
Rise and Fall of SARS-CoV-2 Lineage A.27 in Germany

 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Reconstruction
2.2. Detecting Increase in Proportion
2.3. Phylogenetic Reconstruction
2.4. Three-Dimensional Structure of the S Protein
3. Results and Discussion
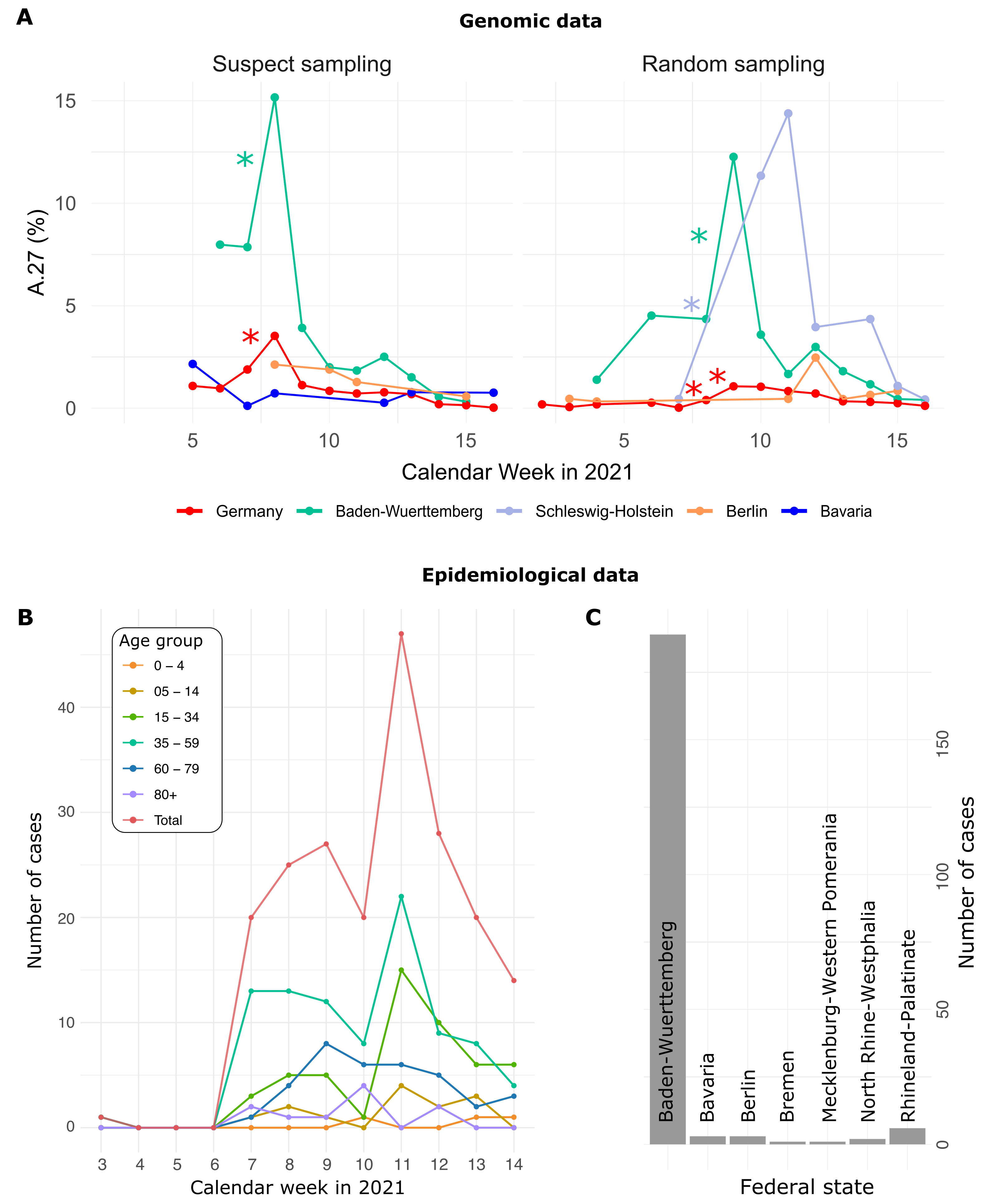
3.1. Increase of A.27 Cases in Baden-Wuerttemberg, Germany
3.2. Epidemiological Data
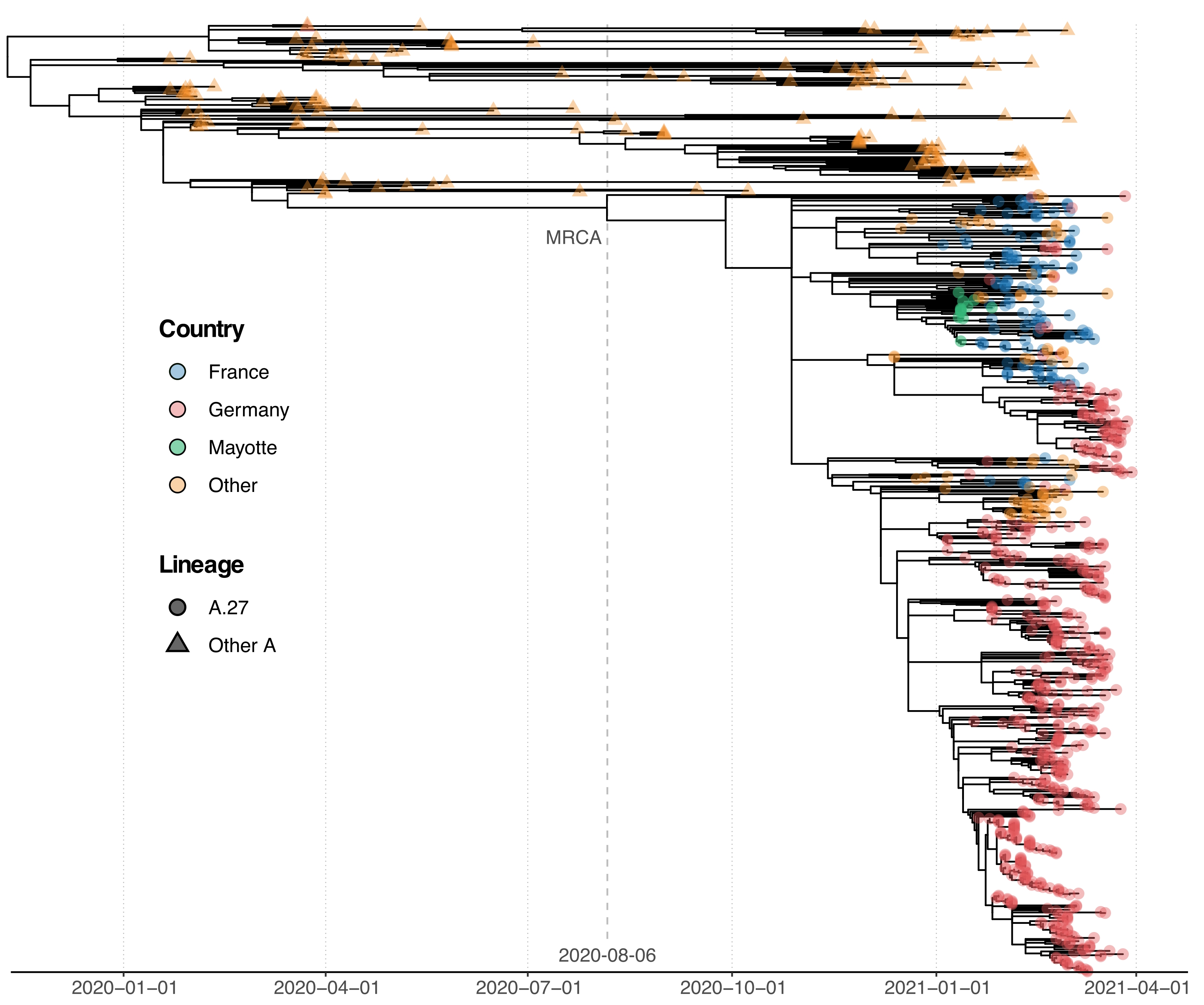
3.3. Phylogenetic Analysis of A.27 Genomes
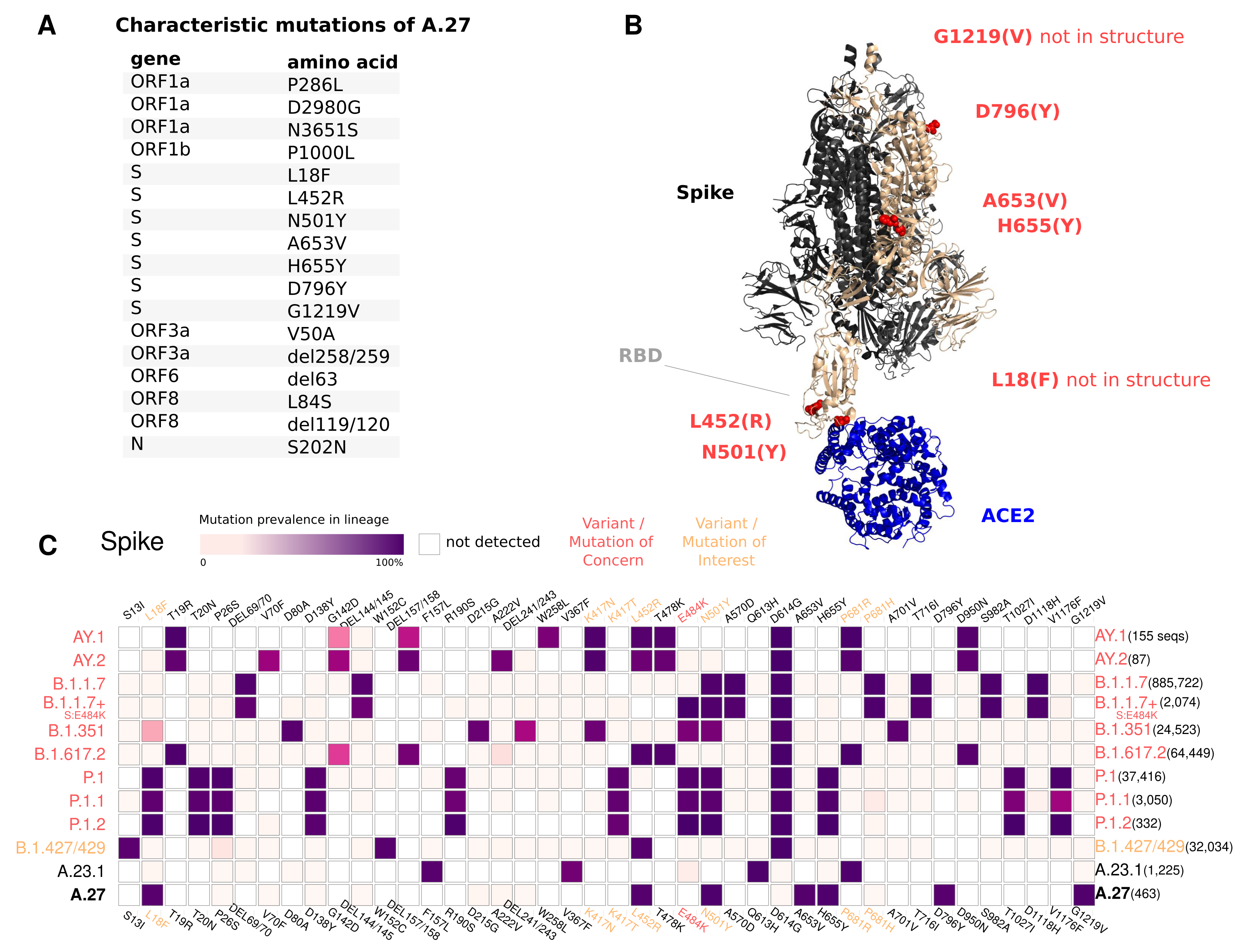
3.4. Spike Mutations of Interest and Concern in A.27
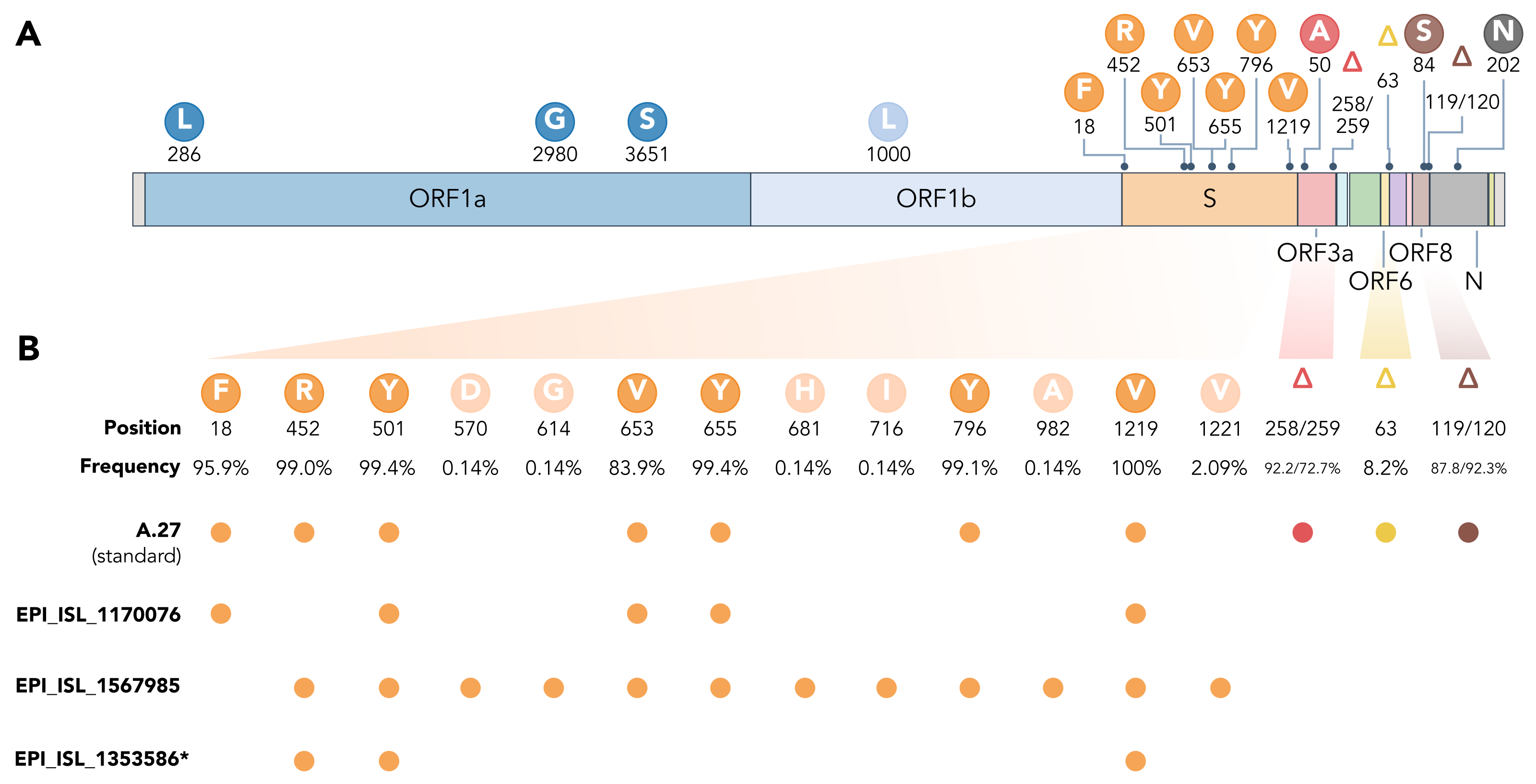
3.5. Spike Mutations of Interest and Concern in Basal A.27 Lineages
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Briefings Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef] [PubMed]
- COG-UK. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 2020, 1, e99. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kröger, S.; Wedde, M.; Hartkopf, F.; Budt, M.; Seifried, J.; Radonic, A.; Belarbi, E.; Hölzer, M.; Böttcher, S.; et al. SARS-CoV-2-Varianten: Evolution im Zeitraffer. Dtsch. Arztebl. 2021, 118, 388–394. [Google Scholar]
- Vavrek, D.; Speroni, L.; Curnow, K.J.; Oberholzer, M.; Moeder, V.; Febbo, P.G. Genomic surveillance at scale is required to detect newly emerging strains at an early timepoint. medRxiv 2021. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 transmission. BioRxiv 2021. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.W.; Lopez, R.; Rahman, N.; Allen, S.G.; Aslam, R.; Buso, N.; Cummins, C.; Fathy, Y.; Felix, E.; Glont, M.; et al. The COVID-19 Data Portal: Accelerating SARS-CoV-2 and COVID-19 research through rapid open access data sharing. Nucleic Acids Res. 2021, 49, W619–W623. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Levasseur, A.; Delerce, J.; Pinault, L.; Dudouet, P.; Devaux, C.; Fournier, P.E.; La Scola, B.; Lagier, J.C.; Raoult, D. Spreading of a new SARS-CoV-2 N501Y spike variant in a new lineage. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef]
- Mallm, J.P.; Bundschuh, C.; Kim, H.; Weidner, N.; Steiger, S.; Lander, I.; Börner, K.; Bauer, K.; Hübschmann, D.; Benes, V.; et al. Local emergence and decline of a SARS-CoV-2 variant with mutations L452R and N501Y in the spike protein. medRxiv 2021. [Google Scholar] [CrossRef]
- Anoh, E.A.; Schubert, G.; Wayoro, O.; Pacôme, M.; Belarbi, E.; Sachse, A.; Calvignac-Spencer, S.; Leendertz, F.; Diané, B.; Akoua-Koffi, C. SARS-CoV-2 variants of concern, variants of interest and lineage A.27 are on the rise in Côte d’Ivoire. medRxiv 2021. [Google Scholar] [CrossRef]
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Hölzer, M. poreCov—An easy to use, fast, and robust workflow for SARS-CoV-2 genome reconstruction via nanopore sequencing. Front. Genet. 2021. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Xiao, T.; Lu, J.; Zhang, J.; Johnson, R.I.; McKay, L.G.A.; Storm, N.; Lavine, C.L.; Peng, H.; Cai, Y.; Rits-Volloch, S.; et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat. Struct. Mol. Biol. 2021, 28, 202–209. [Google Scholar] [CrossRef]
- Pirnay, J.P.; Selhorst, P.; Hong, S.L.; Cochez, C.; Potter, B.; Maes, P.; Petrillo, M.; Dudas, G.; Claes, V.; Van der Beken, Y.; et al. Variant Analysis of SARS-CoV-2 Genomes from Belgian Military Personnel Engaged in Overseas Missions and Operations. MDPI Viruses 2021, 13, 1359. [Google Scholar]
- Krause, G.; Altmann, D.; Faensen, D.; Porten, K.; Benzler, J.; Pfoch, T.; Ammon, A.; Kramer, M.H.; Claus, H. SurvNet electronic surveillance system for infectious disease outbreaks, Germany. Emerg. Infect. Dis. 2007, 13, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Tang, H.; Pajon, R.; Smith, G.; Glenn, G.M.; Shi, W.; Korber, B.; Montefiori, D.C. Neutralization of SARS-CoV-2 Variants B.1.429 and B.1.351. N. Engl. J. Med. 2021, 384, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Ikeda, T.; Saito, A.; Tan, T.S.; Ngare, I.; Nasser, H.; Kimura, I.; Uriu, K.; et al. An emerging SARS-CoV-2 mutant evading cellular immunity and increasing viral infectivity. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kuzmina, A.; Khalaila, Y.; Voloshin, O.; Keren-Naus, A.; Boehm-Cohen, L.; Raviv, Y.; Shemer-Avni, Y.; Rosenberg, E.; Taube, R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe 2021, 29, 522–528.e2. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.; Lutucuta, S.; Nkengasong, J.; Morais, J.; Paixão, J.P.; Neto, Z.; Afonso, P.; Miranda, J.; David, K.; Inglês, L.; et al. A novel variant of interest of SARS-CoV-2 with multiple spike mutations detected through travel surveillance in Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Bugembe, D.L.; V.T.Phan, M.; Ssewanyana, I.; Semanda, P.; Nansumba, H.; Dhaala, B.; Nabadda, S.; O’Toole, Á.N.; Rambaut, A.; Kaleebu, P.; et al. A SARS-CoV-2 lineage A variant (A.23.1) with altered spike has emerged and is dominating the current Uganda epidemic. MedRxiv 2021. [Google Scholar] [CrossRef]
- Mullen, J.L.; Tsueng, G.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Matteson, N.; Andersen, K.G.; et al. SARS-CoV-2 Lineage Comparison from outbreak.info. Available online: https://outbreak.info/compare-lineages (accessed on 23 June 2021).
- Mullen, J.L.; Tsueng, G.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Matteson, N.; Andersen, K.G.; et al. A.27 Lineage Report from outbreak.info. Available online: https://outbreak.info/situation-reports?pango=A.27 (accessed on 25 April 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CW01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 2021 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GER | 3 | 2 | 7 | 29 | 40 | 53 | 61 | 96 | 79 | 84 | 86 | 65 | 52 | 27 | 18 | 8 | 710 |
| (1.07) | (0.22) | (0.32) | (0.82) | (0.92) | (0.89) | (0.91) | (1.35) | (1.02) | (0.91) | (0.81) | (0.62) | (0.48) | (0.25) | (0.14) | (0.10) | ||
| BW | 3 | 0 | 6 | 28 | 35 | 52 | 52 | 83 | 76 | 54 | 53 | 51 | 47 | 21 | 9 | 2 | 572 |
| (2.75) | (0.00) | (1.34) | (4.21) | (4.69) | (4.47) | (4.12) | (6.12) | (3.98) | (2.06) | (1.72) | (1.64) | (1.21) | (0.59) | (0.29) | (0.22) | ||
| SH | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 3 | 0 | 22 | 23 | 4 | 0 | 2 | 3 | 1 | 60 |
| (0.00) | (0.00) | (0.00) | (0.00) | (0.00) | (0.00) | (0.60) | (1.07) | (0.00) | (6.02) | (5.37) | (0.99) | (0.00) | (0.51) | (0.67) | (0.43) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvignac-Spencer, S.; Budt, M.; Huska, M.; Richard, H.; Leipold, L.; Grabenhenrich, L.; Semmler, T.; von Kleist, M.; Kröger, S.; Wolff, T.; et al. Rise and Fall of SARS-CoV-2 Lineage A.27 in Germany. Viruses 2021, 13, 1491. https://doi.org/10.3390/v13081491
Calvignac-Spencer S, Budt M, Huska M, Richard H, Leipold L, Grabenhenrich L, Semmler T, von Kleist M, Kröger S, Wolff T, et al. Rise and Fall of SARS-CoV-2 Lineage A.27 in Germany. Viruses. 2021; 13(8):1491. https://doi.org/10.3390/v13081491
Chicago/Turabian StyleCalvignac-Spencer, Sébastien, Matthias Budt, Matthew Huska, Hugues Richard, Luca Leipold, Linus Grabenhenrich, Torsten Semmler, Max von Kleist, Stefan Kröger, Thorsten Wolff, and et al. 2021. "Rise and Fall of SARS-CoV-2 Lineage A.27 in Germany" Viruses 13, no. 8: 1491. https://doi.org/10.3390/v13081491