
Viral and Prion Infections Associated with Central Nervous System Syndromes in Brazil

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Viruses Commonly Associated with CNS Infection in Brazil
2.1. Enteroviruses and Enterovirus Transmission
2.1.1. Epidemiology of the EV Associated with CNS
2.1.2. Enteroviruses Associated with CNS in Brazil
2.1.3. Diagnosis of Enteroviruses
2.2. Arboviruses
2.2.1. Flaviviruses
Dengue
Zika
Yellow Fever
2.2.2. Alphaviruses
Chikungunya
Other Alphaviruses
2.3. Alphaherpesviruses
Human Betaherpesvirus 5
2.4. Transmissible Spongiform Encephalopathies or Prion Diseases
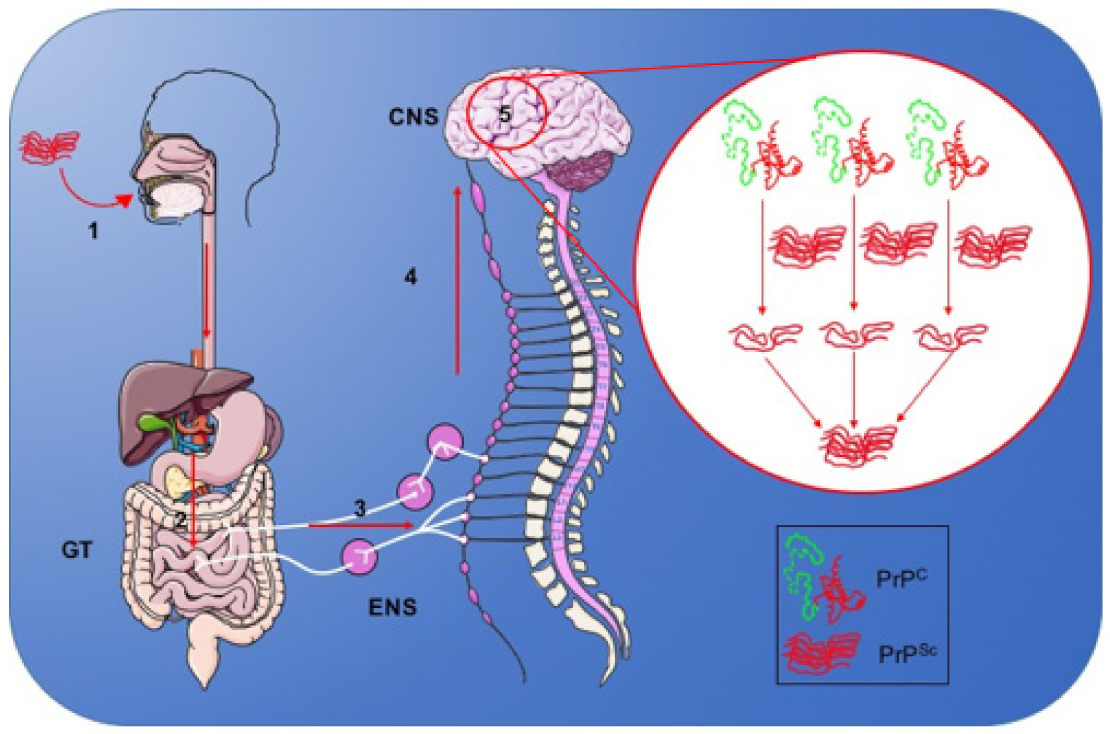
2.4.1. Mechanism of Infection of the Nervous System
2.4.2. Molecular Aspects of Prion Diseases
2.4.3. Epidemiological Aspects of Prion Diseases
Prion Strains
2.4.4. Clinical Aspects of CJD Associated with Alterations in the Central Nervous System
2.4.5. Diagnosis of Prion Diseases
2.4.6. Treatment of Prion Diseases
2.5. SARS-CoV-2
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muñoz, L.S.; Garcia, M.A.; Gordon-Lipkin, E.; Parra, B.; Pardo, C.A. Emerging viral infections and their impact on the global burden of neurological disease. Semin. Neurol. 2018, 38, 163–175. [Google Scholar] [CrossRef]
- Robertson, F.C.; Lepard, J.R.; Mekary, R.A.; Davis, M.C.; Yunusa, I.; Gormley, W.B.; Baticulon, R.E.; Mahmud, M.R.; Misra, B.K.; Rattani, A.; et al. Epidemiology of central nervous system infectious diseases: A meta-analysis and systematic review with implications for neurosurgeons worldwide. J. Neurosurg. 2018, 1, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef]
- Olival, K.J.; Daszak, P. The ecology of emerging neurotropic viruses. J. Neurovirol. 2005, 11, 441–446. [Google Scholar] [CrossRef]
- Clé, M.; Eldin, P.; Briant, L.; Lannuzel, A.; Simonin, Y.; van de Perre, P.; Cabié, A.; Salinas, S. Neurocognitive impacts of arbovirus infections. J. Neuroinflammation 2020, 17, 233. [Google Scholar] [CrossRef] [PubMed]
- Dalman, C.; Allebeck, P.; Gunnell, D.; Harrison, G.; Kristensson, K.; Lewis, G.; Lofving, S.; Rasmussen, F.; Wicks, S.; Karlsson, H. Infections in the CNS during childhood and the risk of subsequent psychotic illness: A cohort study of more than one million Swedish subjects. Am. J. Psychiatry 2008, 165, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Naughton, S.X.; Raval, U.; Pasinetti, G.M. The viral hypothesis in Alzheimer’s disease: Novel insights and pathogen-based biomarkers. J. Pers. Med. 2020, 10, 74. [Google Scholar] [CrossRef]
- Alfahad, T.; Nath, A. Retroviruses and amyotrophic lateral sclerosis. Antivir. Res. 2013, 99, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouk, J.; Rechenchoski, D.Z.; Rodrigues, B.C.D.; Ribelato, E.V.; Faccin-Galhardi, L.C. Viral infections and their relationship to neurological disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Salinas, F.J.; Mestre, L.; Mecha, M.; Feliú, A.; del Campo, R.; Villarrubia, N.; Espejo, C.; Montalbán, X.; Álvarez-Cermeño, J.C.; Villar, L.M.; et al. Gut dysbiosis and neuroimmune responses to brain infection with Theiler’s murine encephalomyelitis virus. Sci. Rep. 2017, 14, 44377. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The commensal microbiota and viral infection: A comprehensive review. Front. Immunol. 2019, 4, 1551. [Google Scholar] [CrossRef] [PubMed]
- Yarandi, S.S.; Peterson, D.A.; Treisman, G.J.; Moran, T.H.; Pasricha, P.J. Modulatory effects of gut microbiota on the central nervous system: How gut could play a role in neuropsychiatric health and diseases. J. Neurogastroenterol. Motil. 2016, 22, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetz, G.; Tetz, V. Prion-like domains in eukaryotic viruses. Sci. Rep. 2018, 8, 8931. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.E.; Carette, J.E. Return of the neurotropic enteroviruses: Co-opting cellular pathways for infection. Viruses 2021, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Chen, B.S.; Lee, H.C.; Lee, K.M.; Gong, Y.N.; Shih, S.R. Enterovirus and encephalitis. Front. Microbiol. 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; McGreevy, A.; Booth, T.F. Molecular pathogenicity of enteroviruses causing neurological disease. Front. Microbiol. 2020, 11, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasbun, R.; Rosenthal, N.; Balada-Llasat, J.M.; Chung, J.; Duff, S.; Bozzette, S.; Zimmer, L.; Ginocchio, C.C. Epidemiology of meningitis and encephalitis in the United States, 2011–2014. Clin. Infect. Dis. 2017, 65, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posnakoglou, L.; Tatsi, E.-B.; Chatzichristou, P.; Siahanidou, T.; Kanaka-Gantenbein, C.; Syriopoulou, V.; Michos, A. Molecular epidemiology of enterovirus in children with central nervous system infections. Viruses 2021, 13, 100. [Google Scholar] [CrossRef]
- Suresh, S.; Forgie, S.; Robinson, J. Non-polio enterovirus detection with acute flaccid paralysis: A systematic review. J. Med. Virol. 2018, 90, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Cassemiro, K.M.S.M.; Burlandy, F.M.; Silva, E.E. Rare natural type 3/type 2 intertypic capsid recombinant vaccine-related poliovirus isolated from a case of acute flaccid paralysis in Brazil, 2015. J. Gen. Virol. 2016, 97, 1545–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho, E.; Sousa, I., Jr.; Burlandy, F.; Costa, E.; Dias, A.; Serrano, R.; Oliveira, M.; Lopes, R.; Debur, M.; Burger, M.; et al. Identification and phylogenetic characterization of human enteroviruses isolated from cases of aseptic meningitis in Brazil, 2013–2017. Viruses 2019, 11, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, I.P., Jr.; Oliveira, M.L.A.; Burlandy, F.M.; Machado, R.S.; Oliveira, S.S.; Tavares, F.N.; Gomes-Neto, F.; da Costa, E.V.; da Silva, E.E. Molecular characterization and epidemiological aspects of non-polio enteroviruses isolated from acute flaccid paralysis in Brazil: A historical series (2005–2017). Emerg. Microbes Infect. 2020, 9, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Compagnoli-Carmona, R.C.; Caetano-Machado, B.; Aparecida de Sousa, C.; Vieira, H.R.; Moraes Alves, M.R.; Farias de Souza, K.A.; de Souza-Gregório, D.; Costa-Vilanova, B.; Sampaio-Tavares-Timenetsky, M.D.C. Distribution of species enterovirus B in patients with central nervous system infections in São Paulo State, Brazil. J. Med. Virol. 2020, 92, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.d.L.; de Castro, C.M.; Oliveira, M.J.; da Silva, E.E. Neutralizing antibodies to enterovirus 71 in Belém, Brazil. Mem. Inst. Oswaldo Cruz 2002, 97, 47–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, M.S.; Lessa, N.; Naveca, F.G.; Monte, R.L.; Braga, W.S.; Figueiredo, L.T.; Ramasawmy, R.; Mourão, M.P. Detection of Herpesvirus, Enterovirus, and Arbovirus infection in patients with suspected central nervous system viral infection in the Western Brazilian Amazon. J. Med. Virol. 2014, 86, 1522–1527. [Google Scholar] [CrossRef]
- De Oliveira, D.B.; Candiani, T.M.; Franco-Luiz, A.P.M.; Almeida, G.M.F.; Abrahão, J.S.; Rios, M.; Coimbra, R.S.; Kroon, E.G. Etiological agents of viral meningitis in children from a dengue-endemic area, Southeast region of Brazil. J. Neurol. Sci. 2017, 375, 390–394. [Google Scholar] [CrossRef]
- Suresh, S.; Rawlinson, W.D.; Andrews, P.I.; Stelzer-Braid, S. Global epidemiology of nonpolio enteroviruses causing severe neurological complications: A systematic review and meta-analysis. Rev. Med. Virol. 2020, 30, e2082. [Google Scholar] [CrossRef]
- Dos Santos, G.P.L.; Skraba, I.; Oliveira, D.; Lima, A.A.F.; de Melo, M.M.M.; Kmetzsch, C.I.; Costa, E.V.; da Silva, E.E. Enterovirus meningitis in Brazil, 1998–2003. J. Med. Virol. 2006, 78, 98–104. [Google Scholar] [CrossRef]
- Pinto-Junior, V.L.; Rebelo, M.C.; Costa, E.V.; Silva, E.E.; Bóia, M.N. Description of a widespread outbreak of aseptic meningitis due to echovirus 30 in Rio de Janeiro state, Brazil. Braz. J. Infect. Dis. 2009, 13, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Luchs, A.; Russo, D.H.; Cilli, A.; Costa, F.F.; Morillo, S.G.; Machado, B.C.; Pellini, A.C.; Carmona, R.C.C.; Timenetsky, M.C. Echovirus 6 associated to aseptic meningitis outbreak, in São Joaquim da Barra, São Paulo, Brazil. Braz. J. Microbiol. 2008, 39, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Kmetzsch, C.I.; Balkie, E.M.; Monteiro, A.; Costa, E.V.; dos Santos, G.P.; da Silva, E.E. Echovirus 13 aseptic meningitis, Brazil. Emerg. Infect. Dis. 2006, 12, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Kebe, O.; Fall, A.D.; Ndiaye, K. Identification and molecular characterization of non-polio enteroviruses from children with acute flaccid paralysis in West Africa, 2013–2014. Sci. Rep. 2017, 7, 3808. [Google Scholar] [CrossRef]
- Tseng, F.-C.; Huang, H.-C.; Chi, C.-Y.; Lin, T.-L.; Liu, C.-C.; Jian, J.-W.; Hsu, L.-C.; Wu, H.-S.; Yang, J.-Y.; Chang, Y.-W.; et al. Epidemiological survey of enterovirus infections occurring in Taiwan between 2000 and 2005: Analysis of sentinel physician surveillance data. J. Med. Virol. 2007, 79, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.K.D.; Sato, D.K. Viral encephalitis: A practical review on diagnostic approach and treatment. J. Pediatr. 2020, 96 (Suppl. 1), 12–19. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Chen, B.S.; Shih, S.R. Editorial: Viral encephalitis. Front. Microbiol. 2020, 11, 599257. [Google Scholar] [CrossRef] [PubMed]
- Valle, D.A.D.; Santos, M.L.S.F.; Giamberardino, H.I.G.; Raboni, S.M.; Scola, R.H. Acute childhood viral encephalitis in southern Brazil. Pediatric Infect. Dis. J. 2020, 39, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.P., Jr.; Machado, R.S.; Burlandy, F.M.; Silva, E.E.D. Detection and characterization of a coxsackievirus B2 strain associated with acute meningoencephalitis, Brazil, 2018. Rev. Soc. Bras. Med. Trop. 2020, 54, e20190499. [Google Scholar] [CrossRef] [PubMed]
- WHO. Enterovirus Surveillance Guidelines—Guidelines for Enterovirus Surveillance in Support of the Polio Eradication; World Health Organization, Regional Office for Europe: Geneva, Switzerland, 2015. [Google Scholar]
- Wilder-Smith, A.; Gubler, D.J.; Weaver, S.C.; Monath, T.P.; Heymann, D.L.; Scott, T.W. Epidemic arboviral diseases: Priorities for research and public health. Lancet Infect. Dis. 2017, 17, e101–e106. [Google Scholar] [CrossRef] [Green Version]
- Harapan, H.; Michie, A.; Sasmono, R.T.; Imrie, A. Dengue: A minireview. Viruses 2020, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.A.D.C.; Costa, C.H.N.; Linhares, A.D.C.; Borba, A.S.; Henriques, D.F.; Silva, E.V.P.D.; Tavares, F.N.; Batista, F.M.A.; Guimarães, H.C.L.; Martins, L.C.; et al. Potential role of dengue virus, chikungunya virus and Zika virus in neurological diseases. Mem. Inst. Oswaldo Cruz 2018, 113, e170538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ministério da Saúde. Manual de Vigilância Sentinela de Doenças Neuroinvasivas por Arbovírus, 1st ed.; Ministério da Saúde: Brasília, Brazil, 2017; Volume 1, p. 44. [Google Scholar]
- Vasilakis, N.; Weaver, S.C. Flavivirus transmission focusing on Zika. Curr. Opin. Virol. 2017, 22, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Heinz, F.X.; Stiasny, K. Flaviviruses and flavivirus vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Samy, A.M.; Alkishe, A.A.; Thomas, S.M.; Wang, L.; Zhang, W. Mapping the potential distributions of etiological agent, vectors, and reservoirs of Japanese Encephalitis in Asia and Australia. Acta Trop. 2018, 188, 108–117. [Google Scholar] [CrossRef]
- Melo, A.S.; Aguiar, R.S.; Amorim, M.M.; Arruda, M.B.; Melo, F.O.; Ribeiro, S.T.; Batista, A.G.; Ferreira, T.; dos Santos, M.P.; Sampaio, V.V.; et al. Congenital Zika virus infection: Beyond neonatal microcephaly. JAMA Neurol. 2016, 73, 1407–1416. [Google Scholar] [CrossRef]
- Monath, T.P.; Barrett, A.D. Pathogenesis and pathophysiology of yellow fever. Adv. Virus Res. 2003, 60, 343–395. [Google Scholar]
- Beck, A.S.; Barrett, A.D. Current status and future prospects of yellow fever vaccines. Expert Rev. Vaccines 2015, 14, 1479–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Ning, Z.J.; Liu, Y.M.; Li, X.H. Neurological manifestations of dengue infection. Front. Cell. Infect. Microbiol. 2017, 7, 449. [Google Scholar] [CrossRef] [Green Version]
- Mustafá, Y.M.; Meuren, L.M.; Coelho, S.V.A.; de Arruda, L.B. Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front. Microbiol. 2019, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Soares, C.N.; Medialdea-Carrera, R.; Ellul, M.; da Silva, M.T.T.; Rosala-Hallas, A.; Jardim, M.R.; Burnside, G.; Pamplona, L.; Bhojak, M.; et al. The spectrum of neurological disease associated with Zika and chikungunya viruses in adults in Rio de Janeiro, Brazil: A case series. PLoS Negl. Trop. Dis. 2018, 12, e0006212. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Kumar, M.; Gurjav, U.; Lum, S.; Nerurkar, V.R. Reversal junction proteins degradation by matrix metalloproteinases inhibitor. Virology 2010, 397, 30–138. [Google Scholar] [CrossRef] [Green Version]
- Jurado, K.A.; Yockey, L.J.; Wong, P.W.; Lee, S.; Huttner, A.J.; Iwasaki, A. Antiviral CD8 T cells induce Zika-virus-associated paralysis in mice. Nat. Microbiol. 2018, 3, 141–147. [Google Scholar] [CrossRef]
- Wang, K.; Wang, H.; Lou, W.; Ma, L.; Li, Y.; Zhang, N.; Wang, C.; Li, F.; Awais, M.; Cao, S.; et al. IP-10 promotes blood-brain barrier damage by inducing tumor necrosis factor alpha production in Japanese encephalitis. Front. Immunol. 2018, 9, 1148. [Google Scholar] [CrossRef]
- Hussmann, K.L.; Samuel, M.A.; Kim, K.S.; Diamond, M.S.; Fredericksen, B.L. Differential replication of pathogenic and nonpathogenic strains of West Nile virus within astrocytes. J. Virol. 2013, 87, 2814–2822. [Google Scholar] [CrossRef] [Green Version]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Y.; Yu, L.; Cao, S.; Wang, K.; Yuan, J.; Wang, C.; Cui, M.; Fu, Z.F. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J. Virol. 2015, 89, 5602–5614. [Google Scholar] [CrossRef] [Green Version]
- Salomão, N.G.; Rabelo, K.; Póvoa, T.F.; Alves, A.M.B.; da Costa, S.M.; Gonçalves, A.J.S.; Amorim, J.F.; Azevedo, A.S.; Nunes, P.C.G.; Basílio-de-Oliveira, C.A.; et al. BALB/c mice infected with DENV-2 strain 66985 by the intravenous route display injury in the central nervous system. Sci. Rep. 2018, 8, 9754. [Google Scholar] [CrossRef] [PubMed]
- Marinho, P.E.S.; Kroon, E.G. Flaviviruses as agents of childhood central nervous system infections in Brazil. New Microbes New Infect. 2019, 31, 100572. [Google Scholar] [CrossRef]
- Chen, R.; Vasilakis, N. Dengue—Quo tu et quo vadis? Viruses 2011, 3, 1562–1608. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Vasilakis, N. Molecular evolution of dengue viruses: Contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 2009, 9, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385453–385465. [Google Scholar] [CrossRef]
- Solomon, T.; Dung, N.M.; Vaughn, D.W.; Kneen, R.; Thao, L.T.; Raengsakulrach, B.; Loan, H.T.; Day, N.P.; Farrar, J.; Myint, K.S.; et al. Neurological manifestations of dengue infection. Lancet 2000, 355, 1053–1059. [Google Scholar] [CrossRef]
- Carod-Artal, F.J.; Wichmann, O.; Farrar, J.; Gascón, J. Neurological complications of dengue virus infection. Lancet Neurol. 2013, 12, 906–919. [Google Scholar] [CrossRef]
- Gupta, M.; Nayak, R.; Khwaja, G.A.; Chowdhury, D. Acute disseminated encephalomyelitis associated with dengue infection: A case report with literature review. J. Neurol. Sci. 2013, 335, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Mello, C.D.S.; Cabral-Castro, M.J.; Silva de Faria, L.C.; Peralta, J.M.; Puccioni-Sohler, M. Dengue and chikungunya infection in neurologic disorders from endemic areas in Brazil. Neurol. Clin. Pract. 2020, 10, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.N.; Cabral-Castro, M.J.; Peralta, J.M.; de Freitas, M.R.; Zalis, M.; Puccioni-Sohler, M. Review of the etiologies of viral meningitis and encephalitis in a dengue endemic region. J. Neurol. Sci. 2011, 303, 75–79. [Google Scholar] [CrossRef]
- Baldaçara, L.; Ferreira, J.R.; Filho, L.C.; Venturini, R.R.; Coutinho, O.M.; Camarço, W.C.; Fernandes, C.C.; Júnior, E.V. Behavior disorder after encephalitis caused by dengue. J. Neuropsychiatry Clin. Neurosci. 2013, 25, E44. [Google Scholar] [CrossRef] [PubMed]
- Madi, D.; Achappa, B.; Ramapuram, J.T.; Chowta, N.; Laxman, M.; Mahalingam, S. Dengue encephalitis—A rare manifestation of dengue fever. Asian Pac. J. Trop. Biomed. 2014, 4, S70–S72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, T.; Badachi, S.; Sarma, G.R.; Nadig, R. “Dot sign” in dengue encephalitis. Ann. Indian Acad. Neurol. 2015, 18, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Thisyakorn, U.; Thisyakorn, C.; Limpitikul, W.; Nisalak, A. Dengue infection with central nervous system manifestations. Southeast Asian J. Trop. Med. Public Health 1999, 30, 504–506. [Google Scholar]
- Misra, U.; Kalita, J.; Syam, U.; Dhole, T. Neurological manifestations of dengue virus infection. J. Neurol. Sci. 2006, 244, 117–122. [Google Scholar] [CrossRef]
- Liou, L.; Lan, S.; Lai, C. Electroencephalography burst suppression in a patient with dengue encephalopathy: A case report. Clin. Neurophysiol. 2008, 119, 2205–2208. [Google Scholar] [CrossRef]
- Oehler, E.; le Hénaff, O.; Ghawche, F. Neurological manifestations of dengue. Presse Med. 2012, 41, e547–e552. [Google Scholar] [CrossRef]
- Soares, C.; Faria, L.; Peralta, J.; de Freitas, M.; Puccioni-Sohler, M. Dengue infection: Neurological manifestations and cerebrospinal fluid (CSF) analysis. J. Neurol. Sci. 2006, 249, 19–24. [Google Scholar] [CrossRef]
- Araújo, F.; Nogueira, R.; Araújo Me, S.; Perdigão, A.; Cavalcanti, L.; Brilhante, R.; Rocha, M.; Vilar, D.F.; Holanda, S.S.; Braga, M.; et al. Dengue in patients with central nervous system manifestations, Brazil. Emerg. Infect. Dis. 2012, 18, 677–679. [Google Scholar] [CrossRef]
- Marinho, P.E.; Bretas de Oliveira, D.; Candiani, T.M.; Crispim, A.P.; Alvarenga, P.P.; Castro, F.C.; Abrahão, J.S.; Rios, M.; Coimbra, R.S.; Kroon, E.G. Meningitis associated with simultaneous infection by multiple dengue virus serotypes in children, Brazil. Emerg. Infect. Dis. 2017, 23, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, L.; Lan, S.; Lai, C. Dengue fever with ischemic stroke: A case report. Neurologist 2008, 14, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Sahu, R.; Holla, V. Neurological manifestations of dengue infection: A review. J. Neurol. Sci. 2014, 346, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Sánchez, A.; Chiquete, E.; Gutiérrez-Plascencia, P.; Castañeda-Moreno, V.; Alfaro-Castellanos, D.; Paredes-Casillas, P.; Ruiz-Sandovala, J.L. Cerebellar hemorrhage in a patient during the convalescent phase of dengue fever. J. Stroke 2014, 16, 202–204. [Google Scholar] [CrossRef] [Green Version]
- Kunishige, M.; Mitsui, T.; Tan, B.; Leong, H.; Takasaki, T.; Kurane, I.; Mihara, A.; Matsumoto, T. Preferential gray matter involvement in dengue myelitis. Neurology 2004, 63, 1980–1981. [Google Scholar] [CrossRef] [PubMed]
- Chanthamat, N.; Sathirapanya, P. Acute transverse myelitis associated with dengue viral infection. J. Spinal Cord Med. 2010, 33, 425–427. [Google Scholar] [CrossRef] [Green Version]
- Weeratunga, P.N.; Caldera, M.C.; Gooneratne, I.K.; Gamage, R.; Perera, P. Neurological manifestations of dengue: A cross sectional study. Travel Med. Infect. Dis. 2014, 12, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Hlaing, C.S.; Tay, C.G.; Kadir, K.A.; Goh, K.J.; Ong, L.C. Longitudinal extensive transverse myelitis with cervical epidural haematoma following dengue virus infection. Eur. J. Paediatr. Neurol. 2016, 20, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.T.; Estofolete, C.F.; Zini, N.; Terzian, A.C.; Gongora, D.V.; Maia, I.L.; Nogueira, M.L. Transverse myelitis as an unusual complication of dengue fever. Am. J. Trop. Med. Hyg. 2017, 96, 380–381. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.M.; Kumawat, B.L.; Ralot, T.; Tripathi, G.; Dixit, S. Guillain-Barre syndrome occurring during dengue fever. J. Indian Med. Assoc. 2011, 109, 675–682. [Google Scholar]
- Jain, R.S.; Handa, R.; Prakash, S.; Nagpal, K.; Gupta, P. Acute hypokalemic quadriparesis: An atypical neurological manifestation of dengue virus. J. Neurovirol. 2014, 20, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Langerak, T.; van Rooij, I.; Doornekamp, L.; Chandler, F.; Baptista, M.; Yang, H.; Koopmans, M.P.G.; GeurtsvanKessel, C.H.; Jacobs, B.C.; Rockx, B.; et al. Guillain-Barré syndrome in Suriname; clinical presentation and identification of preceding infections. Front. Neurol. 2021, 12, 635753. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, I.; Grajales-Muñiz, C.; González-Bonilla, C.; Borja-Aburto, V.H.; Paredes-Cruz, M.; Guerrero-Cantera, J.; González-Ibarra, J.; Vallejos-Parás, A.; Rojas-Mendoza, T.; Santacruz-Tinoco, C.E.; et al. Zika and dengue but not chikungunya are associated with Guillain-Barré syndrome in Mexico: A case-control study. PLoS Negl. Trop. Dis. 2020, 14, e0008032. [Google Scholar] [CrossRef]
- Prateek, S.V.; Paliwal, N.; Tak, H. Dengue, Guillain-Barré syndrome, and cerebral infarction: A case of rare complication. Indian J. Crit. Care Med. 2019, 23, 533–535. [Google Scholar]
- Asbury, A.K.; Cornblath, D.R. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann. Neurol. 1990, 27 (Suppl. 1), S21–S24. [Google Scholar] [CrossRef] [PubMed]
- Shahrizaila, N.; Lehmann, H.C.; Kuwabara, S. Guillain-Barré syndrome. Lancet 2021, 397, 1214–1228. [Google Scholar] [CrossRef]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Heymann, D.L.; Hodgson, A.; Sall, A.A.; Freedman, D.O.; Staples, J.E.; Althabe, F.; Baruah, K.; Mahmud, G.; Kandun, N.; Vasconcelos, P.F.; et al. Zika virus and microcephaly: Why is this situation a PHEIC? Lancet 2016, 387, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Freitas, D.A.; Souza-Santos, R.; Carvalho, L.M.A.; Barros, W.B.; Neves, L.M.; Brasil, P.; Wakimoto, M.D. Congenital Zika syndrome: A systematic review. PLoS ONE 2020, 15, e0242367. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Zammarchi, L.; Tappe, D.; Fortuna, C.; Remoli, M.E.; Günther, S.; Venturi, G.; Bartoloni, A.; Schmidt-Chanasit, J. Zika virus infection in a traveller returning to Europe from Brazil, March 2015. Eurosurveillance 2015, 20, 21153. [Google Scholar] [CrossRef] [Green Version]
- Brasil, P.; Calvet, G.A.; Siqueira, A.M.; Wakimoto, M.; de Sequeira, P.C.; Nobre, A.; Quintana, M.e.S.; Mendonça, M.C.; Lupi, O.; de Souza, R.V.; et al. Zika virus outbreak in Rio de Janeiro, Brazil: Clinical characterization, epidemiological and virological aspects. PLoS Negl. Trop. Dis. 2016, 10, e0004636. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome—Case report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, S.E.; Halstead, S.; Lant, S.B.; Militão de Albuquerque, M.F.P.; de Brito, C.A.A.; de Albuquerque, L.B.B.; Ellul, M.A.; de Oliveira França, R.F.; Gourlay, D.; Griffiths, M.J.; et al. Guillain-Barré syndrome during the Zika virus outbreak in Northeast Brazil: An observational cohort study. J. Neurol. Sci. 2021, 420, 117272. [Google Scholar] [CrossRef]
- Brasil, P.; Sequeira, P.C.; Freitas, A.D.; Zogbi, H.E.; Calvet, G.A.; de Souza, R.V.; Siqueira, A.M.; de Mendonca, M.C.; Nogueira, R.M.; de Filippis, A.M.; et al. Guillain-Barré syndrome associated with Zika virus infection. Lancet 2016, 387, 1482. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, H.M.; Romero, G.A.S.; de Araújo, W.N.; de Oliveira, M.R.F. Guillain-Barré syndrome associated with Zika virus infection in Brazil: A cost-of-illness study. Trans. R. Soc. Trop. Med. Hyg. 2019, 113, 252–258. [Google Scholar] [CrossRef]
- Brito Ferreira, M.L.; Antunes de Brito, C.A.; Moreira, Á.; de Morais Machado, M.; Henriques-Souza, A.; Cordeiro, M.T.; de Azevedo Marques, E.T.; Pena, L.J. Guillain-Barré syndrome, acute disseminated encephalomyelitis and encephalitis associated with Zika virus infection in Brazil: Detection of viral RNA and isolation of virus during late infection. Am. J. Trop. Med. Hyg. 2017, 97, 1405–1409. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, L.S.; Parra, B.; Pardo, C.A. Study NEitA: Neurological implications of Zika virus infection in adults. J. Infect. Dis. 2017, 216, S897–S905. [Google Scholar] [CrossRef]
- Da Silva, I.R.F.; Frontera, J.A.; Bispo de Filippis, A.M.; Nascimento, O.J.M.D. Group R-G-ZR: Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol. 2017, 74, 1190–1198. [Google Scholar] [CrossRef]
- Mécharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef] [Green Version]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika virus associated with meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef] [PubMed]
- Rozé, B.; Najioullah, F.; Fergé, J.L.; Dorléans, F.; Apetse, K.; Barnay, J.L.; Daudens-Vaysse, E.; Brouste, Y.; Césaire, R.; Fagour, L.; et al. Guillain-Barré syndrome associated with Zika virus infection in Martinique in 2016: A prospective study. Clin. Infect. Dis. 2017, 65, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.; Tyshkov, C.; Thakur, K.; Vargas, W. Encephalomyelitis following definitive Zika virus infection. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douam, F.; Ploss, A. Yellow fever virus: Knowledge gaps impeding the fight against an old foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.F. Yellow fever. Rev. Soc. Bras. Med. Trop. 2003, 36, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Possas, C.; Lourenço-de-Oliveira, R.; Tauil, P.L.; Pinheiro, F.P.; Pissinatti, A.; Cunha, R.V.D.; Freire, M.; Martins, R.M.; Homma, A. Yellow fever outbreak in Brazil: The puzzle of rapid viral spread and challenges for immunisation. Mem. Inst. Oswaldo Cruz 2018, 113, e180278. [Google Scholar] [CrossRef] [Green Version]
- Giovanetti, M.; de Mendonça, M.C.L.; Fonseca, V.; Mares-Guia, M.A.; Fabri, A.; Xavier, J.; de Jesus, J.G.; Gräf, T.; dos Santos Rodrigues, C.D.; dos Santos, C.C.; et al. Yellow fever virus reemergence and spread in southeast Brazil, 2016–2019. J. Virol. 2019, 94, e01623-19. [Google Scholar] [CrossRef]
- Rezende, I.M.; Sacchetto, L.; Munhoz de Mello, É.; Alves, P.A.; Iani, F.C.M.; Adelino, T.R.; Duarte, M.M.; Cury, A.L.F.; Bernardes, A.F.L.; Santos, T.A.; et al. Persistence of Yellow fever virus outside the Amazon Basin, causing epidemics in southeast Brazil, from 2016 to 2018. PLoS Negl. Trop. Dis. 2018, 12, e0006538. [Google Scholar] [CrossRef] [Green Version]
- Cunha, M.S.; da Costa, A.C.; de Azevedo Fernandes, N.C.C.; Guerra, J.M.; dos Santos, F.C.P.; Nogueira, J.S.; D’Agostino, L.G.; Komninakis, S.V.; Witkin, S.S.; Ressio, R.A.; et al. Epizootics due to Yellow fever virus in São Paulo State, Brazil: Viral dissemination to new areas (2016–2017). Sci. Rep. 2019, 9, 5474. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, L.; Drumond, B.P.; Han, B.A.; Nogueira, M.L.; Vasilakis, N. Re-emergence of yellow fever in the neotropics—Quo vadis? Emerg. Top. Life Sci. 2020, 4, 399–410. [Google Scholar] [PubMed]
- De Oliveira Figueiredo, P.; Stoffella-Dutra, A.G.; Barbosa Costa, G.; Silva de Oliveira, J.; Dourado Amaral, C.; Duarte Santos, J.; Soares Rocha, K.L.; Araújo Júnior, J.P.; Lacerda Nogueira, M.; Zazá Borges, M.A.; et al. Re-emergence of yellow fever in Brazil during 2016–2019: Challenges, lessons learned, and perspectives. Viruses 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.E.; Holmes, E.C.; Barrett, A.D. Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007, 3, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatorre, E.; de Abreu, F.V.S.; Ribeiro, I.P.; Gómez, M.M.; dos Santos, A.A.C.; Ferreira-de-Brito, A.; Neves, M.S.A.S.; Bonelly, I.; de Miranda, R.M.; Furtado, N.D.; et al. Distinct YFV lineages co-circulated in the central-western and southeastern Brazilian regions from 2015 to 2018. Front. Microbiol. 2019, 10, 1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paules, C.I.; Fauci, A.S. Yellow fever—Once again on the radar screen in the Americas. N. Engl. J. Med. 2017, 376, 1397–1399. [Google Scholar] [CrossRef]
- Jones, E.M.; Wilson, D.C. Clinical features of yellow fever cases at Vom Christian Hospital during the 1969 epidemic on the Jos Plateau, Nigeria. Bull. World Health Organ. 1972, 46, 653–657. [Google Scholar]
- Martin, M.; Tsai, T.F.; Cropp, B.; Chang, G.J.; Holmes, D.A.; Tseng, J.; Shieh, W.; Zaki, S.R.; Al-Sanouri, I.; Cutrona, A.F.; et al. Fever and multisystem organ failure associated with 17D-204 yellow fever vaccination: A report of four cases. Lancet 2001, 358, 98–104. [Google Scholar] [CrossRef]
- Vasconcelos, P.F.; Luna, E.J.; Galler, R.; Silva, L.J.; Coimbra, T.L.; Barros, V.L.; Monath, T.P.; Rodigues, S.G.; Laval, C.; Costa, Z.G.; et al. Serious adverse events associated with yellow fever 17DD vaccine in Brazil: A report of two cases. Lancet 2001, 358, 91–97. [Google Scholar] [CrossRef]
- Kengsakul, K.; Sathirapongsasuti, K.; Punyagupta, S. Fatal myeloencephalitis following yellow fever vaccination in a case with HIV infection. J. Med. Assoc Thail. 2002, 85, 131–134. [Google Scholar]
- Gardner, C.L.; Ryman, K.D. Yellow fever: A reemerging threat. Clin. Lab. Med. 2010, 30, 237–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, A.N.; Miyaji, K.T.; Ibrahim, K.Y.; Lopes, M.H.; Sartori, A.M.C. Adverse events following yellow fever vaccination in immunocompromised persons. Rev. Inst. Med. Trop. Sao Paulo 2021, 63, e13. [Google Scholar] [CrossRef] [PubMed]
- Juan-Giner, A.; Kimathi, D.; Grantz, K.H.; Hamaluba, M.; Kazooba, P.; Njuguna, P.; Fall, G.; Dia, M.; Bob, N.S.; Monath, T.P.; et al. Immunogenicity and safety of fractional doses of yellow fever vaccines: A randomised, double-blind, non-inferiority trial. Lancet 2021, 397, 119–127. [Google Scholar] [CrossRef]
- Martins, R.M.; Pavão, A.L.; de Oliveira, P.M.; dos Santos, P.R.; Carvalho, S.M.; Mohrdieck, R.; Fernandes, A.R.; Sato, H.K.; de Figueiredo, P.M.; von Doellinger, V.R.; et al. Adverse events following yellow fever immunization: Report and analysis of 67 neurological cases in Brazil. Vaccine 2014, 32, 6676–6682. [Google Scholar] [CrossRef] [PubMed]
- De Menezes Martins, R.; Fernandes Leal, M.L.; Homma, A. Serious adverse events associated with yellow fever vaccine. Hum. Vaccines Immunother. 2015, 11, 2183–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, E.D. Yellow fever: Epidemiology and prevention. Clin. Infect. Dis. 2007, 44, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.S.B.; Araujo, P.P.; Sousa, J.R.P.; Donis, A.C.G.; Moreira, D.; Makssoudian, A. Serious adverse event: Late neurotropic disease associated with yellow fever vaccine. Einstein 2020, 18, eRC5041. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.; Geocadin, R.G. Diagnosis and management of acute encephalitis: A practical approach. Neurol. Clin. Pract. 2014, 4, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arboviral Diseases, Neuroinvasive and Non-Neuroinvasive 2015 Case Definition. Available online: wwwn.cdc.gov/nndss/conditions/arboviral-diseases-neuroinvasive-and-non neuroinvasive/case-definition/2015/ (accessed on 31 January 2021).
- Tyler, K.L.; Roos, K.L. The expanding spectrum of Zika virus infections of the nervous system. JAMA Neurol. 2017, 74, 1169–1171. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Baxter, V.K.; Heise, M.T. Immunopathogenesis of alphaviruses. Adv. Virus Res. 2020, 107, 315–382. [Google Scholar] [PubMed]
- Zeller, H.; van Bortel, W.; Sudre, B. Chikungunya: Its history in Africa and Asia and its spread to new regions in 2013–2014. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; de Oliveira, L.F.; Azevedo, R.S.; da Silva, D.E.; da Silva, E.V.; da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, T.M.A.; Ribeiro, E.D.; Corrêa, V.C.E.; Damasco, P.V.; Santos, C.C.; de Bruycker-Nogueira, F.; Chouin-Carneiro, T.; Faria, N.R.D.C.; Nunes, P.C.G.; Heringer, M.; et al. Following in the footsteps of the Chikungunya virus in Brazil: The first autochthonous cases in Amapá in 2014 and its emergence in Rio de Janeiro during 2016. Viruses 2018, 10, 623. [Google Scholar] [CrossRef] [Green Version]
- Lopes, N.; Nozawa, C.; Linhares, R.E.C. Características gerais e epidemiologia dos arbovírus emergentes no Brasil. Inst. Evandro Chagas Pará Rev. Pan-Amaz. Saude 2014, 5, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Bandeira, A.C.; Campos, G.S.; Sardi, S.I.; Rocha, V.F.; Rocha, G.C. Neonatal encephalitis due to Chikungunya vertical transmission: First report in Brazil. IDCases 2016, 5, 57–59. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.; Gerardin, P.; de Brito, C.A.A.; Soares, C.N.; Ferreira, M.L.B.; Solomon, T. The neurological complications of chikungunya virus: A systematic review. Rev. Med. Virol. 2018, 28, e1978. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, D.G.; di Maio Ferreira, F.C.P.A.; Hygino da Cruz, L.C.; Brasil, P.; Rueda Lopes, F.C. Longitudinal brain magnetic resonance imaging of children with perinatal Chikungunya encephalitis. Neuroradiol. J. 2020, 33, 532–537. [Google Scholar] [CrossRef]
- Lima, S.T.S.; Souza, W.M.; Cavalcante, J.W.; da Silva Candido, D.; Fumagalli, M.J.; Carrera, J.P.; Simões Mello, L.M.; de Carvalho Araújo, F.M.; Cavalcante Ramalho, I.L.; de Almeida Barreto, F.K.; et al. Fatal outcome of chikungunya virus infection in Brazil. Clin. Infect. Dis. 2020, ciaa1038. [Google Scholar] [CrossRef]
- Chatterjee, S.N.; Chakravarti, S.K.; Mitra, A.C.; Sarkar, J.K. Virological investigation of cases with neurological complications during the outbreak of haemorrhagic fever in Calcutta. J. Indian Med. Assoc. 1965, 4, 314–316. [Google Scholar]
- Thiruvengadam, K.V.; Kalyanasundaram, V.; Rajgopal, J. Clinical and pathological studies on chikungunya fever in Madras city. Indian J. Med. Res. 1965, 53, 729–744. [Google Scholar] [PubMed]
- Couderc, T.; Chrétien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraisier, C.; Koraka, P.; Belghazi, M.; Bakli, M.; Granjeaud, S.; Pophillat, M.; Lim, S.M.; Osterhaus, A.; Martina, B.; Camoin, L.; et al. Kinetic analysis of mouse brain proteome alterations following Chikungunya virus infection before and after appearance of clinical symptoms. PLoS ONE 2014, 9, e91397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passoni, G.; Langevin, C.; Palha, N.; Mounce, B.C.; Briolat, V.; Affaticati, P.; De Job, E.; Joly, J.S.; Vignuzzi, M.; Saleh, M.C.; et al. Imaging of viral neuroinvasion in the zebrafish reveals that Sindbis and chikungunya viruses favour different entry routes. Dis. Models Mech. 2017, 10, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Economopoulou, A.; Dominguez, M.; Helynck, B.; Sissoko, D.; Wichmann, O.; Quenel, P.; Germonneau, P.; Quatresous, I. Atypical Chikungunya virus infections: Clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Réunion. Epidemiol. Infect. 2009, 137, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandale, B.V.; Sathe, P.S.; Arankalle, V.A.; Wadia, R.S.; Kulkarni, R.; Shah, S.V.; Shah, S.K.; Sheth, J.K.; Sudeep, A.B.; Tripathy, A.S.; et al. Systemic involvements and fatalities during Chikungunya epidemic in India, 2006. J. Clin. Virol. 2009, 46, 145–149. [Google Scholar] [CrossRef]
- Kashyap, R.S.; Morey, S.; Bhullar, S.; Baheti, N.; Chandak, N.; Purohit, H.; Taori, G.; Daginawala, H. Determination of Toll-like receptor-induced cytokine profiles in the blood and cerebrospinal fluid of Chikungunya patients. Neuroimmunomodulation 2014, 21, 338–346. [Google Scholar] [CrossRef]
- Azevedo, M.B.; Coutinho, M.S.C.; Silva, M.A.D.; Arduini, D.B.; Lima, J.D.V.; Monteiro, R.; Mendes, B.N.B.; Lemos, M.C.F.; Noronha, C.P.; Saraceni, V. Neurologic manifestations in emerging arboviral diseases in Rio de Janeiro City, Brazil, 2015–2016. Rev. Soc. Bras. Med. Trop. 2018, 51, 347–351. [Google Scholar] [CrossRef]
- Martins, H.A.; Bernardino, S.N.; Santos, C.C.; Ribas, V.R. Chikungunya and myositis: A case report in Brazil. J. Clin. Diagn. Res. 2016, 10, OD05–OD06. [Google Scholar] [CrossRef]
- Rocha, V.F.D.; de Oliveira, A.H.P.; Bandeira, A.C.; Sardi, S.I.; Garcia, R.F.; Magalhães, S.A.; Sampaio, C.A.; Campos Soares, G. Chikungunya virus infection associated with encephalitis and anterior uveitis. Ocul. Immunol. Inflamm. 2018, 26, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Sá, P.K.O.; Nunes, M.M.; Leite, I.R.; Campelo, M.D.G.L.; Leão, C.F.R.; Souza, J.R.; Castellano, L.R.; Fernandes, A.I.V. Chikungunya virus infection with severe neurologic manifestations: Report of four fatal cases. Rev. Soc. Bras. Med. Trop. 2017, 50, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Brito Ferreira, M.L.; Militão de Albuquerque, M.F.P.; de Brito, C.A.A.; de Oliveira França, R.F.; Porto Moreira, Á.; de Morais Machado, M.; da Paz Melo, R.; Medialdea-Carrera, R.; Dornelas Mesquita, S.; Lopes Santos, M.; et al. Neurological disease in adults with Zika and chikungunya virus infection in Northeast Brazil: A prospective observational study. Lancet Neurol. 2020, 19, 826–839. [Google Scholar] [CrossRef]
- Soares, D.S.; Fortaleza, L.Y.; Melo, M.C. Chikungunya-induced manic episode in a patient with no psychiatric history: A case report. Braz. J. Psychiatry 2020, 42, 687. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.P.; Villas-Bôas, R.; Scott, S.S.O.; Nóbrega, P.R.; Sobreira-Neto, M.A.; Castro, J.D.V.; Cavalcante, B.; Braga-Neto, P. Encephalitis associated with the chikungunya epidemic outbreak in Brazil: Report of 2 cases with neuroimaging findings. Rev. Soc. Bras. Med. Trop. 2017, 50, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puccioni-Sohler, M.; Farias, L.C.; Cabral-Castro, M.J.; Zalis, M.G.; Kalil, R.S.; Salgado, M.C.F. Cerebrospinal fluid immunoglobulins as potential biomarkers of Chikungunya encephalitis. Emerg. Infect. Dis. 2018, 24, 939–941. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.S.O.; Braga-Neto, P.; Pereira, L.P.; Nóbrega, P.R.; de Assis Aquino Gondim, F.; Sobreira-Neto, M.A.; Schiavon, C.C.M. Immunoglobulin-responsive chikungunya encephalitis: Two case reports. J. Neurovirol. 2017, 23, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.M.; Santos, N.C.; Martins, I.C. Dengue and Zika viruses: Epidemiological history, potential therapies, and promising vaccines. Trop. Med. Infect. Dis. 2020, 5, 150. [Google Scholar] [CrossRef]
- Alice, F. Infecção humana pelo vírus “leste” da encefalite equina. Bol. Inst. Biol. Bahia 1956, 3, 3–9. [Google Scholar]
- Casseb, A.R.; Brito, T.C.; Silva, M.R.M.; Chiang, J.O.; Martins, L.C.; Silva, S.P.; Henriques, D.F.; Casseb, L.M.N.; Vasconcelos, P.F.C. Prevalence of antibodies to equine alphaviruses in the State of Pará, Brazil. Arq. Inst. Biol. 2016, 83, e0202014. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, V.G.; de Rezende Féres, V.C.; Saivish, M.V.; de Lima Gimaque, J.B.; Moreli, M.L. Silent emergence of Mayaro and Oropouche viruses in humans in Central Brazil. Int. J. Infect. Dis. 2017, 62, 84–85. [Google Scholar] [CrossRef] [Green Version]
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kawada, J.I. Neurological disorders associated with human alphaherpesviruses. Adv. Exp. Med. Biol. 2018, 1045, 85–102. [Google Scholar] [PubMed]
- Gnann, J.W., Jr.; Whitley, R.J. Herpes simplex encephalitis: An update. Curr Infect Dis Rep. 2017, 19, 13. [Google Scholar] [CrossRef]
- Levitz, R.E. Herpes simplex encephalitis: A review. Heart Lung 1998, 27, 209–212. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex virus infections of the central nervous system. Continuum (Minneap Minn) 2015, 21, 1704–1713. [Google Scholar] [CrossRef]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes simplex virus type 1 infection of the central nervous system: Insights into proposed interrelationships with neurodegenerative disorders. Front. Cell. Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Nagel, M.A.; Niemeyer, C.S.; Bubak, A.N. Central nervous system infections produced by varicella zoster virus. Curr. Opin. Infect. Dis. 2020, 33, 273–278. [Google Scholar] [CrossRef]
- Mendoza, L.P.; Bronzoni, R.V.; Takayanagui, O.M.; Aquino, V.H.; Figueiredo, L.T. Viral infections of the central nervous system in Brazil. J. Infect. 2007, 54, 589–596. [Google Scholar] [CrossRef]
- Pfaff, F.; Groth, M.; Sauerbrei, A.; Zell, R. Genotyping of herpes simplex virus type 1 by whole-genome sequencing. J. Gen. Virol. 2016, 97, 2732–2741. [Google Scholar] [CrossRef]
- Kolb, A.W.; Adams, M.; Cabot, E.L.; Craven, M.; Brandt, C.R. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: Phylogeny, sequence variability, and SNP distribution. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9061–9073. [Google Scholar] [CrossRef] [Green Version]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [Green Version]
- Parsons, L.R.; Tafuri, Y.R.; Shreve, J.T.; Bowen, C.D.; Shipley, M.M.; Enquist, L.W.; Szpara, M.L. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. mBio 2015, 6, e02213-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, R.M.; Lamers, S.L.; Weiner, B.; Ray, S.C.; Colgrove, R.C.; Diaz, F.; Jing, L.; Wang, K.; Saif, S.; Young, S.; et al. Genome sequencing and analysis of geographically diverse clinical isolates of herpes simplex virus 2. J. Virol. 2015, 89, 8219–8232. [Google Scholar] [CrossRef] [Green Version]
- Zell, R.; Taudien, S.; Pfaff, F.; Wutzler, P.; Platzer, M.; Sauerbrei, A. Sequencing of 21 varicella-zoster virus genomes reveals two novel genotypes and evidence of recombination. J. Virol. 2012, 86, 1608–1622. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.W.; Ané, C.; Brandt, C.R. Using HSV-1 genome phylogenetics to track past human migrations. PLoS ONE 2013, 8, e76267. [Google Scholar] [CrossRef] [Green Version]
- Piret, J.; Boivin, G. Antiviral resistance in herpes simplex virus and varicella-zoster virus infections: Diagnosis and management. Curr. Opin. Infect. Dis. 2016, 29, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Whitley, R.J.; Stone, E.F.; Mohan, K. Difference between herpes simplex virus type 1 and type 2 neonatal encephalitis in neurological outcome. Lancet 1988, 1, 1–4. [Google Scholar] [CrossRef]
- Raschilas, F.; Wolf, M.; Delatour, F.; Chaffaut, C.; De Broucker, T.; Chevret, S.; Lebon, P.; Canton, P.; Rozenberg, F. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: Results of a multicenter study. Clin. Infect. Dis. 2002, 35, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Michael, B.D.; Smith, P.E.; Sanderson, F.; Davies, N.W.; Hart, I.J.; Holland, M.; Easton, A.; Buckley, C.; Kneen, R.; et al. Management of suspected viral encephalitis in adults—Association of British Neurologists and British Infection Association National Guidelines. J. Infect. 2012, 64, 347–373. [Google Scholar] [CrossRef] [PubMed]
- Schleede, L.; Bueter, W.; Baumgartner-Sigl, S.; Opladen, T.; Weigt-Usinger, K.; Stephan, S.; Smitka, M.; Leiz, S.; Kaiser, O.; Kraus, V.; et al. Pediatric herpes simplex virus encephalitis: A retrospective multicenter experience. J. Child Neurol. 2013, 28, 321–331. [Google Scholar] [CrossRef]
- Aurelius, E.; Johansson, B.; Skoldenberg, B.; Staland, A.; Forsgren, M. Rapid diagnosis of herpes simplex encephalitis by nested polymerase chain reaction assay of cerebrospinal fluid. Lancet 1991, 337, 189–192. [Google Scholar] [CrossRef]
- Bhullar, S.S.; Chandak, N.H.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F.; Kashyap, R.S. Determination of viral load by quantitative real-time PCR in herpes simplex encephalitis patients. Intervirology 2014, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Poissy, J.; Champenois, K.; Dewilde, A.; Melliez, H.; Georges, H.; Senneville, E.; Yazdanpanah, Y. Impact of herpes sim- plex virus load and red blood cells in cerebrospinal fluid upon herpes simplex meningo-encephalitis outcome. BMC Infect. Dis. 2012, 12, 356. [Google Scholar] [CrossRef]
- Bhullar, S.S.; Chandak, N.H.; Baheti, N.N.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F.; Kashyap, R.S. Diagnosis of herpes simplex encephalitis by ELISA using antipeptide antibodies against type-common epitopes of glycoprotein B of herpes simplex viruses. J. Immunoass. Immunochem. 2016, 37, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Skoldenberg, B.; Forsgren, M.; Alestig, K.; Forkman, A.; Lövgren, K.; Norrby, R.; Stiernstedt, G.; Forsgren, M.; Bergström, T.; Dahlqvist, E.; et al. Acyclovir versus vidarabine in herpes simplex encephalitis. Randomised multicentre study in consecutive Swedish patients. Lancet 1984, 2, 707–711. [Google Scholar] [CrossRef]
- Whitley, R.J.; Alford, C.A.; Hirsch, M.S.; Schooley, R.T.; Luby, J.P.; Aoki, F.Y.; Hanley, D.; Nahmias, A.J.; Soong, S.-J.; The NIAID Collaborative Antiviral Study Group. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N. Engl. J. Med. 1986, 314, 144–149. [Google Scholar] [CrossRef]
- Elbers, J.M.; Bitnun, A.; Richardson, S.E.; Ford-Jones, E.L.; Tellier, R.; Wald, R.M.; Petric, M.; Kolski, H.; Heurter, H.; MacGregor, D. A 12-year prospective study of childhood herpes simplex encephalitis: Is there a broader spectrum of disease? Pediatrics 2007, 119, e399–e407. [Google Scholar] [CrossRef]
- Aksamit, A.J., Jr. Treatment of viral encephalitis. Neurol. Clin. 2021, 39, 197–207. [Google Scholar] [CrossRef]
- Dioverti, M.V.; Razonable, R.R. Cytomegalovirus. Microbiol. Spectr. 2016, 4, 1–26. [Google Scholar] [CrossRef]
- Davis, N.L.; King, C.C.; Kourtis, A.P. Cytomegalovirus infection in pregnancy. Birth Defects Res. 2017, 109, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Bowen, L.N.; Smith, B.; Reich, D.; Quezado, M.; Nath, A. HIV-associated opportunistic CNS infections: Pathophysiology, diagnosis and treatment. Nat. Rev. Neurol. 2016, 12, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.R.; Patel, E.U.; Abraham, A.G.; Quinn, T.C.; Tobian, A.A.R. Changes in cytomegalovirus seroprevalence among U.S. children aged 1 to 5 years: The national health and nutrition examination surveys. Clin. Infect. Dis. 2020, 10, ciaa1168. [Google Scholar] [CrossRef] [PubMed]
- Chimelli, L.; Rosemberg, S.; Hahn, M.D.; Lopes, M.B.; Netto, M.B. Pathology of the central nervous system in patients infected with the human immunodeficiency virus (HIV): A report of 252 autopsy cases from Brazil. Neuropathol. Appl. Neurobiol. 1992, 18, 478–488. [Google Scholar] [CrossRef]
- Vilas Boas, L.S.; de Souza, V.A.; Penalva de Oliveira, A.C.; Rodriguez Viso, A.T.; Nascimento Filho, A.M.; Nascimento, M.C.; Pannuti, C.S. Cytomegalovirus glycoprotein B genotypes and central nervous system disease in AIDS patients. J. Med. Virol. 2003, 71, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carreras, J.; Maes, P. Human cytomegalovirus genomics and transcriptomics through the lens of next-generation sequencing: Revision and future challenges. Virus Genes 2019, 55, 138–164. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Pati, P.; Jensen, T.L.; Goll, J.B.; Gelber, C.E.; Singh, A.; McNeal, M.; Boppana, S.B.; Bernstein, D.I. Cytomegalovirus genetic diversity following primary infection. J. Infect. Dis. 2020, 221, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Barrado, L.; Prieto, C.; Hernando, S.; Folgueira, L. Detection of glycoproteins B and H geno- types to predict the development of Cytomegalovirus disease in solid organ transplant recipients. J. Clin. Virol. 2018, 109, 50–56. [Google Scholar] [CrossRef]
- Hu, H.; Peng, W.; Peng, Q.; Cheng, Y. Cytomegalovirus genotype distribution among congenital and perinatal infected patients with HCMV-associated thrombocytopenia. Fetal Pediatric Pathol. 2020, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human cytomegalovirus genomes sequenced directly from clinical material: Variation, multiple-strain infection, recombination, and gene loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüttmann, S.; Husstedt, I.W.; Lügering, N.; Heese, C.; Stoll, R.; Domschke, W.; Evers, S.; Kuchelmeister, K.; Gullotta, F. Cytomegalovirus encephalomyelomeningoradiculitis in acquired immunodeficiency syndrome (AIDS). J. Infect. 1997, 35, 78–81. [Google Scholar] [CrossRef]
- Kawasaki, H.; Kosugi, I.; Meguro, S.; Iwashita, T. Pathogenesis of developmental anomalies of the central nervous system induced by congenital cytomegalovirus infection. Pathol. Int. 2017, 67, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Maschke, M.; Kastrup, O.; Diener, H.C. CNS manifestations of cytomegalovirus infections: Diagnosis and treatment. CNS Drugs 2002, 16, 303–315. [Google Scholar]
- Bookstaver, P.B.; Mohorn, P.L.; Shah, A.; Tesh, L.D.; Quidley, A.M.; Kothari, R.; Bland, C.M.; Weissman, S. Management of viral central nervous system infections: A primer for clinicians. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517703342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Zabel, M.D.; Reid, C. A brief history of prions. Pathog. Dis. 2015, 73, ftv087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Mahmood, S. An overview of animal prion diseases. Virol. J. 2011, 8, 493. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, T.; Sakai, K.; Kobayashi, A.; Kitamoto, T.; Ae, R.; Nakamura, Y.; Sanjo, N.; Arai, K.; Koide, M.; Katada, F.; et al. Characterization of sporadic Creutzfeldt-Jakob disease and history of neurosurgery to identify potential iatrogenic cases. Emerg. Infect. Dis. 2020, 26, 1140–1146. [Google Scholar] [CrossRef]
- Brown, P.; Brandel, J.-P.; Sato, T.; Nakamura, Y.; MacKenzie, J.; Will, R.G.; Ladogana, A.; Pocchiari, M.; Leschek, E.W.; Schonberger, L.B. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg. Infect. Dis. 2012, 18, 901–907. [Google Scholar] [CrossRef]
- Collinge, J.; Whitfield, J.; McKintosh, E.; Beck, J.; Mead, S.; Thomas, D.J.; Alpers, M.P. Kuru in the 21st century—An acquired human prion disease with very long incubation periods. Lancet 2006, 367, 2068–2074. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Sisó, S.; González, L.; Jeffrey, M. Neuroinvasion in prion diseases: The roles of ascending neural infection and blood dissemination. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schätzl, H.M.; da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef]
- Silva, J.L.; Gomes, M.P.B.M.P.B.; Vieira, T.C.R.G.; Cordeiro, Y. PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. 2010, 15, 132–150. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, I.; Udgaonkar, J.B. Structural mechanisms of oligomer and amyloid fibril formation by the prion protein. Chem. Commun. 2018, 54, 6230–6242. [Google Scholar] [CrossRef]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [Green Version]
- Taneja, V.; Verma, M.; Vats, A. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138. [Google Scholar] [CrossRef]
- Solforosi, L.; Milani, M.; Mancini, N.; Clementi, M.; Burioni, R. A closer look at prion strains. Prion 2013, 7, 99–108. [Google Scholar] [CrossRef]
- Rossi, M.; Baiardi, S.; Parchi, P. Understanding prion strains: Evidence from studies of the disease forms affecting humans. Viruses 2019, 11, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, M.; Kitamoto, T.; Doh-Ura, K.; Iwaki, T.; Tateishi, J. Creutzfeldt-Jakob disease with codon 129 polymorphism (Valine): A comparative study of patients with codon 102 point mutation or without mutations. Acta Neuropathol. 1992, 84, 349–354. [Google Scholar] [CrossRef]
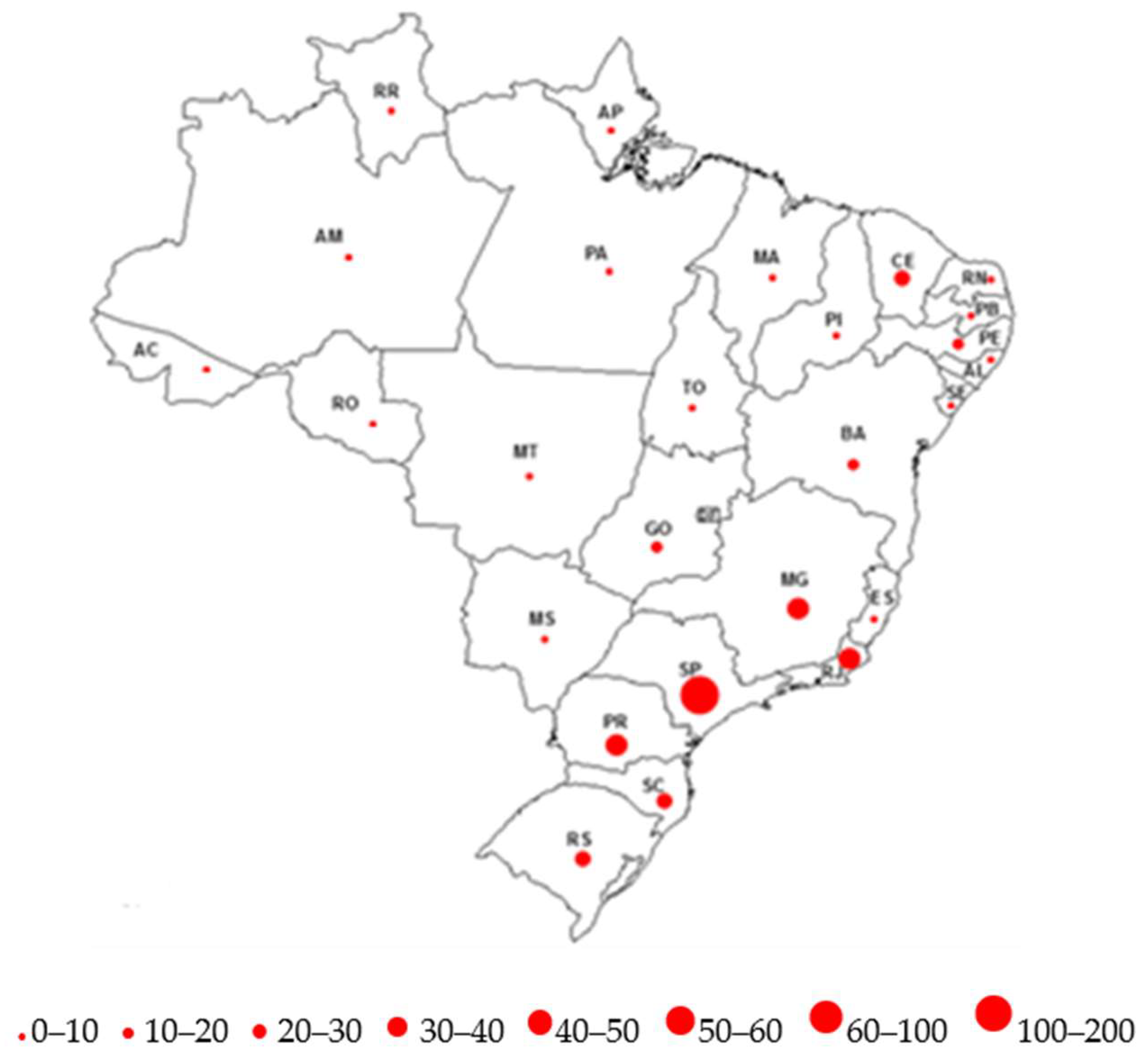
- Gattás, V.L.; Lima Neto, A.S.; Dimech, G.S.; Mancini, D.; Cantarino, L.M.; Marins, J.R.P.; Luna, E.J.A. New variant of Creutzfeldt-Jakob (vCJD) disease and other human prion diseases under epidemiological surveillance in Brazil. Dement. Neuropsychol. 2007, 1, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, J.E.G. DCJ EpiShiny. Available online: https://epicjd.shinyapps.io/dcjBRASIL/ (accessed on 2 January 2021).
- De Paula, E.V.; Addas-Carvalho, M.; Costa, D.S.P.; Saad, S.T.O.; Gilli, S.C.O. Genotype frequencies at codon 129 of the Prion Protein Gene in Brazil: Implications in susceptibility to variant Creutzfeldt-Jakob disease compared to European and Asian populations. Eur. J. Epidemiol. 2005, 20, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Nitrini, R.; Rosemberg, S.; Passos-Bueno, M.R.; Teixeira Da Silva, L.S.; Iughetti, P.; Papadopoulos, M.; Carrilho, P.M.; Caramelli, P.; Albrecht, S.; Zatz, M.; et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann. Neurol. 1997, 42, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Marie, S.K.N.; Kok, F.; Nitrini, R. Familial Creutzfeldt-Jakob disease associated with a point mutation at codon 210 of the prion protein gene. Arq. Neuropsiquiatr. 2001, 59, 932–935. [Google Scholar] [CrossRef] [Green Version]
- Smid, J.; Martins, V.R.; Landemberger, M.C.; Riva, D.; Anghinah, R.; Nitrini, R. Creutzfeldt-Jakob disease associated with a missense mutation at codon 200 of the prion protein gene in Brazil. Dement. Neuropsychol. 2007, 1, 222–224. [Google Scholar] [CrossRef] [Green Version]
- De Souza, R.K.M.; Josviak, N.D.; Batistela, M.S.; Santos, P.S.F.; Landemberger, M.C.; Ramina, R. First case of V180I rare mutation in a Brazilian patient with Creutzfeldt-Jakob disease. Prion 2017, 11, 465–468. [Google Scholar] [CrossRef]
- Ferreira Caboclo, L.O.S.; Huang, N.; Lepski, G.A.; Livramento, J.A.; Buchpiguel, C.A.; Porto, C.S.; Nitrini, R. Iatrogenic Creutzfeldt-Jakob disease following human growth hormone therapy: Case report. Arq. Neuropsiquiatr. 2002, 60, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Macario, M.E.; Moura-Neto, V.; Vaisman, M.; Araujo, H.M.M.; Buescu, A.; Cordeiro, J.G.H.; Chagas, C. Abnormal proteins in the cerebrospinal fluid of a patient with Creutzfeldt-Jakob disease following administration of human pituitary growth hormone. Braz. J. Med. Biol. Res. 1992, 25, 1127–1130. [Google Scholar]
- Smid, J.; Neto, A.S.; Landemberger, M.C.; Machado, C.F.; Nóbrega, P.R.; Canedo, N.H.S.; Schultz, R.R.; Naslavsky, M.S.; Rosemberg, S.; Kok, F.; et al. High phenotypic variability in gerstmann-sträussler-scheinker disease. Arq. Neuropsiquiatr. 2017, 75, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, D.C.; Gonçalves Filho, A.L.d.M.; Pacheco, F.T.; Barros, B.R.; Littig, I.A.; Nunes, R.H.; Maia Júnior, A.C.M.; da Rocha, A.J. Imaging of Creutzfeldt-Jakob disease: Imaging patterns and their differential diagnosis. Radiographics 2017, 37(1), 234–257. [Google Scholar] [CrossRef] [Green Version]
- Ascari, L.M.; Rocha, S.C.; Gonçalves, P.B.; Vieira, T.C.R.G.; Cordeiro, Y. Challenges and advances in antemortem diagnosis of human transmissible spongiform encephalopathies. Front. Bioeng. Biotechnol. 2020, 8, 585896. [Google Scholar] [CrossRef] [PubMed]
- CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD). Available online: https://www.cdc.gov/prions/cjd/diagnostic-criteria.html (accessed on 2 January 2021).
- Gibson, L.M.; Chappell, F.M.; Summers, D.; Collie, D.A.; Sellar, R.; Best, J.; Knight, R.; Ironside, J.W.; Wardlaw, J.M. Post-mortem magnetic resonance imaging in patients with suspected prion disease: Pathological confirmation, sensitivity, specificity and observer reliability. A national registry. PLoS ONE 2018, 13, e0201434. [Google Scholar] [CrossRef] [Green Version]
- Fiorini, M.; Iselle, G.; Perra, D.; Bongianni, M.; Capaldi, S.; Sacchetto, L.; Ferrari, S.; Mombello, A.; Vascellari, S.; Testi, S.; et al. High diagnostic accuracy of RT-QuIC assay in a prospective study of patients with suspected sCJD. Int. J. Mol. Sci. 2020, 21, 880. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Brunk, C.; Budka, H.; Cervenakova, L.; Collie, D.; Green, A.; Ironside, J.; Knight, R.; MacKenzie, J.; Pergami, P.; et al. WHO Manual for Surveillance of Human Transmissible Spongiform Encephalopathies, Including Variant Creutzfeldt-Jakob Disease; World Health Organization: Geneva, Switzerland, 2003; ISBN 9241545887. [Google Scholar]
- Barbosa, B.J.A.P.; Castrillo, B.B.; Alvim, R.P.; de Brito, M.H.; Gomes, H.R.; Brucki, S.M.D.; Smid, J.; Nitrini, R.; Landemberger, M.C.; Martins, V.R.; et al. Second-generation RT-QuIC assay for the diagnosis of Creutzfeldt-Jakob disease patients in Brazil. Front. Bioeng. Biotechnol. 2020, 8, 929. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Tagliavini, F. Clinical trials. Handb. Clin. Neurol. 2018, 153, 431–444. [Google Scholar]
- Raymond, G.J.; Zhao, H.T.; Race, B.; Raymond, L.D.; Williams, K.; Swayze, E.E.; Graffam, S.; Le, J.; Caron, T.; Stathopoulos, J.; et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight 2019, 5, e131175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Ma, J. Immunotherapy against prion disease. Pathogens 2020, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Gao, X.Q.; Yang, C.X.; Tan, S.K.; Sun, Z.L.; Yan, N.H.; Pang, Y.G.; Yuan, M.; Chen, G.J.; Xu, G.T.; et al. Neuroprotective effect of grafting GDNF gene-modified neural stem cells on cerebral ischemia in rats. Brain Res. 2009, 1284, 1–11. [Google Scholar] [CrossRef]
- Frid, K.; Binyamin, O.; Fainstein, N.; Keller, G.; Ben-Hur, T.; Gabizon, R. Autologous neural progenitor cell transplantation into newborn mice modeling for E200K genetic prion disease delays disease progression. Neurobiol. Aging 2018, 65, 192–200. [Google Scholar] [CrossRef]
- Relaño-Ginès, A.; Gabelle, A.; Hamela, C.; Belondrade, M.; Casanova, D.; Mourton-Gilles, C.; Lehmann, S.; Crozet, C. Prion replication occurs in endogenous adult neural stem cells and alters their neuronal fate: Involvement of endogenous neural stem cells in prion diseases. PLoS Pathog. 2013, 9, e1003485. [Google Scholar] [CrossRef] [PubMed]
- Relaño-Ginés, A.; Lehmann, S.; Bencsik, A.; Herva, M.E.; Torres, J.M.; Crozet, C.A. Stem cell therapy extends incubation and survival time in prion-infected mice in a time window-dependant manner. J. Infect. Dis. 2011, 204, 1038–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavarpour-Bali, H.; Ghasemi-Kasman, M. Update on neurological manifestations of COVID-19. Life Sci. 2020, 257, 118063. [Google Scholar] [CrossRef]
- Espíndola, O.M.; Brandão, C.O.; Gomes, Y.C.P.; Siqueira, M.; Soares, C.N.; Lima, M.A.S.D.; Leite, A.C.C.B.; Torezani, G.; Araujo, A.Q.C.; Silva, M.T.T. Cerebrospinal fluid findings in neurological diseases associated with COVID-19 and insights into mechanisms of disease development. Int. J. Infect. Dis. 2021, 102, 155–162. [Google Scholar] [CrossRef]
- Domingues, R.B.; Mendes-Correa, M.C.; de Moura Leite, F.B.V.; Sabino, E.C.; Salarini, D.Z.; Claro, I.; Santos, D.W.; de Jesus, J.G.; Ferreira, N.E.; Romano, C.M.; et al. First case of SARS-COV-2 sequencing in cerebrospinal fluid of a patient with suspected demyelinating disease. J. Neurol. 2020, 267, 3154–3156. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
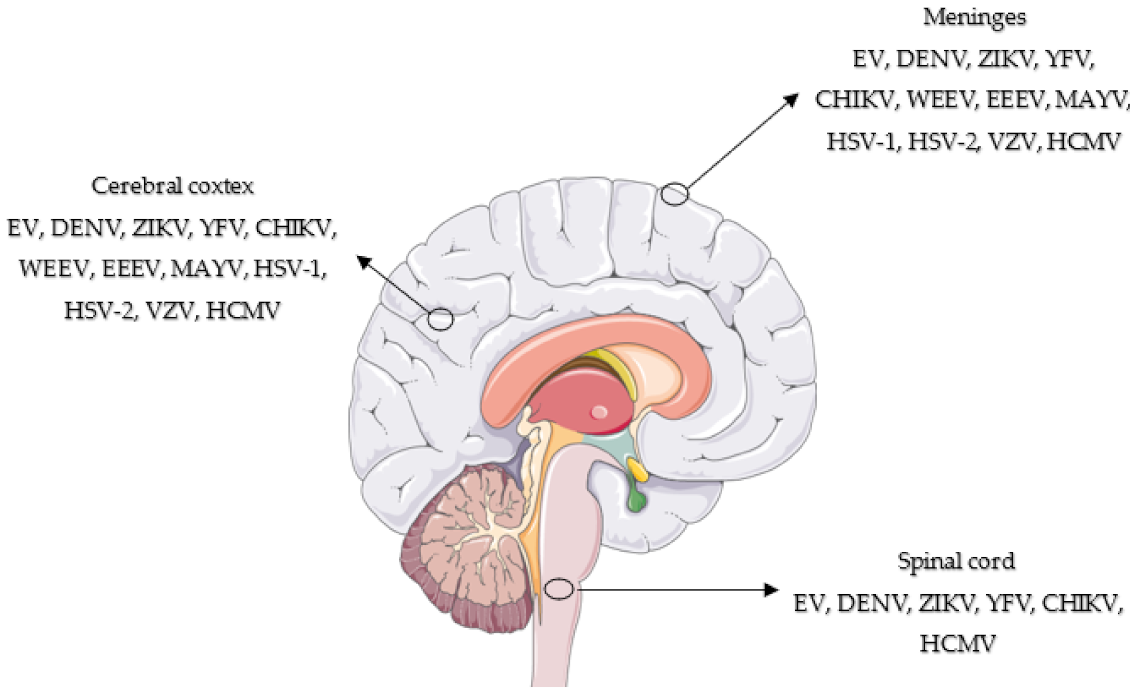
| Genus/Causative Agents | Structure | Transmission | Route to Brain | Neurological Disorders | Types Commonly CNS-Associated Infections |
|---|---|---|---|---|---|
| Enterovirus | Single-stranded RNA Nonenveloped Icosahedral symmetry | Fecal–oral, respiratory | Infected endothelial cells; retrograde axonal transport; Trojan Horse within infected monocytes | Aseptic meningitis, acute flaccid paralysis, encephalitis/meningoencephalitis | E6, E7, E11, E18, E30, CVB5, CVA2, EV-A71, CVB5, E6, E7, E11, CVA13, EV-C99, CVB2, E6, E18, E30 |
| Flavivirus | Single-stranded RNA Enveloped Icosahedral symmetry | Via arthropod vectors | Hematogenous; axonal transport; Trojan Horse within infected leukocytes | Encephalitis, microencephaly (Zika) | Dengue, Yellow fever, Zika |
| Alphavirus | Single-stranded RNA Enveloped Icosahedral symmetry | Via arthropod vectors | Anterograde axonal transport | Encephalitis | Chikungunya |
| Simplexvirus | Double-stranded DNA Enveloped Icosahedral symmetry | Oral-to-oral, oral–genital contact with sores | Primary infections: olfactory, hematogenous and genital (HSV-2), retrograde axonal transport Reactivation: from neurons trigeminal or sacral (HSV-2) ganglia, anterograde transport to reach CNS | Encephalitis, meningoencephalitis | HSV-1 HSV-2 |
| Varicellovirus | Double-stranded DNA Enveloped Icosahedral symmetry | Via droplets, aerosol, direct contact | Peripheral spread VZV- reactivation from sensory ganglia- central spread to brain or to temporal arteritis or to spinal cord arteries | Meningoencephalitis, myelitis, cranial neuropathies | VZV |
| Cytomegalovirus | Double-stranded DNA Enveloped Icosahedral symmetry | Contact with saliva or urine; sexual; contact breast milk; transplanted organs; blood transfusions | Hematopoietic cells monocytes—systemic spread- brain, spinal cord, meninges, nerve roots | Encephalitis, myelitis, polyradiculopathy, multifocal neuropathy | CMV |
| Prion scrapie | Beta structure enriched form of cellular prion protein | Iatrogenic, oral, blood | Infected epithelium, lymphoid tissues | Rapidly progressive cognitive impairment with behavioral and visual disturbances, ataxia, and myoclonus. | BSE, vCJD, iCJD |
| sCJD Diagnostic | Signals and Symptoms |
|---|---|
| Possible | Progressive dementia AND at least 2 out of the following 4 clinical features: Myoclonus, cerebellar or visual disturbance, pyramidal and extrapyramidal dysfunction, akinetic mutism; AND the absence of a positive result for any of the 4 tests that would classify a case as “probable”; AND duration of illness less than 2 years; AND without routine investigations indicating an alternative diagnosis. |
| Probable | Neuropsychiatric disorder plus positive RT-QuIC in cerebrospinal fluid (CSF) or other tissues OR rapidly progressive dementia; AND at least 2 out of the following 4 clinical features: Myoclonus, cerebellar or visual disorder, pyramidal and extrapyramidal dysfunction, akinetic mutism; AND a positive result on at least 1 of the following laboratory tests: A typical EEG (periodic sharp wave complexes) during an illness of any duration, a positive 14-3-3 CSF assay in patients with a disease duration of less than 2 years, high signal in caudate/putamen on magnetic resonance imaging (MRI) brain scan or at least 2 cortical regions (temporal, parietal, occipital) either on diffusion-weighted imaging (DWI) or fluid-attenuated inversion recovery (FLAIR); AND without routine investigations indicating an alternative diagnosis. |
| Definite | Neuropathological confirmation and/or confirmation of PrPres by immunocytochemistry or Western blot and/or presence of scrapie fibers. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, I.P., Jr.; dos Santos, F.B.; de Paula, V.S.; Vieira, T.C.R.G.; Dias, H.G.; Barros, C.A.; da Silva, E.E. Viral and Prion Infections Associated with Central Nervous System Syndromes in Brazil. Viruses 2021, 13, 1370. https://doi.org/10.3390/v13071370
Sousa IP Jr., dos Santos FB, de Paula VS, Vieira TCRG, Dias HG, Barros CA, da Silva EE. Viral and Prion Infections Associated with Central Nervous System Syndromes in Brazil. Viruses. 2021; 13(7):1370. https://doi.org/10.3390/v13071370
Chicago/Turabian StyleSousa, Ivanildo P., Jr., Flavia B. dos Santos, Vanessa S. de Paula, Tuane C.R.G. Vieira, Helver G. Dias, Caroline A. Barros, and Edson E. da Silva. 2021. "Viral and Prion Infections Associated with Central Nervous System Syndromes in Brazil" Viruses 13, no. 7: 1370. https://doi.org/10.3390/v13071370