
Emergence of a Novel Dengue Virus 3 (DENV-3) Genotype-I Coincident with Increased DENV-3 Cases in Yangon, Myanmar between 2017 and 2019

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Sample Collection
2.2. Serological Confirmation of DENV Infection Status
2.3. Virus Isolation, RNA Extraction, and DENV Serotyping by RT-PCR
2.4. Whole-Genome Sequencing
2.5. Phylogenetic Analysis and Amino Acid Variant Analysis
2.6. Sanger Sequencing
2.7. Quantification of DENV in Serum Samples
2.8. Statistical Analyses
3. Results
3.1. Clinicopathological Characteristics and Serostatus of Patients
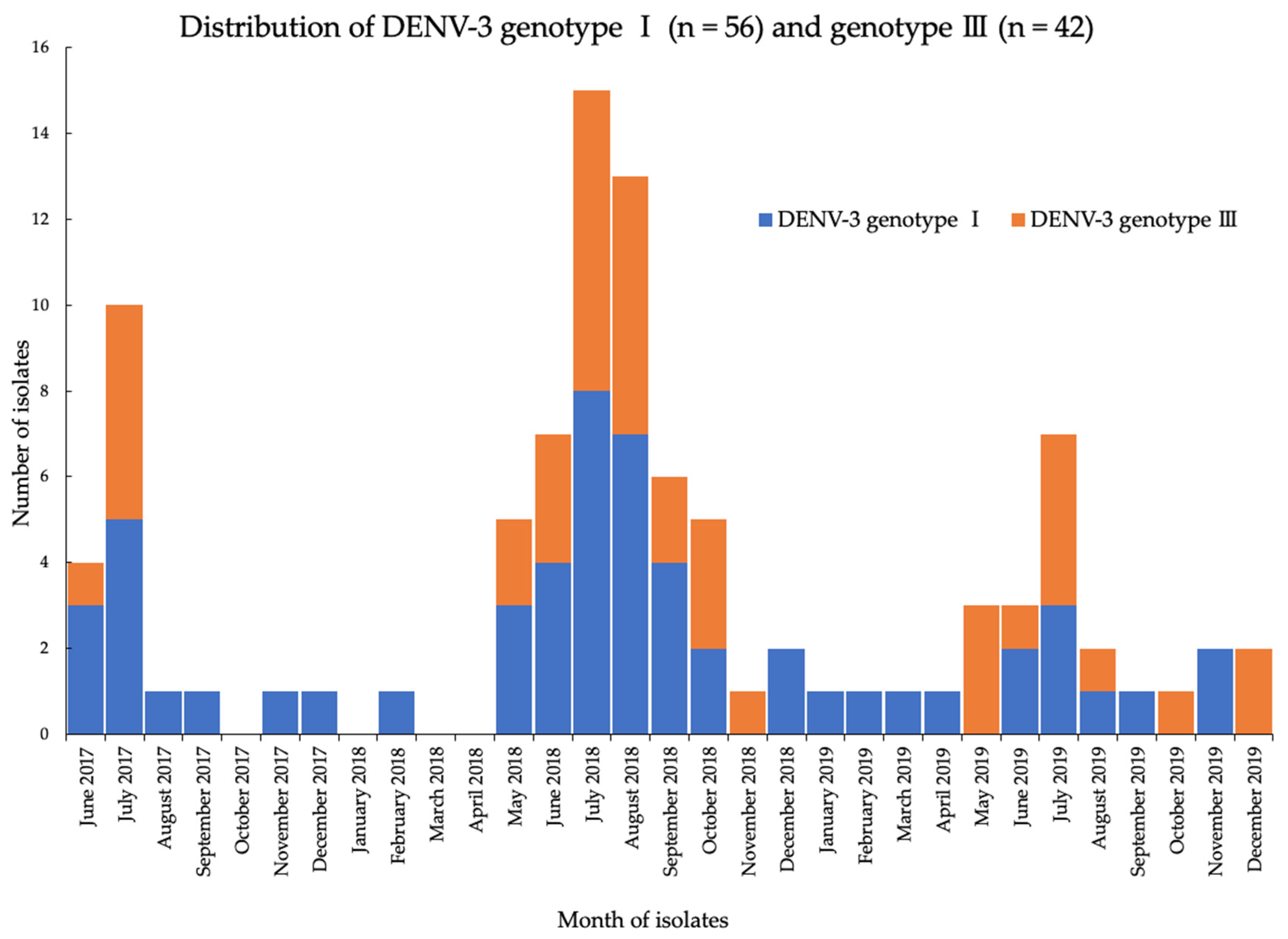
3.2. Distribution of Circulating DENV Serotypes
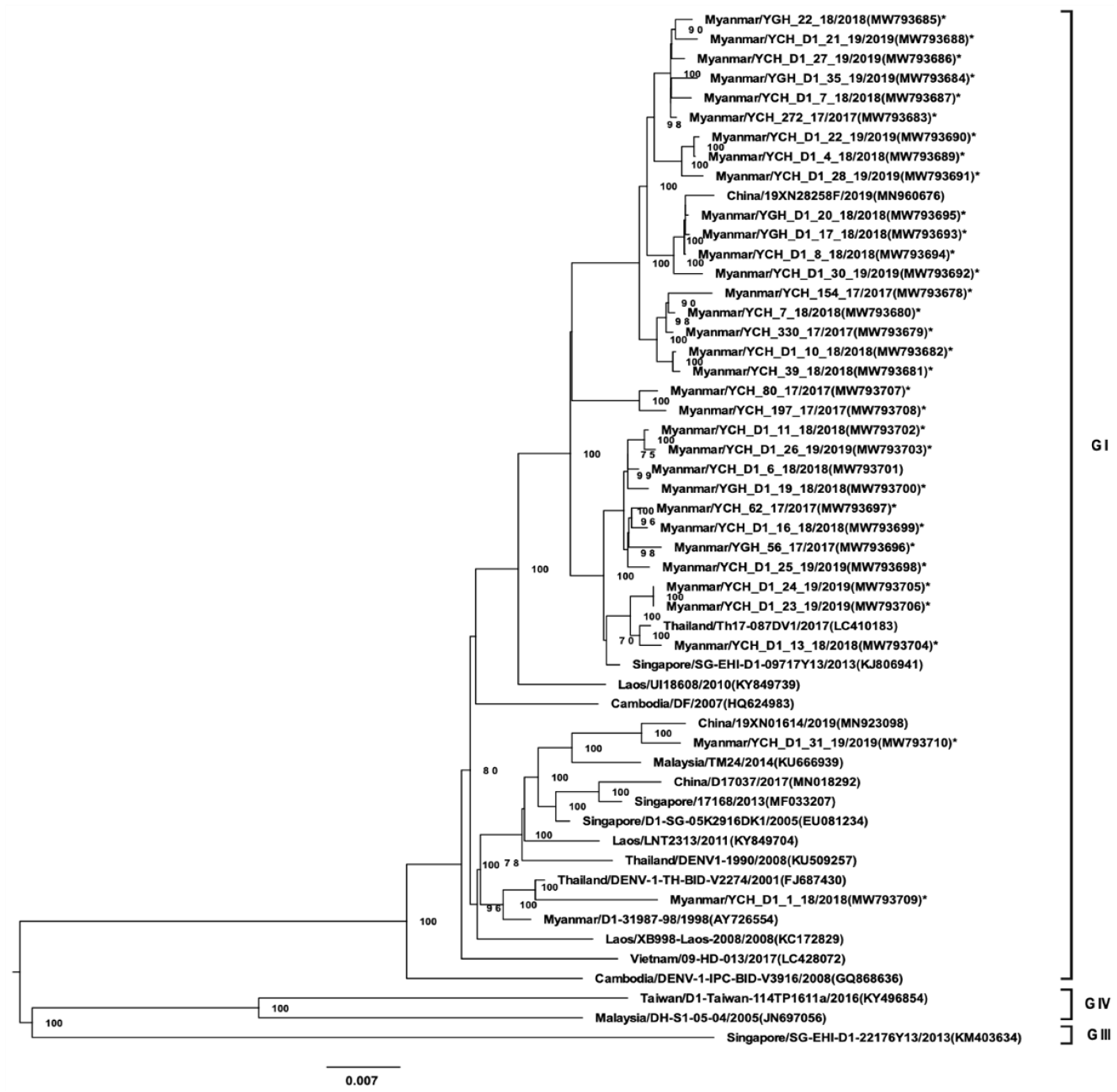
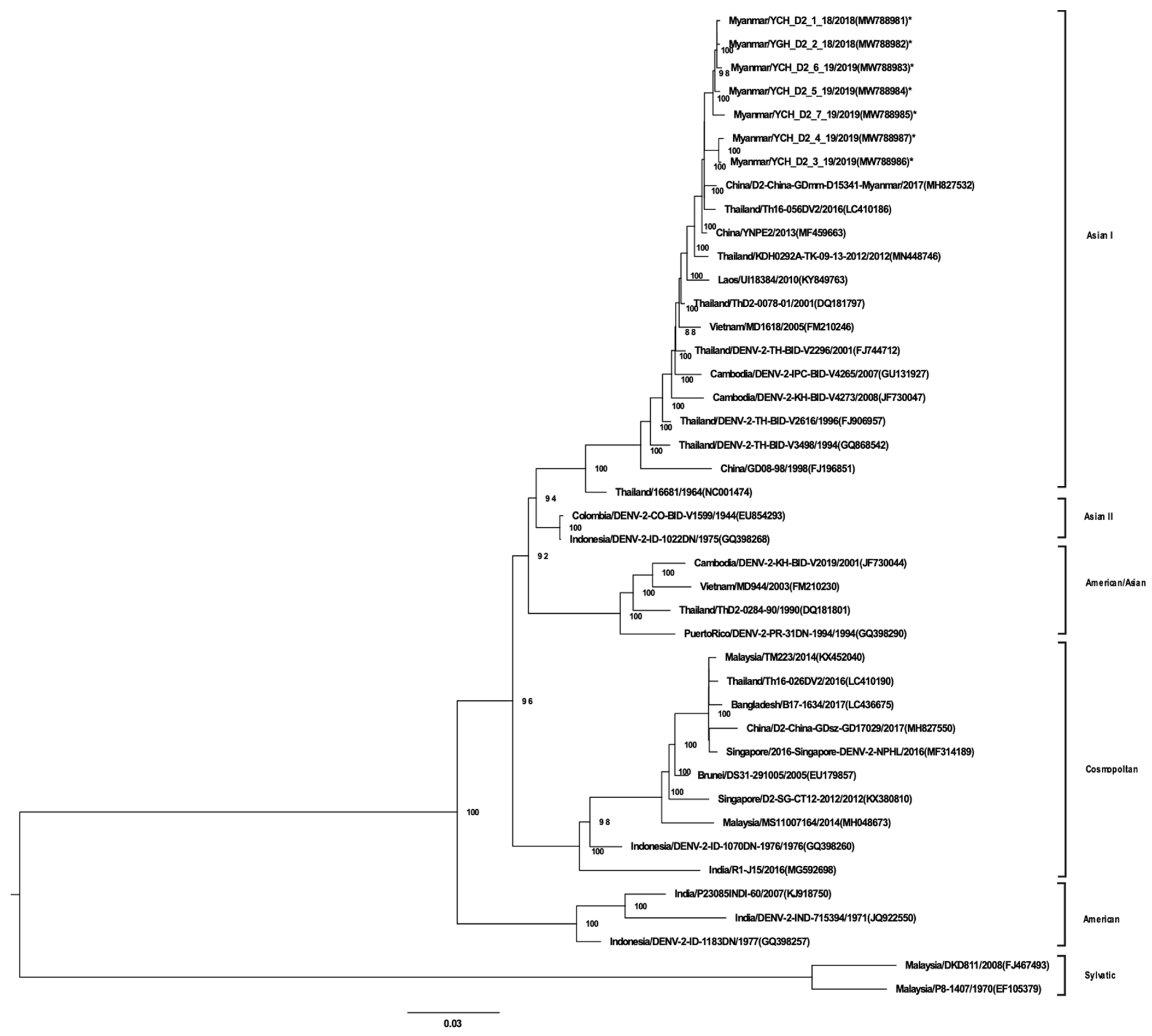
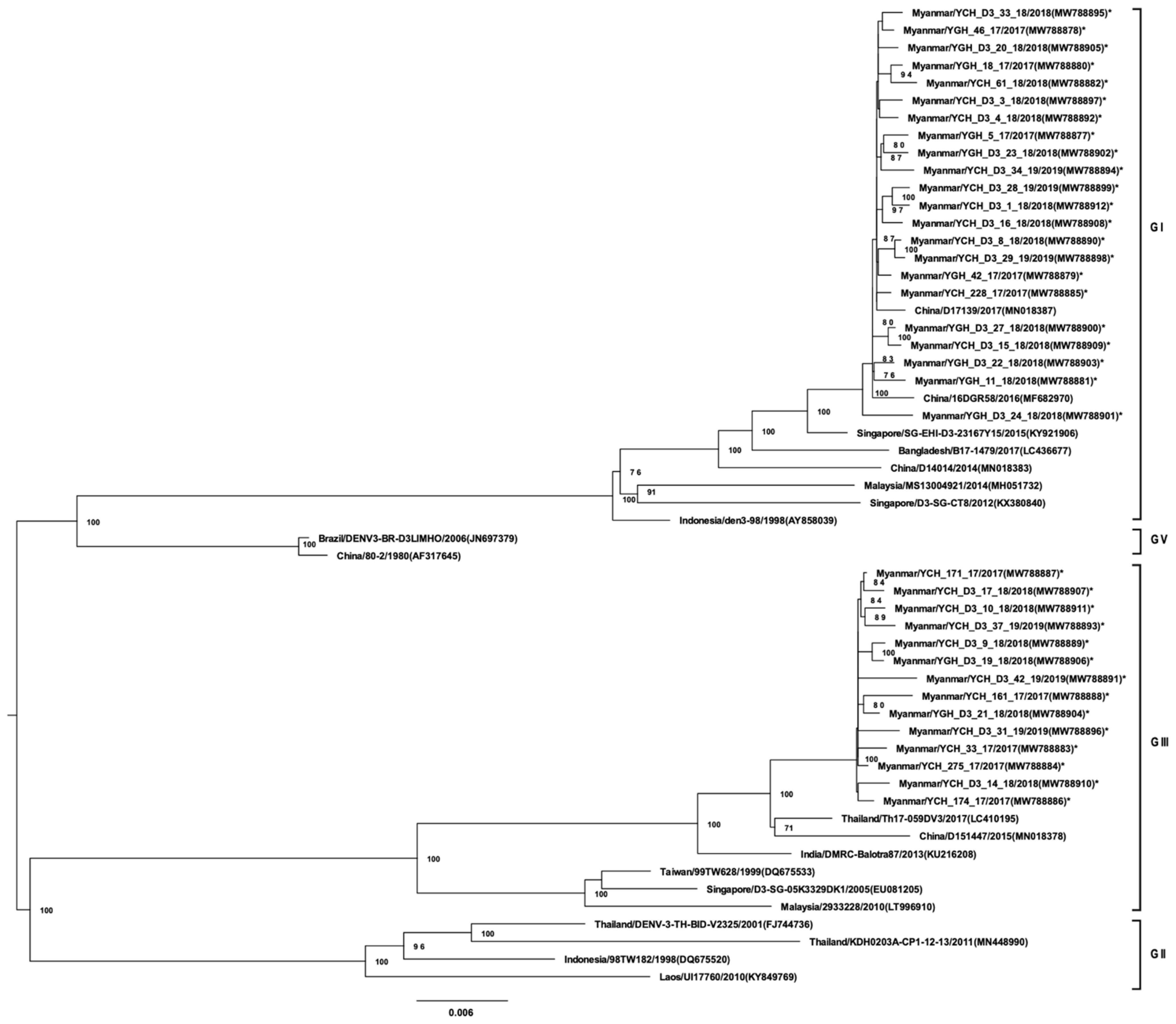
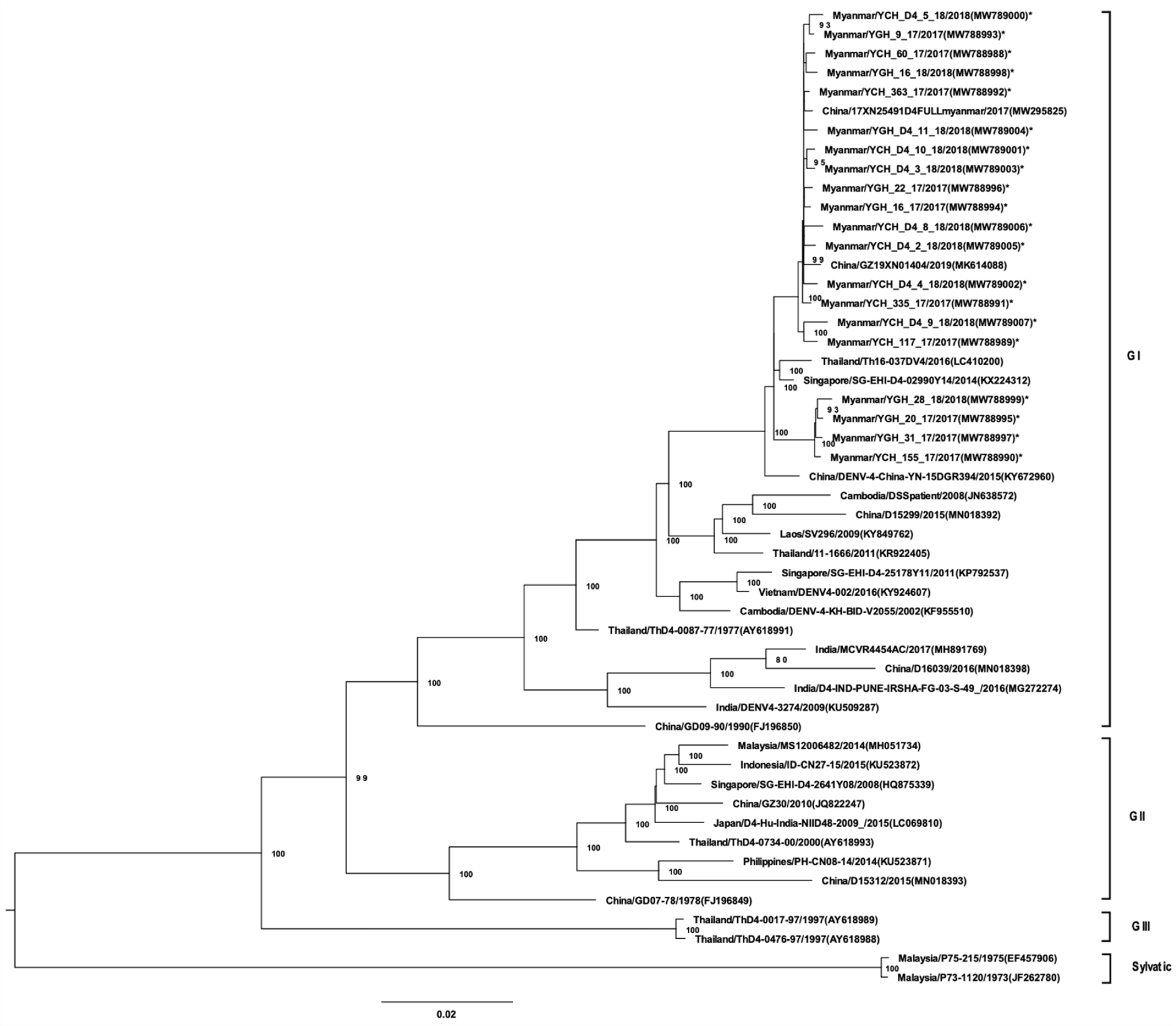
3.3. Whole-Genome Sequencing and Phylogenetic Analysis
3.4. DENV Amino Acid Variants
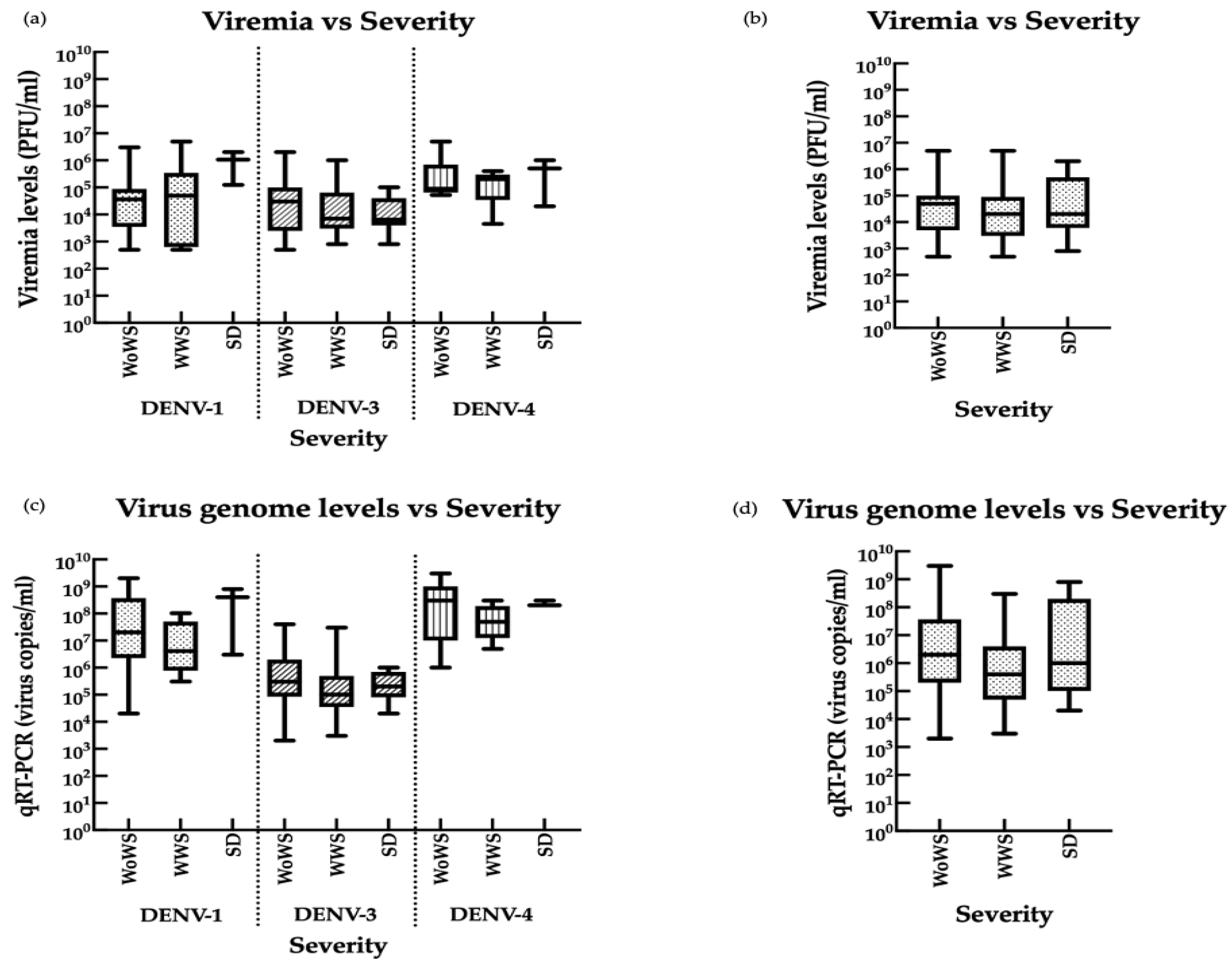
3.5. Associations between Viremia, Disease Severity, and Infection Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- About Dengue: What You Need to Know|Dengue|CDC. Available online: https://www.cdc.gov/dengue/about/index.html (accessed on 14 March 2021).
- WHO|Dengue and Severe Dengue. Available online: https://apps.who.int/mediacentre/factsheets/fs117/en/index.html (accessed on 14 March 2021).
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The Global Distribution and Burden of Dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Dengue Fever—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/immunology-and-microbiology/dengue-fever (accessed on 16 March 2021).
- Murugesan, A.; Manoharan, M. Dengue Virus. In Emerging and Reemerging Viral Pathogens: Volume 1: Fundamental and Basic Virology Aspects of Human, Animal and Plant Pathogens; Elsevier: London, UK, 2020; pp. 281–359. [Google Scholar] [CrossRef]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control HHS Public Access. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of Dengue Virus: Implications for Flavivirus Organization, Maturation, and Fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Oo, P.M.; Wai, K.T.; Harries, A.D.; Shewade, H.D.; Oo, T.; Thi, A.; Lin, Z. The Burden of Dengue, Source Reduction Measures, and Serotype Patterns in Myanmar, 2011 to 2015-R2. Trop. Med. Health 2017, 45, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlaing Myat, T.; Lowry, K.; Thein Thein, M.; Than Nu, S.; Aye Maung, H.; Kyu Kyu, K.; Kyaw Zin, T.; Soe, T.; Aaskov, J. Myanmar Dengue Outbreak Associated with Displacement of Serotypes 2, 3, and 4 by Dengue 1. Emerg. Infect. Dis. 2004, 10, 593–597. [Google Scholar]
- Ngwe Tun, M.M.; Kyaw, A.K.; Makki, N.; Muthugala, R.; Nabeshima, T.; Inoue, S.; Hayasaka, D.; Moi, M.L.; Buerano, C.C.; Thwe, S.M.; et al. Characterization of the 2013 Dengue Epidemic in Myanmar with Dengue Virus 1 as the Dominant Serotype. Infect. Genet. Evol. 2016, 43, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, A.K.; Ngwe Tun, M.M.; Moi, M.L.; Nabeshima, T.; Soe, K.T.; Thwe, S.M.; Myint, A.A.; Maung, K.T.T.; Aung, W.; Hayasaka, D.; et al. Clinical, Virological and Epidemiological Characterization of Dengue Outbreak in Myanmar, 2015. Epidemiol. Infect. 2017, 145, 1886–1897. [Google Scholar] [CrossRef] [Green Version]
- Rico-Hesse, R. Microevolution and Virulence of Dengue Viruses. In Advances in Virus Research; Academic Press: San Diego, CA, USA, 2003; Volume 59, pp. 315–341. [Google Scholar] [CrossRef] [Green Version]
- Halstead, S.B. The Alexander D. Langmuir Lecture the Pathogenesis of Dengue. Am. J. Epidemiol. 1981, 114, 632–648. [Google Scholar] [CrossRef]
- Murray, N.E.A.; Quam, M.B.; Wilder-Smith, A. Epidemiology of Dengue: Past, Present and Future Prospects. Clin. Epidemiol. 2013, 5, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Alonzo, M.T.G.; Kurosawa, Y.; Mapua, C.A.; Reyes, J.D.; Dimaano, E.M.; Alera, M.T.P.; Saito, M.; Oishi, K.; Hasebe, F.; et al. Evaluation of a Dengue IgG Indirect Enzyme-Linked Immunosorbent Assay and a Japanese Encephalitis IgG Indirect Enzyme-Linked Immunosorbent Assay for Diagnosis of Secondary Dengue Virus Infection. Vector Borne Zoonotic Dis. 2010, 10, 143–150. [Google Scholar] [CrossRef]
- Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/23762963/ (accessed on 16 March 2021).
- Ngwe Tun, M.M.; Thant, K.Z.; Inoue, S.; Nabeshima, T.; Aoki, K.; Kyaw, A.K.; Myint, T.; Tar, T.; Maung, K.T.T.; Hayasaka, D.; et al. Detection of East/Central/South African Genotype of Chikungunya Virus in Myanmar, 2010. Emerg. Infect. Dis. 2014, 20, 1378–1381. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, R.S.; Calisher, C.H.; Gubler, D.J.; Chang, G.J.; Vorndam, A.V. Rapid Detection and Typing of Dengue Viruses from Clinical Samples by Using Reverse Transcriptase-Polymerase Chain Reaction. J. Clin. Microbiol. 1992, 30, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Tanaka, M.; Igarashi, A. Rapid Identification of Dengue Virus Serotypes by Using Polymerase Chain Reaction. J. Clin. Microbiol. 1991, 29, 2107–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudigyo, D.; Rahmawati, G.; Setiasari, D.W.; Poluan, R.H.; Sesotyosari, S.L.; Wardana, T.; Herawati, C.; Heriyanto, D.S.; Indrasari, S.R.; Afiahayati; et al. Transcriptome Profile of next Generation Sequence Data Related to Inflammation on Nasopharyngeal Carcinoma Cases in Indonesia. Asian Pac. J. Cancer Prev. 2020, 21, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Yang, X.; Charlebois, P.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant Inference for Viral Populations. BMC Genomics 2013, 14, 674. [Google Scholar] [CrossRef] [Green Version]
- Sandmann, S.; De Graaf, A.O.; Karimi, M.; Van Der Reijden, B.A.; Hellström-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ngwe Tun, M.M.; Muthugala, R.; Nabeshima, T.; Soe, A.M.; Dumre, S.P.; Rajamanthri, L.; Jayawardana, D.; Attanayake, S.; Inoue, S.; Morita, K. Complete Genome Analysis and Characterization of Neurotropic Dengue Virus 2 Cosmopolitan Genotype Isolated from the Cerebrospinal Fluid of Encephalitis Patients. PLoS ONE 2020, 15, e0234508. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Thant, K.Z.; Ngwe Tun, M.M.; del Carmen Parquet, M.; Inoue, S.; Lwin, Y.Y.; Lin, S.; Aye, K.T.; Khin, P.T.; Myint, T.; Htwe, K.; et al. Molecular Epidemiology of Dengue Viruses Co-Circulating in Upper Myanmar in 2006. Trop. Med. Health 2015, 43, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moi, M.L.; Lim, C.K.; Kotaki, A.; Takasaki, T.; Kurane, I. Detection of Higher Levels of Dengue Viremia Using FcγR-Expressing BHK-21 Cells than FcγR-Negative Cells in Secondary Infection but Not in Primary Infection. J. Infect. Dis. 2011, 203, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Takasaki, T.; Yamada, K.I.; Nerome, R.; Tajima, S.; Kurane, I. Development and Evaluation of Fluorogenic TaqMan Reverse Transcriptase PCR Assays for Detection of Dengue Virus Types 1 to 4. J. Clin. Microbiol. 2004, 42, 5935–5937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittke, V.; Robb, T.E.; Thu, H.M.; Nisalak, A.; Nimmannitya, S.; Kalayanrooj, S.; Vaughn, D.W.; Endy, T.P.; Holmes, E.C.; Aaskov, J.G. Extinction and Rapid Emergence of Strains of Dengue 3 Virus during an Interepidemic Period. Virology 2002, 301, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamel, R.; Surasombatpattana, P.; Wichit, S.; Dauvé, A.; Donato, C.; Pompon, J.; Vijaykrishna, D.; Liegeois, F.; Vargas, R.M.; Luplertlop, N.; et al. Phylogenetic Analysis Revealed the Co-Circulation of Four Dengue Virus Serotypes in Southern Thailand. PLoS ONE 2019, 14, e0221179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, P.Y.; Su, C.L.; Liao, T.L.; Yang, C.F.; Chang, S.F.; Lin, C.C.; Chang, M.C.; Hu, H.C.; Huang, J.H. Molecular Characterization of Dengue Viruses Imported into Taiwan during 2003–2007: Geographic Distribution and Genotype Shift. Am. J. Trop. Med. Hyg. 2009, 80, 1039–1046. [Google Scholar] [CrossRef]
- Ong, S.H.; Yip, J.T.; Chen, Y.L.; Liu, W.; Harun, S.; Lystiyaningsih, E.; Heriyanto, B.; Beckett, C.G.; Mitchell, W.P.; Hibberd, M.L.; et al. Periodic Re-Emergence of Endemic Strains with Strong Epidemic Potential-A Proposed Explanation for the 2004 Indonesian Dengue Epidemic. Infect. Genet. Evol. 2008, 8, 191–204. [Google Scholar] [CrossRef]
- Sasmono, R.T.; Wahid, I.; Trimarsanto, H.; Yohan, B.; Wahyuni, S.; Hertanto, M.; Yusuf, I.; Mubin, H.; Ganda, I.J.; Latief, R.; et al. Genomic Analysis and Growth Characteristic of Dengue Viruses from Makassar, Indonesia. Infect. Genet. Evol. 2015, 32, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Sucipto, T.H.; Kotaki, T.; Mulyatno, K.C.; Churrotin, S.; Labiqah, A.; Soegijanto, S.; Kameoka, M. Phylogenetic Analysis of Dengue Virus in Bangkalan, Madura Island, East Java Province, Indonesia. J. Trop. Med. 2020, 2018, 8127093. [Google Scholar] [CrossRef] [Green Version]
- Amir, M.; Hussain, A.; Asif, M.; Ahmed, S.; Alam, H.; Moga, M.A.; Cocuz, M.E.; Marceanu, L.; Blidaru, A. Full-Length Genome and Partial Viral Genes Phylogenetic and Geographical Analysis of Dengue Serotype 3 Isolates. Microorganisms 2021, 9, 323. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Isolates Per Year | ||||
|---|---|---|---|---|
| Serotype | 2017 | 2018 | 2019 | Total |
| DENV-1 | 14 | 30 | 20 | 64 |
| DENV-2 | 0 | 3 | 5 | 8 |
| DENV-3 | 18 | 55 | 25 | 98 |
| DENV-4 | 20 | 18 | 4 | 42 |
| 52 | 106 | 54 | 212 | |
| Patients Positive for Each DENV Serotype (N, %) | ||||||
|---|---|---|---|---|---|---|
| Patient Characteristic | DENV-1 (n = 64, 30.1%) | DENV-2 (n = 8, 3.8%) | DENV-3 (n = 98, 46.2%) | DENV-4 (n = 42, 19.8%) | Total (n = 212, 100%) | p Value |
| Age (years) | 8.8 (±5.1) 2 | 8.0 (±4.6) | 9.6 (±5.4) | 11.7 (±5.4) | 9.7 (±6.3) | |
| Age group (years) | ||||||
| ≤12 | 52 (32.5%) | 7 (4.4%) | 75 (46.8%) | 26 (16.3%) | 160 (100%) | 0.111 |
| >12 | 12 (23%) | 1 (2%) | 23 (44.2%) | 16 (30.8) | 52 (100%) | |
| Gender | ||||||
| Male | 30 (46.8%) | 6 (75%) | 64 (65.3%) | 20 (47.6%) | 120 (56.7%) | 0.047 |
| Female | 34 (53.2%) | 2 (25%) | 34 (34.7%) | 22 (52.4%) | 92 (43.3%) | |
| Signs and Symptoms | ||||||
| Rash | 3 | 0 | 8 | 3 | 14 | 0.717 |
| Hess test | 62 | 7 | 94 | 38 | 201 | 0.038 |
| Coffee ground vomiting | 4 | 1 | 13 | 1 | 19 | 0.161 |
| Muscle pain | 6 | 1 | 19 | 12 | 38 | 0.081 |
| Joint pain | 6 | 1 | 17 | 10 | 34 | 0.241 |
| Abdominal pain | 21 | 2 | 31 | 15 | 69 | 0.932 |
| Hepatomegaly | 41 | 5 | 39 | 20 | 105 | 0.027 |
| Splenomegaly | 0 | 0 | 1 | 1 | 2 | 0.632 |
| Drowsiness | 17 | 2 | 31 | 8 | 58 | 0.493 |
| Thrombocytopenia. (<150,000/μL platelets) | 26 | 2 | 32 | 20 | 80 | 0.308 |
| Epistaxis | 11 | 2 | 19 | 5 | 37 | 0.689 |
| Melena | 0 | 0 | 7 | 1 | 8 | 0.106 |
| Symptoms 3 | ||||||
| Without warning signs | 27 (42.2%) | 2 (25%) | 27 (27.5%) | 18 (42.8%) | 74 (34.9%) | |
| With warning signs | 32 (50%) | 6 (75%) | 62 (63.3%) | 19 (45.2%) | 119 (56.1%) | 0.284 |
| Severe dengue | 5 (7.8%) | 0 | 9 (9.2%) | 5 (12%) | 19 (9%) | |
| Anti-DENV IgM 4 | ||||||
| Negative | 24 (37.5%) | 3 (37.5%) | 43 (43.8%) | 12 (28.6%) | 82 (38.7%) | 0.397 |
| Positive | 40 (62.5%) | 5 (62.5%) | 55 (56.2%) | 30 (71.4%) | 130 (61.3%) | |
| Type of infection 5 | ||||||
| Primary | 31 (48.4%) | 1 (12.5%) | 50 (51%) | 5 (11.9%) | 87 (41%) | <0.001 |
| Secondary | 33 (51.6%) | 7 (87.5%) | 48 (49%) | 37 (88.1%) | 125 (59%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soe, A.M.; Ngwe Tun, M.M.; Nabeshima, T.; Myat, T.W.; Htun, M.M.; Lin, H.; Hom, N.S.; Inoue, S.; Nwe, K.M.; Aye, L.P.P.; et al. Emergence of a Novel Dengue Virus 3 (DENV-3) Genotype-I Coincident with Increased DENV-3 Cases in Yangon, Myanmar between 2017 and 2019. Viruses 2021, 13, 1152. https://doi.org/10.3390/v13061152
Soe AM, Ngwe Tun MM, Nabeshima T, Myat TW, Htun MM, Lin H, Hom NS, Inoue S, Nwe KM, Aye LPP, et al. Emergence of a Novel Dengue Virus 3 (DENV-3) Genotype-I Coincident with Increased DENV-3 Cases in Yangon, Myanmar between 2017 and 2019. Viruses. 2021; 13(6):1152. https://doi.org/10.3390/v13061152
Chicago/Turabian StyleSoe, Aung Min, Mya Myat Ngwe Tun, Takeshi Nabeshima, Theingi Win Myat, Moh Moh Htun, Htin Lin, Nang Sarm Hom, Shingo Inoue, Khine Mya Nwe, Lynn Pa Pa Aye, and et al. 2021. "Emergence of a Novel Dengue Virus 3 (DENV-3) Genotype-I Coincident with Increased DENV-3 Cases in Yangon, Myanmar between 2017 and 2019" Viruses 13, no. 6: 1152. https://doi.org/10.3390/v13061152