
Exogenous Vimentin Supplementation Transiently Affects Early Steps during HPV16 Pseudovirus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. HPV Pseudovirion Preparation and Labelling
2.3. Negative Staining Transmission Electron Microscopy
2.4. HPV Pseudovirus Binding and Internalisation Assays
2.5. HPV Pseudovirus Infection Assays
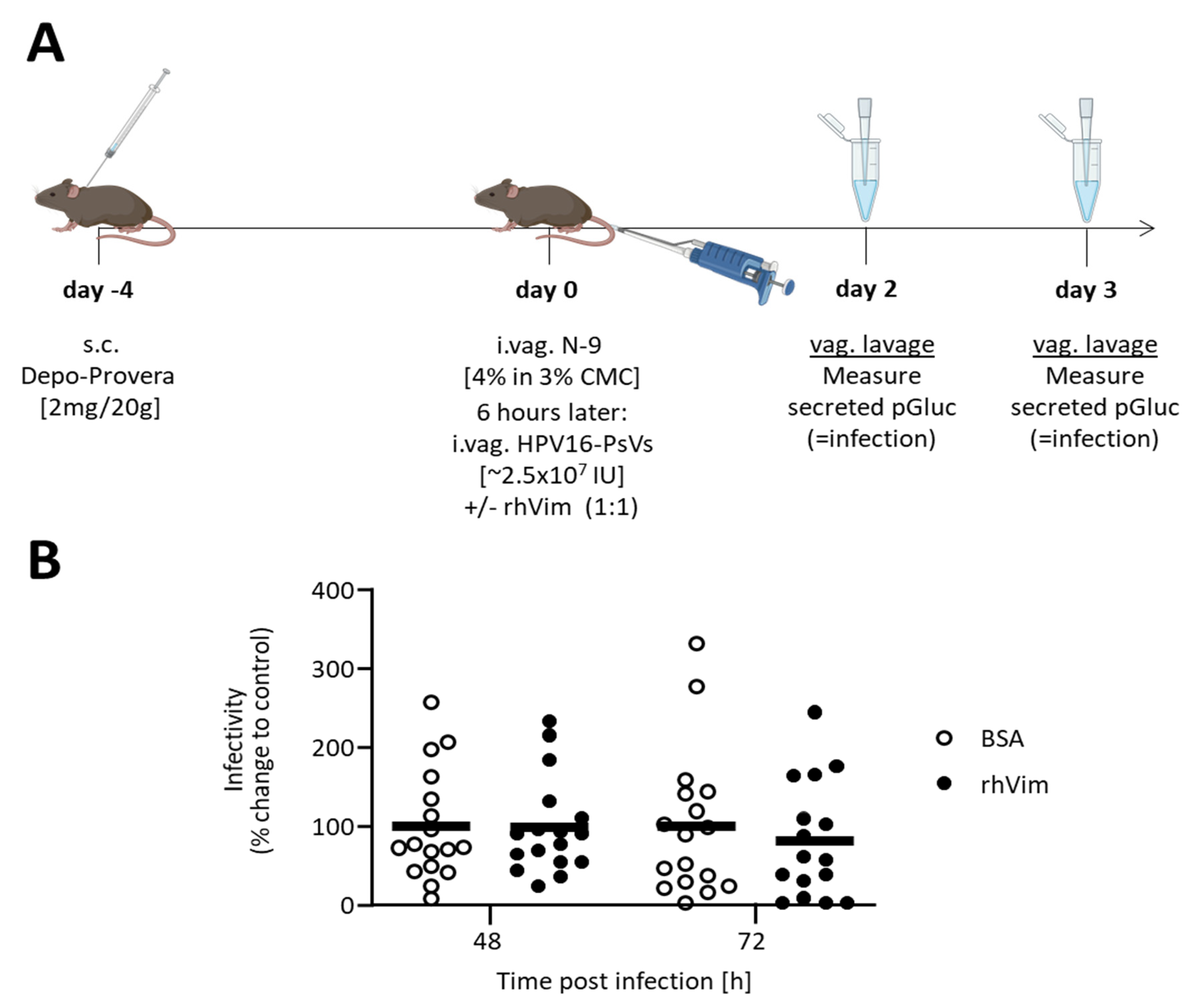
2.6. HPV16-PsVs Murine Cervicovaginal Challenge Model
2.7. Immunofluorescence and Confocal Microscopy
3. Results
3.1. Globular, but Not Filamentous, rhVim Modulates HPV16-PsVs Internalisation into NIKS Cells
3.2. rhVim Transiently Affects Early HPV16-PsVs Infection Steps
3.3. Pre-Incubation with rhVim Affects Both HPV16-PsVs Engagement with Cell Surface HSPGs and the Potential Entry Complex In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ozbun, M.A. Extracellular events impacting human papillomavirus infections: Epithelial wounding to cell signaling involved in virus entry. Papillomavirus Res. 2019, 7, 188–192. [Google Scholar] [CrossRef]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [Green Version]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [Green Version]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [Green Version]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space To Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef] [Green Version]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; Da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: A review of binding and entry. J. Virol. 2013, 87, 6062–6072. [Google Scholar] [CrossRef] [Green Version]
- Siddiqa, A.; Broniarczyk, J.; Banks, L. Papillomaviruses and Endocytic Trafficking. Int. J. Mol. Sci. 2018, 19, 2619. [Google Scholar] [CrossRef] [Green Version]
- Lowy, D.R.; Schiller, J.T. Reducing HPV-associated cancer globally. Cancer Prev. Res. 2012, 5, 18–23. [Google Scholar] [CrossRef] [Green Version]
- WHO/ICO Information Centre on HPV and Cervical Cancer (HPV Information Centre). Human Papillomavirus and Related Cancers in Africa; Summary Report; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- De Vuyst, H.; Alemany, L.; Lacey, C.; Chibwesha, C.J.; Sahasrabuddhe, V.; Banura, C.; Denny, L.; Parham, G.P. The burden of human papillomavirus infections and related diseases in sub-saharan Africa. Vaccine 2013, 31 (Suppl. S5), 32–46. [Google Scholar] [CrossRef] [Green Version]
- Williamson, A.L. The Interaction between Human Immunodeficiency Virus and Human Papillomaviruses in Heterosexuals in Africa. J. Clin. Med. 2015, 4, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Denny, L. Control of Cancer of the Cervix in Low- and Middle-Income Countries. Ann. Surg. Oncol. 2015, 22, 728–733. [Google Scholar] [CrossRef]
- Okoye, J.O.; Ofordile, C.A.; Adeleke, O.K.; Okechi, O. Prevalence of high-risk HPV genotypes in sub-Saharan Africa based on Human immunodeficiency virus status: A 20-year systematic review. Epidemiol. Health 2021, 43, e2021039. [Google Scholar] [CrossRef]
- Taku, O.; Mbulawa, Z.Z.A.; Phohlo, K.; Garcia-Jardon, M.; Businge, C.B.; Williamson, A.-L. Distribution of Human Papillomavirus (HPV) Genotypes in HIV-Negative and HIV-Positive Women with Cervical Intraepithelial Lesions in the Eastern Cape Province, South Africa. Viruses 2021, 13, 280. [Google Scholar] [CrossRef]
- Carse, S.; Bergant, M.; Schäfer, G. Advances in Targeting HPV Infection as Potential Alternative Prophylactic Means. Int. J. Mol. Sci. 2021, 22, 2201. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Muller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.N.; Buck, C.B.; Thompson, C.D.; Kines, R.; Bernardo, M.; Choyke, P.L.; Lowy, D.R.; Schiller, J.T. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat. Med. 2007, 13, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Hua, C.; Zhu, Y.; Wu, C.; Si, L.; Wang, Q.; Sui, L.; Jiang, S. The Underlying Mechanism of 3-Hydroxyphthalic Anhydride-Modified Bovine Beta-Lactoglobulin to Block Human Papillomavirus Entry Into the Host Cell. Front. Microbiol. 2019, 10, 2188. [Google Scholar] [CrossRef]
- Marais, D.; Gawarecki, D.; Allan, B.; Ahmed, K.; Altini, L.; Cassim, N.; Gopolang, F.; Hoffman, M.; Ramjee, G.; Williamson, A.L. The effectiveness of Carraguard, a vaginal microbicide, in protecting women against high-risk human papillomavirus infection. Antivir. Ther. 2011, 16, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Magnan, S.; Tota, J.E.; El-Zein, M.; Burchell, A.N.; Schiller, J.T.; Ferenczy, A.; Tellier, P.P.; Coutlee, F.; Franco, E.L.; Group, C.S. Efficacy of a Carrageenan gel Against Transmission of Cervical HPV (CATCH): Interim analysis of a randomized, double-blind, placebo-controlled, phase 2B trial. Clin. Microbiol. Infect. 2019, 25, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Perino, A.; Consiglio, P.; Maranto, M.; De Franciscis, P.; Marci, R.; Restivo, V.; Manzone, M.; Capra, G.; Cucinella, G.; Calagna, G. Impact of a new carrageenan-based vaginal microbicide in a female population with genital HPV-infection: First experimental results. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6744–6752. [Google Scholar]
- Guo, X.; Qiu, L.; Wang, Y.; Wang, Y.; Wang, Q.; Song, L.; Li, Y.; Huang, K.; Du, X.; Fan, W.; et al. A randomized open-label clinical trial of an anti-HPV biological dressing (JB01-BD) administered intravaginally to treat high-risk HPV infection. Microbes Infect. 2016, 18, 148–1452. [Google Scholar] [CrossRef]
- Li, L.; He, L.; Tan, S.; Guo, X.; Lu, H.; Qi, Z.; Pan, C.; An, X.; Jiang, S.; Liu, S. 3-hydroxyphthalic anhydride-modified chicken ovalbumin exhibits potent and broad anti-HIV-1 activity: A potential microbicide for preventing sexual transmission of HIV-1. Antimicrob. Agents Chemother. 2010, 54, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Neurath, A.R.; Strick, N.; Li, Y.Y. 3-Hydroxyphthaloyl beta-lactoglobulin. III. Antiviral activity against herpesviruses. Antivir. Chem. Chemother. 1998, 9, 177–184. [Google Scholar] [CrossRef]
- Fernandez-Romero, J.A.; Abraham, C.J.; Rodriguez, A.; Kizima, L.; Jean-Pierre, N.; Menon, R.; Begay, O.; Seidor, S.; Ford, B.E.; Gil, P.I.; et al. Zinc acetate/carrageenan gels exhibit potent activity in vivo against high-dose herpes simplex virus 2 vaginal and rectal challenge. Antimicrob. Agents Chemother. 2012, 56, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Skoler-Karpoff, S.; Ramjee, G.; Ahmed, K.; Altini, L.; Plagianos, M.G.; Friedland, B.; Govender, S.; De Kock, A.; Cassim, N.; Palanee, T.; et al. Efficacy of Carraguard for prevention of HIV infection in women in South Africa: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1977–1987. [Google Scholar] [CrossRef]
- Schäfer, G.; Graham, L.M.; Lang, D.; Blumenthal, M.J.; Bergant Marusic, M.; Katz, A.A. Vimentin modulates infectious internalisation of HPV16 pseudovirions. J. Virol. 2017, 91, e00307-17. [Google Scholar] [CrossRef] [Green Version]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Perez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar] [CrossRef] [Green Version]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 2005, 119, 445–462. [Google Scholar]
- Handisurya, A.; Day, P.M.; Thompson, C.D.; Buck, C.B.; Kwak, K.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Murine skin and vaginal mucosa are similarly susceptible to infection by pseudovirions of different papillomavirus classifications and species. Virology 2012, 433, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Ujma, S.; Carse, S.; Chetty, A.; Horsnell, W.; Clark, H.; Madsen, J.; Mackay, R.M.; Watson, A.; Griffiths, M.; Katz, A.A.; et al. Surfactant Protein A Impairs Genital HPV16 Pseudovirus Infection by Innate Immune Cell Activation in A Murine Model. Pathogens 2019, 8, 288. [Google Scholar] [CrossRef] [Green Version]
- Allen-Hoffmann, B.L.; Schlosser, S.J.; Ivarie, C.A.; Sattler, C.A.; Meisner, L.F.; O’Connor, S.L. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J. Investig. Dermatol. 2000, 114, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culp, T.D.; Budgeon, L.R.; Christensen, N.D. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 2006, 347, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.N. Infection of murine vaginal or endocervical mucosa with human papillomavirus pseudovirions. Nat. Protoc. 2007. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of Colocalization of Objects in Dual-Color Confocal Images. J. Microsc. Oxford 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, F.; Peterson, M.K.; Caldeira Araujo, H.; Lautenschlager, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schopferer, M.; Bär, H.; Hochstein, B.; Sharma, S.; Mücke, N.; Herrmann, H.; Willenbacher, N. Desmin and vimentin intermediate filament networks: Their viscoelastic properties investigated by mechanical rheometry. J. Mol. Biol. 2009, 388, 133–143. [Google Scholar] [CrossRef]
- Giroglou, T.; Florin, L.; Schafer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Structure and Assembly. Cold Spring Harb. Perspect Biol. 2016, 8, 18242. [Google Scholar] [CrossRef]
- Viedma-Poyatos, A.; Pajares, M.A.; Perez-Sala, D. Type III intermediate filaments as targets and effectors of electrophiles and oxidants. Redox Biol. 2020, 36, 101582. [Google Scholar] [CrossRef]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of HPV infection. Gynecol. Oncol. 2010, 118 (Suppl. S1), 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor-Vaknin, N.; Punturieri, A.; Sitwala, K.; Markovitz, D.M. Vimentin is secreted by activated macrophages. Nat. Cell Biol. 2003, 5, 59–63. [Google Scholar] [CrossRef]
- Frescas, D.; Roux, C.M.; Aygun-Sunar, S.; Gleiberman, A.S.; Krasnov, P.; Kurnasov, O.V.; Strom, E.; Virtuoso, L.P.; Wrobel, M.; Osterman, A.L.; et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc. Natl. Acad. Sci. USA 2017, 114, 1668–1677. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Cho, W.; Kim, I.; Lee, S.H.; Oh, G.T.; Park, Y.M. Oxidized LDL induces vimentin secretion by macrophages and contributes to atherosclerotic inflammation. J. Mol. Med. 2020, 98, 973–983. [Google Scholar] [CrossRef]
- Du, N.; Cong, H.; Tian, H.; Zhang, H.; Zhang, W.; Song, L.; Tien, P. Cell surface vimentin is an attachment receptor for enterovirus 71. J. Virol. 2014, 88, 5816–5833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Cao, Y.; Su, W.; Huang, S.; Lu, W.; Zhou, Y.; Gao, J.; Zhao, W.; Zhang, B.; Wu, X. Enterovirus A71 VP1 Variation A289T Decreases the Central Nervous System Infectivity via Attenuation of Interactions between VP1 and Vimentin In Vitro and In Vivo. Viruses 2019, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.J.; Yu, C.Y.; Liao, C.L.; Lin, Y.L. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell. Microbiol. 2011, 13, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zou, L.; Yang, Y.; Yuan, J.; Hu, Z.; Liu, H.; Peng, H.; Shang, W.; Zhang, X.; Zhu, J.; et al. Superficial vimentin mediates DENV-2 infection of vascular endothelial cells. Sci. Rep. 2016, 6, 38372. [Google Scholar] [CrossRef]
- Suprewicz, L.; Swoger, M.; Gupta, S.; Piktel, E.; Byfield, F.J.; Iwamoto, D.V.; Germann, D.; Reszec, J.; Marcinczyk, N.; Carroll, R.J.; et al. Extracellular vimentin as a target against SARS-CoV-2 host cell invasion. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Thalla, D.G.; Jung, P.; Bischoff, M.; Lautenschlager, F. Role of Extracellular Vimentin in Cancer-Cell Functionality and Its Influence on Cell Monolayer Permeability Changes Induced by SARS-CoV-2 Receptor Binding Domain. Int. J. Mol. Sci. 2021, 22, 7469. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carse, S.; Lang, D.; Katz, A.A.; Schäfer, G. Exogenous Vimentin Supplementation Transiently Affects Early Steps during HPV16 Pseudovirus Infection. Viruses 2021, 13, 2471. https://doi.org/10.3390/v13122471
Carse S, Lang D, Katz AA, Schäfer G. Exogenous Vimentin Supplementation Transiently Affects Early Steps during HPV16 Pseudovirus Infection. Viruses. 2021; 13(12):2471. https://doi.org/10.3390/v13122471
Chicago/Turabian StyleCarse, Sinead, Dirk Lang, Arieh A. Katz, and Georgia Schäfer. 2021. "Exogenous Vimentin Supplementation Transiently Affects Early Steps during HPV16 Pseudovirus Infection" Viruses 13, no. 12: 2471. https://doi.org/10.3390/v13122471