossRef]
- Paessler, S.; Weaver, S.C. Vaccines for Venezuelan equine encephalitis. Vaccine 2009, 27 (Suppl. 4), D80–D85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koprowski, H.; Cox, H.R. Human laboratory infection with Venezuelan equine encephalomyelitis virus; report of four cases. N. Engl. J. Med. 1947, 236, 647–654. [Google Scholar] [CrossRef]
- Croddy, E. Chemical and Biological Warfare: A Comprehensive Survey for the Concerned Citizen; Copernicus Books: New York, NY, USA, 2002; Volume xxii, 306p. [Google Scholar]
- Pittman, P.R.; Makuch, R.S.; Mangiafico, J.A.; Cannon, T.L.; Gibbs, P.H.; Peters, C.J. Long-term duration of detectable neutralizing antibodies after administration of live-attenuated VEE vaccine and following booster vaccination with inactivated VEE vaccine. Vaccine 1996, 14, 337–343. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef] [PubMed]
- Gotte, B.; Liu, L.; McInerney, G.M. The Enigmatic Alphavirus Non-Structural Protein 3 (nsP3) Revealing Its Secrets at Last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- LaStarza, M.W.; Lemm, J.A.; Rice, C.M. Genetic analysis of the nsP3 region of Sindbis virus: Evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 1994, 68, 5781–5791. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.F.; Sawicki, S.G.; Sawicki, D.L. Alphavirus nsP3 functions to form replication complexes transcribing negative-strand RNA. J. Virol. 1994, 68, 6466–6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakovic, A.; Bhalla, N.; Kortchak, S.; Sun, C.; Zhou, W.; Ahmed, A.; Risner, K.; Klimstra, W.B.; Narayanan, A. Venezuelan Equine Encephalitis Virus nsP3 Phosphorylation Can Be Mediated by IKKbeta Kinase Activity and Abrogation of Phosphorylation Inhibits Negative-Strand Synthesis. Viruses 2020, 12, 1021. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Kim, D.Y.; Akhrymuk, M.; Mobley, J.A.; Frolova, E.I. Hypervariable Domain of Eastern Equine Encephalitis Virus nsP3 Redundantly Utilizes Multiple Cellular Proteins for Replication Complex Assembly. J. Virol. 2017, 91, e00371-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kaariainen, L. Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef] [Green Version]
- Lastarza, M.W.; Grakoui, A.; Rice, C.M. Deletion and duplication mutations in the C-terminal nonconserved region of Sindbis virus nsP3: Effects on phosphorylation and on virus replication in vertebrate and invertebrate cells. Virology 1994, 202, 224–232. [Google Scholar] [CrossRef]
- Kim, D.Y.; Reynaud, J.M.; Rasalouskaya, A.; Akhrymuk, I.; Mobley, J.A.; Frolov, I.; Frolova, E.I. New World and Old World Alphaviruses Have Evolved to Exploit Different Components of Stress Granules, FXR and G3BP Proteins, for Assembly of Viral Replication Complexes. PLoS Pathog. 2016, 12, e1005810. [Google Scholar] [CrossRef]
- Gorchakov, R.; Garmashova, N.; Frolova, E.; Frolov, I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 2008, 82, 10088–10101. [Google Scholar] [CrossRef] [Green Version]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, S.E.; Sheahan, B.J.; Atkins, G.J. Deletions in the hypervariable domain of the nsP3 gene attenuate Semliki Forest virus virulence. J. Gen. Virol. 2006, 87, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Griffin, D.E. Interaction of Sindbis virus non-structural protein 3 with poly(ADP-ribose) polymerase 1 in neuronal cells. J. Gen. Virol. 2009, 90, 2073–2080. [Google Scholar] [CrossRef]
- Peranen, J.; Kaariainen, L. Biogenesis of type I cytopathic vacuoles in Semliki Forest virus-infected BHK cells. J. Virol. 1991, 65, 1623–1627. [Google Scholar] [CrossRef] [Green Version]
- Salonen, A.; Vasiljeva, L.; Merits, A.; Magden, J.; Jokitalo, E.; Kaariainen, L. Properly folded nonstructural polyprotein directs the semliki forest virus replication complex to the endosomal compartment. J. Virol. 2003, 77, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, N.J.; Akhrymuk, M.; Shustov, A.V.; Frolova, E.I.; Frolov, I. Hypervariable domain of nonstructural protein nsP3 of Venezuelan equine encephalitis virus determines cell-specific mode of virus replication. J. Virol. 2013, 87, 7569–7584. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.D.; Bakovic, A.; Callahan, V.; Narayanan, A.; Kehn-Hall, K. New World alphavirus protein interactomes from a therapeutic perspective. Antiviral Res. 2019, 163, 125–139. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Williamson, P.R.; Zheng, W. Improving therapy of severe infections through drug repurposing of synergistic combinations. Curr. Opin. Pharmacol. 2019, 48, 92–98. [Google Scholar] [CrossRef]
- Li, C.C.; Wang, X.J.; Wang, H.R. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov. Today 2019, 24, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.N.; Roy, I. Drugs, host proteins and viral proteins: How their promiscuities shape antiviral design. Biol. Rev. Camb. Philos Soc. 2021, 96, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Shechter, S.; Thomas, D.R.; Lundberg, L.; Pinkham, C.; Lin, S.C.; Wagstaff, K.M.; Debono, A.; Kehn-Hall, K.; Jans, D.A. Novel inhibitors targeting Venezuelan equine encephalitis virus capsid protein identified using In Silico Structure-Based-Drug-Design. Sci. Rep. 2017, 7, 17705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atasheva, S.; Fish, A.; Fornerod, M.; Frolova, E.I. Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 2010, 84, 4158–4171. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, L.; Brahms, A.; Hooper, I.; Carey, B.; Lin, S.C.; Dahal, B.; Narayanan, A.; Kehn-Hall, K. Repurposed FDA-Approved drug sorafenib reduces replication of Venezuelan equine encephalitis virus and other alphaviruses. Antivir. Res. 2018, 157, 57–67. [Google Scholar] [CrossRef]
- Berge, T.O.; Banks, I.S.; Tigertt, W.D. Attenuation of Venezuelan Equine Encephalomyelitis Virus by in vitro Cultivation in Guinea-Pig Heart Cells. Am. J. Hyg. 1961, 73, 209–218. [Google Scholar] [CrossRef]
- Thomas, J.M.; Klimstra, W.B.; Ryman, K.D.; Heidner, H.W. Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. J. Virol. 2003, 77, 5598–5606. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Gardner, C.L.; Watson, A.M.; Ryman, K.D.; Klimstra, W.B. Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J. Virol. 2014, 88, 2035–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, N.; Sun, C.; Metthew Lam, L.K.; Gardner, C.L.; Ryman, K.D.; Klimstra, W.B. Host translation shutoff mediated by non-structural protein 2 is a critical factor in the antiviral state resistance of Venezuelan equine encephalitis virus. Virology 2016, 496, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.D.; Ammosova, T.; Pinkham, C.; Lin, X.; Zhou, W.; Liotta, L.A.; Nekhai, S.; Kehn-Hall, K. Protein Phosphatase 1alpha Interacts with Venezuelan Equine Encephalitis Virus Capsid Protein and Regulates Viral Replication through Modulation of Capsid Phosphorylation. J. Virol. 2018, 92, e02068-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Amaya, M.; Brooks-Faulconer, T.; Lark, T.; Keck, F.; Bailey, C.; Raman, V.; Narayanan, A. Venezuelan equine encephalitis virus non-structural protein 3 (nsP3) interacts with RNA helicases DDX1 and DDX3 in infected cells. Antiviral Res. 2016, 131, 49–60. [Google Scholar] [CrossRef]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; de la Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of IKKbeta in Venezuelan equine encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Fultz, M.J.; Miller, T.H.; McKinney, L.C.; Grieder, F.B. Astrocytes as targets for Venezuelan equine encephalitis virus infection. J. Neurovirol. 1999, 5, 342–354. [Google Scholar] [CrossRef]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef] [Green Version]
- Pritchett, J.C.; Naesens, L.; Montoya, J. Chapter 19—Treating HHV-6 Infections: The Laboratory Efficacy and Clinical Use of Anti-HHV-6 Agents. In Human Herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed.; Flamand, L., Lautenschlager, I., Krueger, G.R.F., Ablashi, D.V., Eds.; Elsevier: Boston, MA, USA, 2014; pp. 311–331. [Google Scholar] [CrossRef]
- Hyttel, J. Citalopram--pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 1982, 6, 277–295. [Google Scholar] [CrossRef]
- Durham, E.; Jen, S.; Wang, L.; Nasworthy, J.; Elsalanty, M.; Weinberg, S.; Yu, J.; Cray, J. Effects of Citalopram on Sutural and Calvarial Cell Processes. PLoS ONE 2015, 10, e0139719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagneaux, L.; Gillet, N.; Stamatopoulos, B.; Delforge, A.; Dejeneffe, M.; Massy, M.; Meuleman, N.; Kentos, A.; Martiat, P.; Willems, L.; et al. Valproic acid induces apoptosis in chronic lymphocytic leukemia cells through activation of the death receptor pathway and potentiates TRAIL response. Exp. Hematol. 2007, 35, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Khadija, S.; Veluthakal, R.; Sidarala, V.; Kowluru, A. Glucotoxic and diabetic conditions induce caspase 6-mediated degradation of nuclear lamin A in human islets, rodent islets and INS-1 832/13 cells. Apoptosis 2014, 19, 1691–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimball, S.R. Eukaryotic initiation factor eIF2. Int. J. Biochem. Cell Biol. 1999, 31, 25–29. [Google Scholar] [CrossRef]
- Carrasco, L.; Sanz, M.A.; Gonzalez-Almela, E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Moreno, M.; Sanz, M.A.; Carrasco, L. Initiation codon selection is accomplished by a scanning mechanism without crucial initiation factors in Sindbis virus subgenomic mRNA. RNA 2015, 21, 93–112. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Moreno, M.; Sanz, M.A.; Pelletier, J.; Carrasco, L. Requirements for eIF4A and eIF2 during translation of Sindbis virus subgenomic mRNA in vertebrate and invertebrate host cells. Cell Microbiol. 2013, 15, 823–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, M.A.; Gonzalez Almela, E.; Carrasco, L. Translation of Sindbis Subgenomic mRNA is Independent of eIF2, eIF2A and eIF2D. Sci. Rep. 2017, 7, 43876. [Google Scholar] [CrossRef] [Green Version]
- Van Steeg, H.; Thomas, A.; Verbeek, S.; Kasperaitis, M.; Voorma, H.O.; Benne, R. Shutoff of neuroblastoma cell protein synthesis by Semliki Forest virus: Loss of ability of crude initiation factors to recognize early Semliki Forest virus and host mRNAs. J. Virol. 1981, 38, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprioli, G.; Cahill, M.; Logrippo, S.; James, K. Elucidation of the mass fragmentation pathways of tomatidine and beta1-hydroxytomatine using orbitrap mass spectrometry. Nat. Prod. Commun. 2015, 10, 575–576. [Google Scholar] [PubMed] [Green Version]
- Kuo, C.Y.; Huang, W.C.; Liou, C.J.; Chen, L.C.; Shen, J.J.; Kuo, M.L. Tomatidine Attenuates Airway Hyperresponsiveness and Inflammation by Suppressing Th2 Cytokines in a Mouse Model of Asthma. Mediat. Inflamm. 2017, 2017, 5261803. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.; Gattuso, M.; Grondin, G.; Marsault, E.; Bouarab, K.; Malouin, F. Tomatidine inhibits replication of Staphylococcus aureus small-colony variants in cystic fibrosis airway epithelial cells. Antimicrob. Agents Chemother. 2011, 55, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Chiu, F.L.; Lin, J.K. Tomatidine inhibits iNOS and COX-2 through suppression of NF-kappaB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008, 582, 2407–2412. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Yang, Y.P.; Huang, P.I.; Li, W.C.; Huang, M.C.; Kao, C.L.; Chen, Y.J.; Chen, M.T. Exercise suppresses COX-2 pro-inflammatory pathway in vestibular migraine. Brain Res. Bull. 2015, 116, 98–105. [Google Scholar] [CrossRef]
- Keck, F.; Kortchak, S.; Bakovic, A.; Roberts, B.; Agrawal, N.; Narayanan, A. Direct and indirect pro-inflammatory cytokine response resulting from TC-83 infection of glial cells. Virulence 2018, 9, 1403–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of FDA-Approved Anti-Inflammatory Drugs Against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diosa-Toro, M.; Troost, B.; van de Pol, D.; Heberle, A.M.; Urcuqui-Inchima, S.; Thedieck, K.; Smit, J.M. Tomatidine, a novel antiviral compound towards dengue virus. Antivir. Res. 2019, 161, 90–99. [Google Scholar] [CrossRef]
- Troost, B.; Mulder, L.M.; Diosa-Toro, M.; van de Pol, D.; Rodenhuis-Zybert, I.A.; Smit, J.M. Tomatidine, a natural steroidal alkaloid shows antiviral activity towards chikungunya virus in vitro. Sci. Rep. 2020, 10, 6364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toribio, R.; Diaz-Lopez, I.; Boskovic, J.; Ventoso, I. An RNA trapping mechanism in Alphavirus mRNA promotes ribosome stalling and translation initiation. Nucleic Acids Res. 2016, 44, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Grandgirard, D.; Studer, E.; Monney, L.; Belser, T.; Fellay, I.; Borner, C.; Michel, M.R. Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: Evidence for a caspase-mediated, proteolytic inactivation of Bcl-2. EMBO J. 1998, 17, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Nava, V.E.; Rosen, A.; Veliuona, M.A.; Clem, R.J.; Levine, B.; Hardwick, J.M. Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J. Virol. 1998, 72, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Huang, Q.; Isaacs, J.T.; Reed, J.C.; Griffin, D.E.; Hardwick, J.M. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature 1993, 361, 739–742. [Google Scholar] [CrossRef]
- Celexa Tablets (Citalopram Hydrobromide). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/020822a.cfm (accessed on 20 December 2020).
- Benton, T.; Lynch, K.; Dube, B.; Gettes, D.R.; Tustin, N.B.; Ping Lai, J.; Metzger, D.S.; Blume, J.; Douglas, S.D.; Evans, D.L. Selective serotonin reuptake inhibitor suppression of HIV infectivity and replication. Psychosom. Med. 2010, 72, 925–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakovic, A.; Bhalla, N.; Alem, F.; Campbell, C.; Zhou, W.; Narayanan, A. Inhibitors of Venezuelan Equine Encephalitis Virus Identified Based on Host Interaction Partners of Viral Non-Structural Protein 3. Viruses 2021, 13, 1533. https://doi.org/10.3390/v13081533
Bakovic A, Bhalla N, Alem F, Campbell C, Zhou W, Narayanan A. Inhibitors of Venezuelan Equine Encephalitis Virus Identified Based on Host Interaction Partners of Viral Non-Structural Protein 3. Viruses. 2021; 13(8):1533. https://doi.org/10.3390/v13081533
Chicago/Turabian StyleBakovic, Allison, Nishank Bhalla, Farhang Alem, Catherine Campbell, Weidong Zhou, and Aarthi Narayanan. 2021. "Inhibitors of Venezuelan Equine Encephalitis Virus Identified Based on Host Interaction Partners of Viral Non-Structural Protein 3" Viruses 13, no. 8: 1533. https://doi.org/10.3390/v13081533