Structural Insight into Paramyxovirus and Pneumovirus Entry Inhibition


Abstract
:1. Introduction
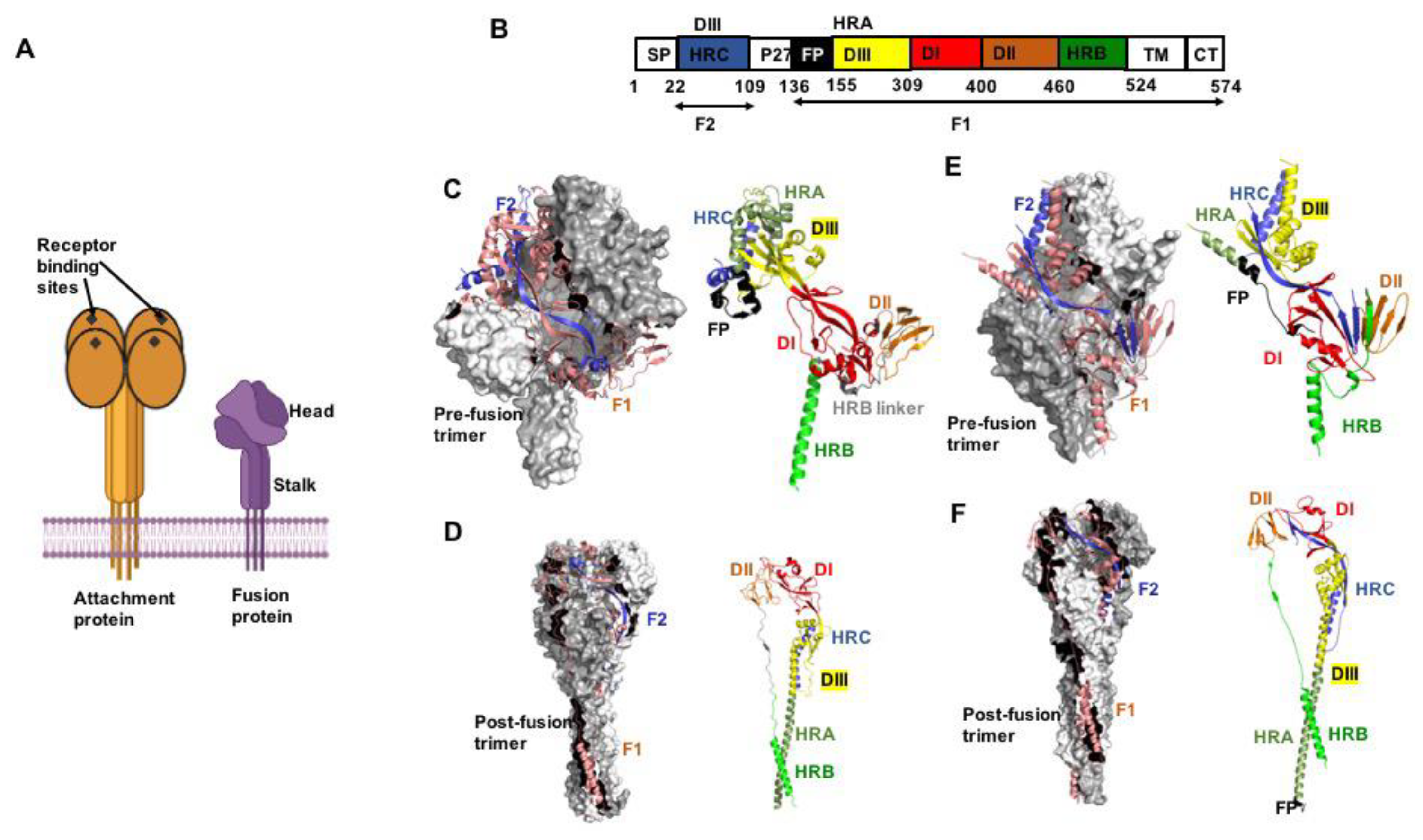
2. The Pneumo- and Paramyxovirus Entry Machinery
2.1. Attachment Proteins
2.2. F Proteins
2.3. Fusion Activation
3. Entry Inhibition
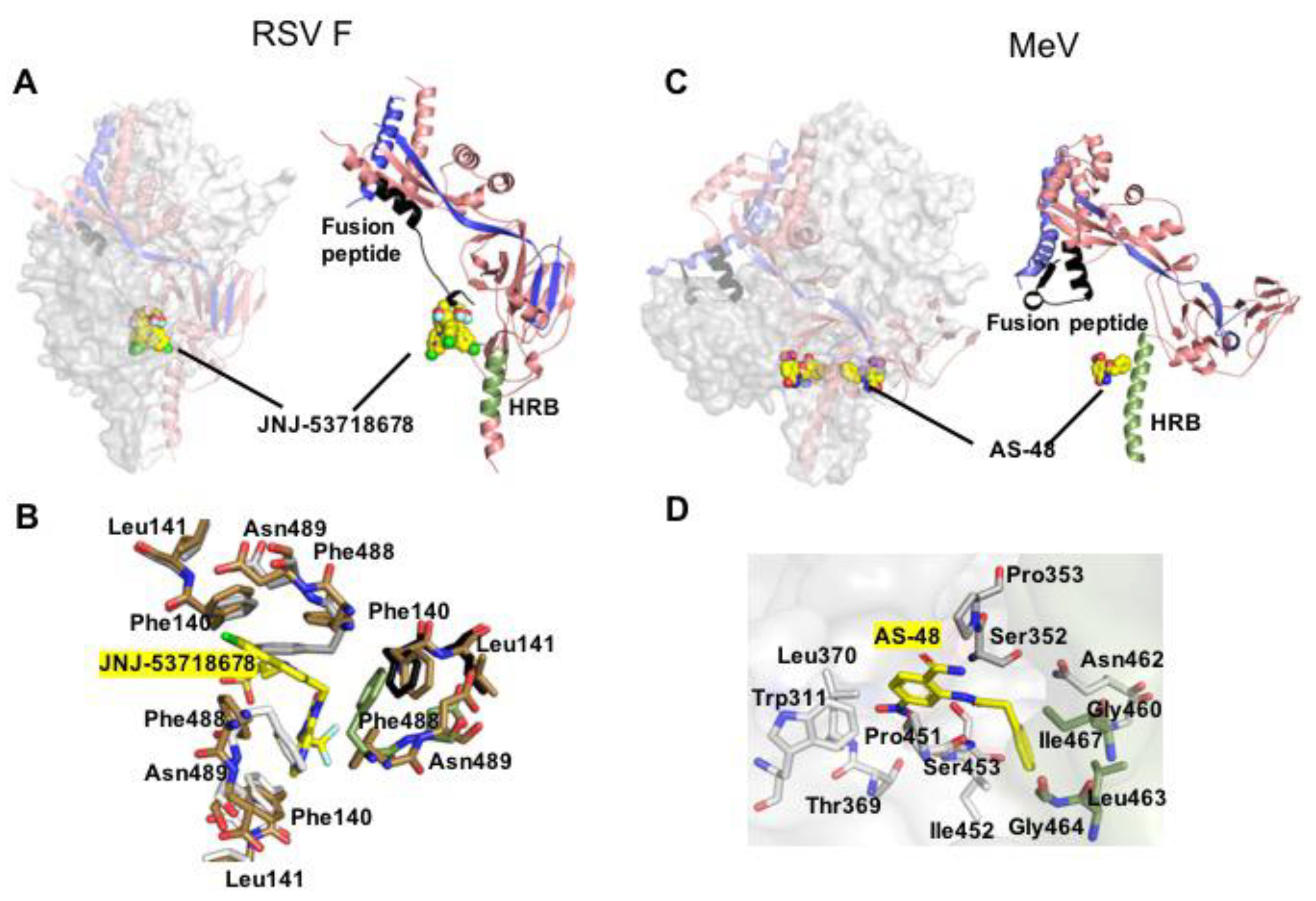
3.1. Synthetic F Protein Blockers
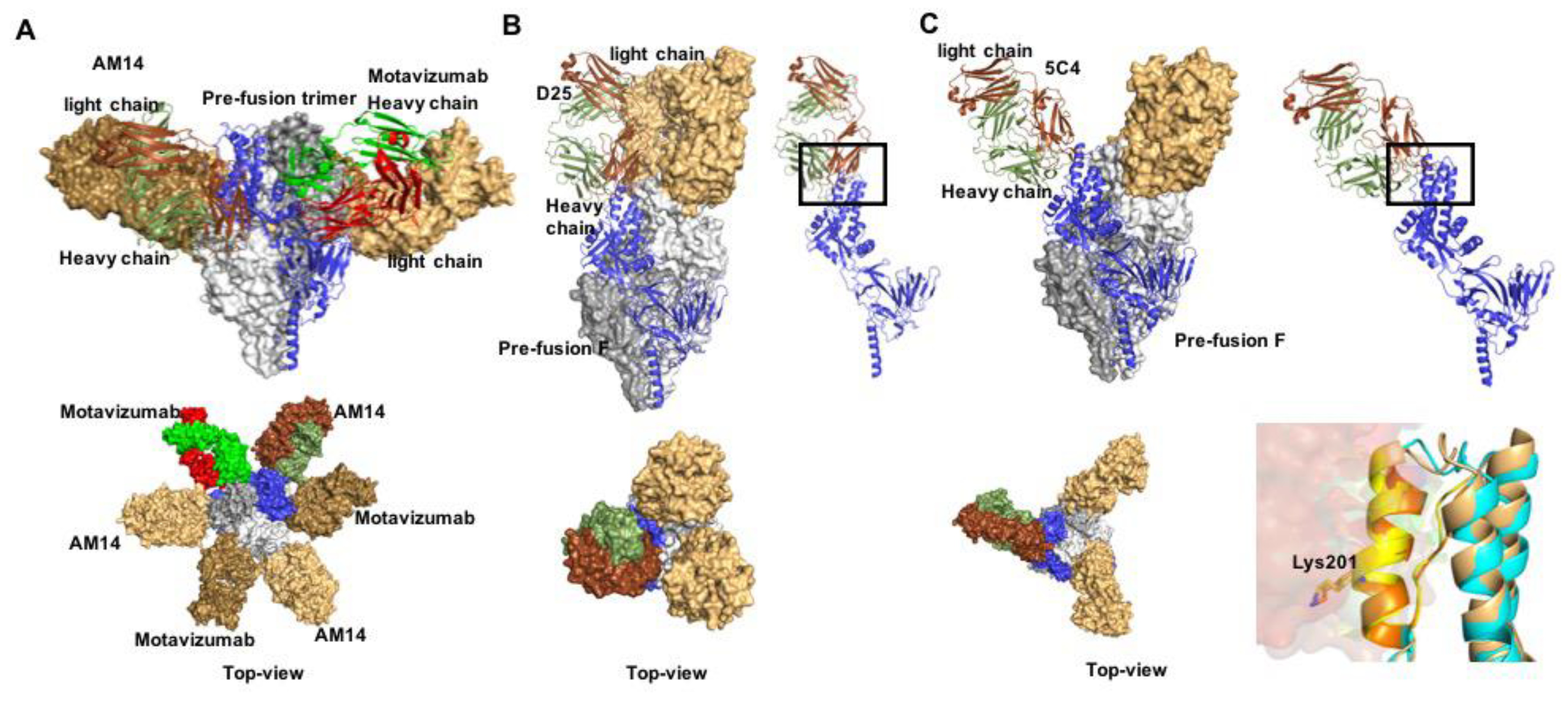
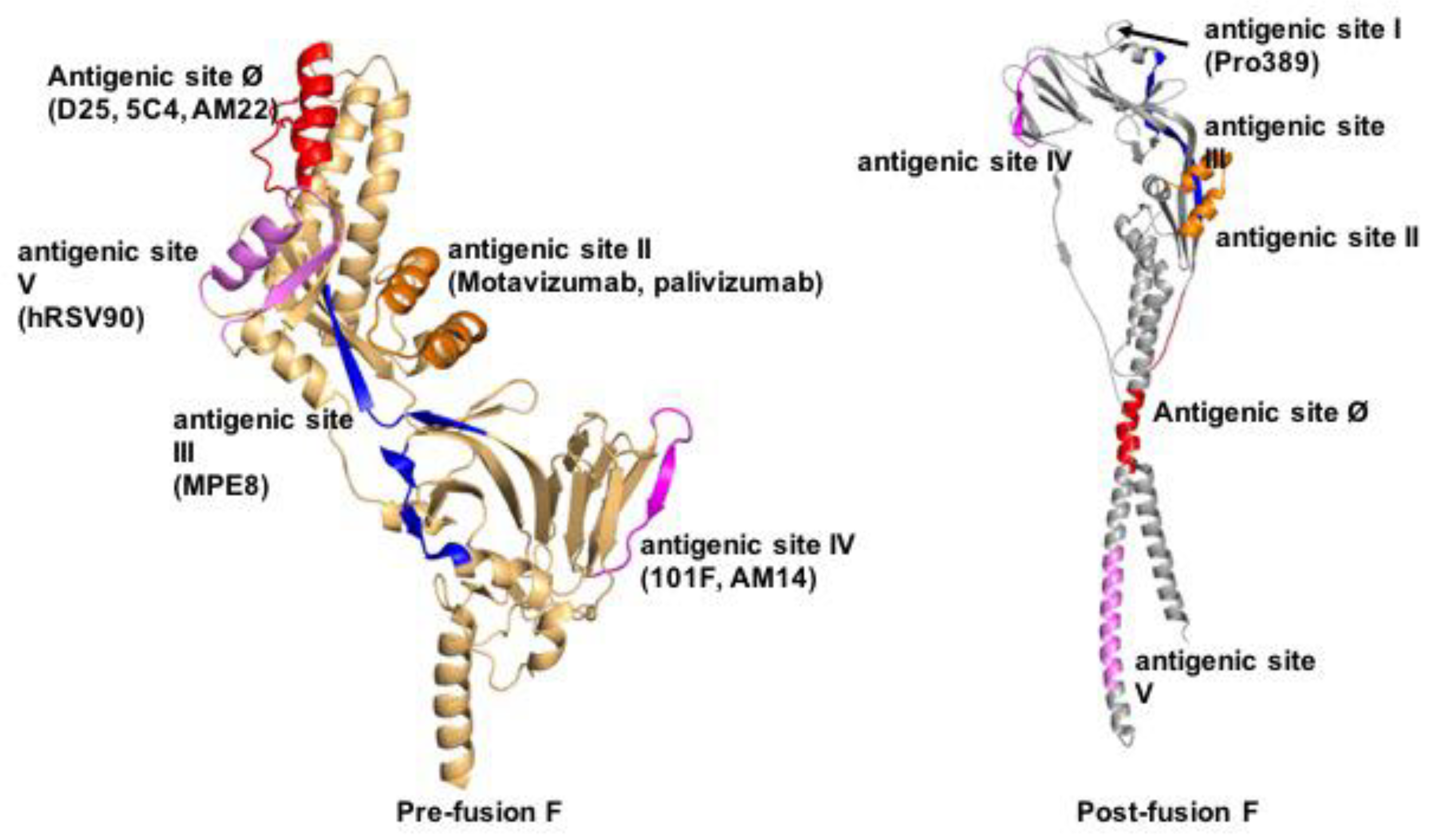
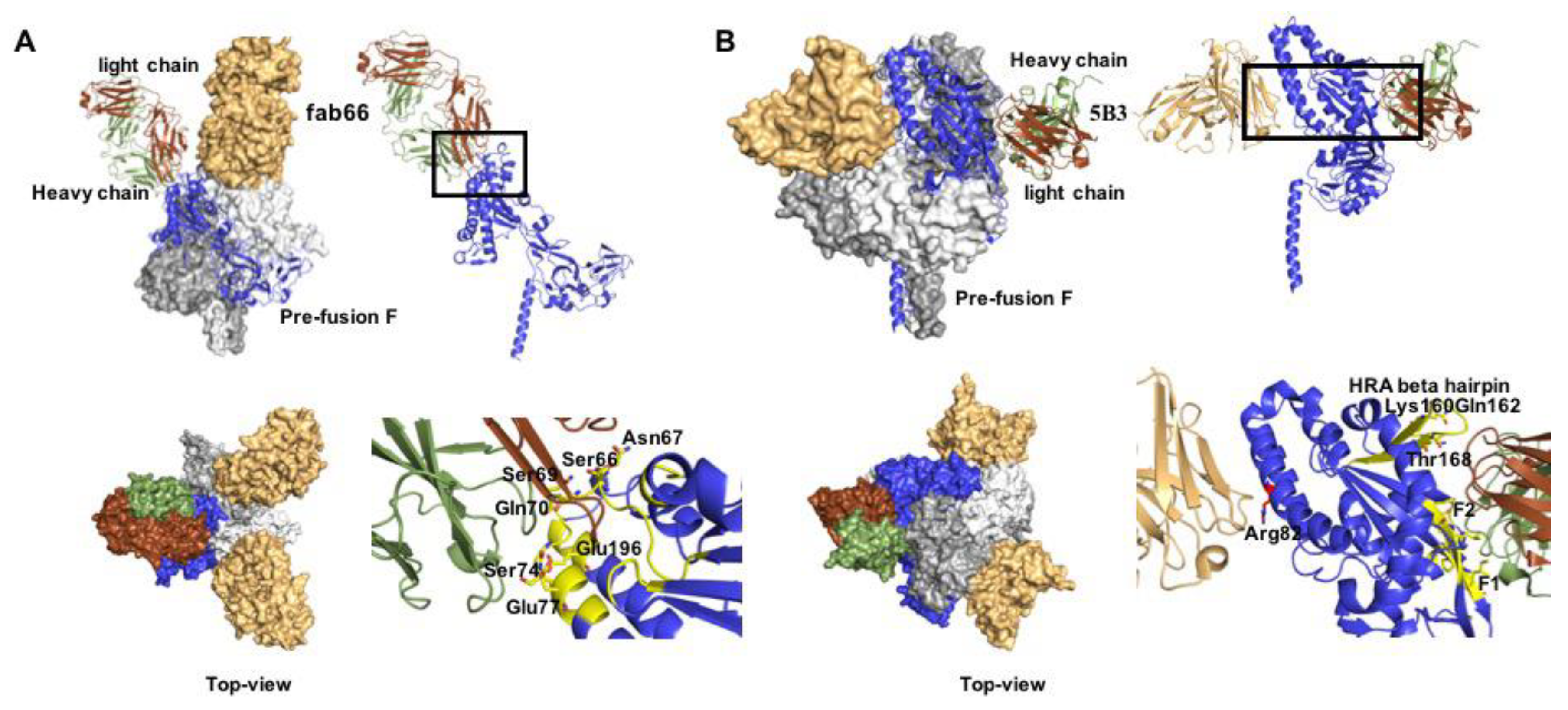
3.2. Druggable Sites and Neutralizing Epitopes
3.3. Clinical Efficacy of Small Molecule F Protein Inhibitors
3.4. Perspectives for Pneumo- and Paramyxovirus Entry Inhibitors
Acknowledgments
Conflicts of Interest
References
- Lamb, R.A.; Parks, G.D. Paramyxoviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 957–995. [Google Scholar]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Abedi, G.R.; Prill, M.M.; Langley, G.E.; Wikswo, M.E.; Weinberg, G.A.; Curns, A.T.; Schneider, E. Estimates of Parainfluenza Virus-Associated Hospitalizations and Cost Among Children Aged Less Than 5 Years in the United States, 1998–2010. J. Pediatric Infect. Dis. Soc. 2016, 5, 7–13. [Google Scholar] [CrossRef]
- Peck, A.J.; Englund, J.A.; Kuypers, J.; Guthrie, K.A.; Corey, L.; Morrow, R.; Hackman, R.C.; Cent, A.; Boeckh, M. Respiratory virus infection among hematopoietic cell transplant recipients: Evidence for asymptomatic parainfluenza virus infection. Blood 2007, 110, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.P.; Shah, P.K.; Azzi, J.M.; Chemaly, R.F. Parainfluenza virus infections in hematopoietic cell transplant recipients and hematologic malignancy patients: A systematic review. Cancer Lett. 2016, 370, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Xie, H.; Campbell, A.P.; Kuypers, J.M.; Leisenring, W.M.; Englund, J.A.; Boeckh, M. Parainfluenza virus lower respiratory tract disease after hematopoietic cell transplant: Viral detection in the lung predicts outcome. Clin. Infect. Dis. 2014, 58, 1357–1368. [Google Scholar] [CrossRef] [Green Version]
- Ustun, C.; Slaby, J.; Shanley, R.M.; Vydra, J.; Smith, A.R.; Wagner, J.E.; Weisdorf, D.J.; Young, J.A. Human parainfluenza virus infection after hematopoietic stem cell transplantation: Risk factors, management, mortality, and changes over time. Biol. Blood Marrow Transplant. 2012, 18, 1580–1588. [Google Scholar] [CrossRef] [Green Version]
- Nichols, W.G.; Corey, L.; Gooley, T.; Davis, C.; Boeckh, M. Parainfluenza virus infections after hematopoietic stem cell transplantation: Risk factors, response to antiviral therapy, and effect on transplant outcome. Blood 2001, 98, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Lee, B. Hendra and nipah infection: Pathology, models and potential therapies. Infect. Disord. Drug Targets 2011, 11, 315–336. [Google Scholar] [CrossRef] [Green Version]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.L.; Kopp, B.T.; Paul, G.; Landgrave, L.C.; Hayes, D., Jr.; Thompson, R. Respiratory syncytial virus: Current and emerging treatment options. Clinicoecon. Outcomes Res. 2014, 6, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein in complex with an amino-terminal peptide of the phosphoprotein. J. Virol. 2018, 92, e01304-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battles, M.B.; Más, V.; Olmedillas, E.; Cano, O.; Vázquez, M.; Rodríguez, L.; Melero, J.A.; McLellan, J.S. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eléouët, J.-F. Structure of the respiratory syncytial virus polymerase complex. Cell 2019, 179, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLellan, J.S.; Yang, Y.; Graham, B.S.; Kwong, P.D. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 2011, 85, 7788–7796. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Qian, X.; Lattmann, S.; El Sahili, A.; Yeo, T.H.; Jia, H.; Cressey, T.; Ludeke, B.; Noton, S.; Kalocsay, M. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 2020, 577, 275–279. [Google Scholar] [CrossRef]
- Swanson, K.; Wen, X.; Leser, G.P.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 2010, 402, 372–379. [Google Scholar] [CrossRef]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagné, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef]
- Yin, H.-S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.-S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef]
- Plemper, R.K. Cell Entry of Enveloped Viruses. Curr. Opin. Virol. 2011, 1, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–533. [Google Scholar] [CrossRef] [Green Version]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.F.; Eaton, B.T.; et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef]
- Techaarpornkul, S.; Barretto, N.; Peeples, M.E. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 2001, 75, 6825–6834. [Google Scholar] [CrossRef] [Green Version]
- Karron, R.A.; Buonagurio, D.A.; Georgiu, A.F.; Whitehead, S.S.; Adamus, J.E.; Clements-Mann, M.L.; Harris, D.O.; Randolph, V.B.; Udem, S.A.; Murphy, B.R.; et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication In Vitro: Clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 1997, 94, 13961–13966. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.N.; Whitehead, S.S.; Collins, P.L. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication In Vitro and In Vivo. Virology 2001, 289, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Maher, C.F.; Hussell, T.; Blair, E.; Ring, C.J.; Openshaw, P.J. Recombinant respiratory syncytial virus lacking secreted glycoprotein G is attenuated, non-pathogenic but induces protective immunity. Microbes Infect. 2004, 6, 1049–1055. [Google Scholar] [CrossRef]
- Widjojoatmodjo, M.N.; Boes, J.; van Bers, M.; van Remmerden, Y.; Roholl, P.J.; Luytjes, W. A highly attenuated recombinant human respiratory syncytial virus lacking the G protein induces long-lasting protection in cotton rats. Virol. J. 2010, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Hotard, A.L.; Currier, M.G.; Lee, S.; Stobart, C.C.; Moore, M.L. Respiratory Syncytial Virus Attachment Glycoprotein Contribution to Infection Depends on the Specific Fusion Protein. J. Virol. 2016, 90, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Chan, Y.P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.F.; Skiniotis, G.; et al. Crystal Structure of the Pre-fusion Nipah Virus Fusion Glycoprotein Reveals a Novel Hexamer-of-Trimers Assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiguchi, T.; Fukuda, Y.; Matsuoka, R.; Kuroda, D.; Kubota, M.; Shirogane, Y.; Watanabe, S.; Tsumoto, K.; Kohda, D.; Plemper, R.K.; et al. Structures of the prefusion form of measles virus fusion protein in complex with inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 2496–2501. [Google Scholar] [CrossRef] [Green Version]
- McLellan, J.S.; Chen, M.; Leung, S.; Graepel, K.W.; Du, X.; Yang, Y.; Zhou, T.; Baxa, U.; Yasuda, E.; Beaumont, T.; et al. Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 2013, 340, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Stewart-Jones, G.B.E.; Chuang, G.Y.; Xu, K.; Zhou, T.; Acharya, P.; Tsybovsky, Y.; Ou, L.; Zhang, B.; Fernandez-Rodriguez, B.; Gilardi, V.; et al. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc. Natl. Acad. Sci. USA 2018, 115, 12265–12270. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.A.; Dutch, R.E.; Lamb, R.A.; Jardetzky, T.S. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell 1999, 3, 309–319. [Google Scholar] [CrossRef]
- Bose, S.; Zokarkar, A.; Welch, B.D.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Nat. Acad. Sci. USA 2012, 109, E2625–E2634. [Google Scholar] [CrossRef] [Green Version]
- Brindley, M.A.; Suter, R.; Schestak, I.; Kiss, G.; Wright, E.R.; Plemper, R.K. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J. Virol. 2013, 87, 11693–11703. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Stone, J.A.; Bradel-Tretheway, B.; Dabundo, J.; Benavides Montano, J.A.; Santos-Montanez, J.; Biering, S.B.; Nicola, A.V.; Iorio, R.M.; Lu, X.; et al. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog. 2013, 9, e1003770. [Google Scholar] [CrossRef]
- Bose, S.; Song, A.S.; Jardetzky, T.S.; Lamb, R.A. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J. Virol. 2014, 88, 3925–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardetzky, T.S.; Lamb, R.A. Activation of paramyxovirus membrane fusion and virus entry. Curr. Opin. Virol. 2014, 5, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, B.D.; Yuan, P.; Bose, S.; Kors, C.A.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog. 2013, 9, e1003534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plemper, R.K.; Hammond, A.L.; Cattaneo, R. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J. Biol. Chem. 2001, 276, 44239–44246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brindley, M.A.; Takeda, M.; Plattet, P.; Plemper, R.K. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. USA 2012, 109, E3018–E3027. [Google Scholar] [CrossRef] [Green Version]
- Brindley, M.A.; Chaudhury, S.; Plemper, R.K. Measles Virus Glycoprotein Complexes Preassemble Intracellularly and Relax during Transport to the Cell Surface in Preparation for Fusion. J. Virol. 2015, 89, 1230–1241. [Google Scholar] [CrossRef] [Green Version]
- Behera, A.K.; Matsuse, H.; Kumar, M.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem. Biophys. Res. Commun. 2001, 280, 188–195. [Google Scholar] [CrossRef]
- Currier, M.G.; Lee, S.; Stobart, C.C.; Hotard, A.L.; Villenave, R.; Meng, J.; Pretto, C.D.; Shields, M.D.; Nguyen, M.T.; Todd, S.O.; et al. EGFR Interacts with the Fusion Protein of Respiratory Syncytial Virus Strain 2–20 and Mediates Infection and Mucin Expression. PLoS Pathog. 2016, 12, e1005622. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, R.; Ward, M.; Bright, H.; Priest, R.; Foster, M.R.; Hurle, M.; Blair, E.; Bird, M. Isolation and characterisation of potential respiratory syncytial virus receptor(s) on epithelial cells. Microbes Infect. 2003, 5, 123–133. [Google Scholar] [CrossRef]
- Beeler, J.A.; van Wyke Coelingh, K. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: Effect of mutation upon fusion function. J. Virol. 1989, 63, 2941–2950. [Google Scholar] [CrossRef] [Green Version]
- Bossart, K.N.; Zhu, Z.; Middleton, D.; Klippel, J.; Crameri, G.; Bingham, J.; McEachern, J.A.; Green, D.; Hancock, T.J.; Chan, Y.P.; et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PLoS Pathog. 2009, 5, e1000642. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Mire, C.E.; Geisbert, J.B.; Chan, Y.P.; Agans, K.N.; Feldmann, F.; Fenton, K.A.; Zhu, Z.; Dimitrov, D.S.; Scott, D.P.; et al. Therapeutic treatment of Nipah virus infection in nonhuman primates with a neutralizing human monoclonal antibody. Sci. Transl. Med. 2014, 6, 242ra282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrandt, N.D.; Ji, H.; Patel, N.K.; Riggs, J.M.; Brewah, Y.A.; Ready, S.; Donacki, N.E.; Folliot, K.; Barnes, A.S.; Senthil, K.; et al. Isolation and characterization of monoclonal antibodies which neutralize human metapneumovirus In Vitro and In Vivo. J. Virol. 2006, 80, 7799–7806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Outlaw, V.K.; Bottom-Tanzer, S.; Kreitler, D.F.; Gellman, S.H.; Porotto, M.; Moscona, A. Dual Inhibition of Human Parainfluenza Type 3 and Respiratory Syncytial Virus Infectivity with a Single Agent. J. Am. Chem. Soc. 2019, 141, 12648–12656. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Yokoyama, C.C.; Palermo, L.M.; Mungall, B.; Aljofan, M.; Cortese, R.; Pessi, A.; Moscona, A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010, 84, 6760–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behzadi, M.A.; Leyva-Grado, V.H. Overview of Current Therapeutics and Novel Candidates Against Influenza, Respiratory Syncytial Virus, and Middle East Respiratory Syndrome Coronavirus Infections. Front. Microbiol. 2019, 10, 1327. [Google Scholar] [CrossRef] [Green Version]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Roymans, D.; De Bondt, H.L.; Arnoult, E.; Geluykens, P.; Gevers, T.; Van Ginderen, M.; Verheyen, N.; Kim, H.; Willebrords, R.; Bonfanti, J.F.; et al. Binding of a potent small-molecule inhibitor of six-helix bundle formation requires interactions with both heptad-repeats of the RSV fusion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Samuel, D.; Xing, W.; Niedziela-Majka, A.; Wong, J.S.; Hung, M.; Brendza, K.M.; Perron, M.; Jordan, R.; Sperandio, D.; Liu, X.; et al. GS-5806 inhibits pre- to postfusion conformational changes of the respiratory syncytial virus fusion protein. Antimicrob. Agents Chemother. 2015, 59, 7109–7112. [Google Scholar] [CrossRef] [Green Version]
- Plemper, R.K.; Doyle, J.; Sun, A.; Prussia, A.; Cheng, L.T.; Rota, P.A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob. Agents Chemother. 2005, 49, 3755–3761. [Google Scholar] [CrossRef] [Green Version]
- Plemper, R.K.; Hammond, A.L. Inhibition of Membrane Fusion as a Target for Antiviral Therapy. Antiinfect. Agents Med. Chem. 2007, 6, 248–262. [Google Scholar] [CrossRef]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Kilby, J.M.; Lalezari, J.P.; Eron, J.J.; Carlson, M.; Cohen, C.; Arduino, R.C.; Goodgame, J.C.; Gallant, J.E.; Volberding, P.; Murphy, R.L.; et al. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retrovir. 2002, 18, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Jamjian, M.C.; McNicholl, I.R. Enfuvirtide: First fusion inhibitor for treatment of HIV infection. Am. J. Health Syst. Pharm. 2004, 61, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Trottier, B.; Walmsley, S.; Reynes, J.; Piliero, P.; O’Hearn, M.; Nelson, M.; Montaner, J.; Lazzarin, A.; Lalezari, J.; Katlama, C.; et al. Safety of enfuvirtide in combination with an optimized background of antiretrovirals in treatment-experienced HIV-1-infected adults over 48 weeks. J. Acquir. Immune Defic. Syndr. 2005, 40, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battles, M.B.; Langedijk, J.P.; Furmanova-Hollenstein, P.; Chaiwatpongsakorn, S.; Costello, H.M.; Kwanten, L.; Vranckx, L.; Vink, P.; Jaensch, S.; Jonckers, T.H.; et al. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat. Chem. Biol. 2016, 12, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Lee, S.; Thakkar, V.D.; Luo, M.; Moore, M.L.; Plemper, R.K. Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E3441–E3449. [Google Scholar] [CrossRef] [Green Version]
- Weisshaar, M.; Cox, R.; Plemper, R.K. Blocking Respiratory Syncytial Virus Entry: A Story with Twists. DNA Cell Biol. 2015, 34, 505–510. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.M.; Toots, M.; Yoon, J.J.; Sourimant, J.; Ludeke, B.; Fearns, R.; Bourque, E.; Patti, J.; Lee, E.; Vernachio, J.; et al. Development of an allosteric inhibitor class blocking RNA elongation by the respiratory syncytial virus polymerase complex. J. Biol. Chem. 2018, 293, 16761–16777. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Weisshaar, M.; Lamb, K.; Chung, H.K.; Lin, M.Z.; Plemper, R.K. Replication-Competent Influenza Virus and Respiratory Syncytial Virus Luciferase Reporter Strains Engineered for Co-Infections Identify Antiviral Compounds in Combination Screens. Biochemistry 2015, 54, 5589–5604. [Google Scholar] [CrossRef] [Green Version]
- Plemper, R.K.; Erlandson, K.J.; Lakdawala, A.S.; Sun, A.; Prussia, A.; Boonsombat, J.; Aki-Sener, E.; Yalcin, I.; Yildiz, I.; Temiz-Arpaci, O.; et al. A target site for template-based design of measles virus entry inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 5628–5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.; Prussia, A.; White, L.K.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Compans, R.W.; Plemper, R.K. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J. Virol. 2006, 80, 1524–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Prussia, A.; Snyder, J.P.; Plemper, R.K. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J. Virol. 2007, 81, 8821–8826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prussia, A.J.; Plemper, R.K.; Snyder, J.P. Measles virus entry inhibitors: A structural proposal for mechanism of action and the development of resistance. Biochemistry 2008, 47, 13573–13583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.G.; Battles, M.B.; Lin, C.C.; Bianchi, S.; Corti, D.; McLellan, J.S. Alternative conformations of a major antigenic site on RSV F. PLoS Pathog. 2019, 15, e1007944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Battles, M.B.; Moin, S.M.; Chen, M.; Modjarrad, K.; Kumar, A.; Kanekiyo, M.; Graepel, K.W.; Taher, N.M.; Hotard, A.L.; et al. Structural basis of respiratory syncytial virus subtype-dependent neutralization by an antibody targeting the fusion glycoprotein. Nat. Commun. 2017, 8, 1877. [Google Scholar] [CrossRef]
- Ngwuta, J.O.; Chen, M.; Modjarrad, K.; Joyce, M.G.; Kanekiyo, M.; Kumar, A.; Yassine, H.M.; Moin, S.M.; Killikelly, A.M.; Chuang, G.Y.; et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci. Transl. Med. 2015, 7, 309ra162. [Google Scholar] [CrossRef] [Green Version]
- Rossey, I.; McLellan, J.S.; Saelens, X.; Schepens, B. Clinical Potential of Prefusion RSV F-specific Antibodies. Trends Microbiol. 2018, 26, 209–219. [Google Scholar] [CrossRef]
- Wen, X.; Mousa, J.J.; Bates, J.T.; Lamb, R.A.; Crowe, J.E., Jr.; Jardetzky, T.S. Structural basis for antibody cross-neutralization of respiratory syncytial virus and human metapneumovirus. Nat. Microbiol. 2017, 2, 16272. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, Y.; Diaz, D.; Pena-Briseno, A.; Murray, J.; Huang, J.; Tripp, R.A.; Mousa, J.J. A Potent Neutralizing Site III-Specific Human Antibody Neutralizes Human Metapneumovirus In Vivo. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Iwuchukwu, O.P.; Avadhanula, V.; Aideyan, L.O.; McBride, T.J.; Ferlic-Stark, L.L.; Patel, K.D.; Piedra, F.A.; Shah, D.P.; Chemaly, R.F.; et al. Antigenic Site-Specific Competitive Antibody Responses to the Fusion Protein of Respiratory Syncytial Virus Were Associated With Viral Clearance in Hematopoietic Cell Transplantation Adults. Front. Immunol. 2019, 10, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousa, J.J.; Kose, N.; Matta, P.; Gilchuk, P.; Crowe, J.E., Jr. A novel pre-fusion conformation-specific neutralizing epitope on the respiratory syncytial virus fusion protein. Nat. Microbiol. 2017, 2, 16271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.J.; Hierholzer, J.C.; Stone, Y.O.; Tsou, C.; Fernie, B.F. Identification of epitopes on respiratory syncytial virus proteins by competitive binding immunoassay. J. Clin. Microbiol. 1986, 23, 475–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.A.; Bustos, R.; Orvell, C.; Berois, M.; Arbiza, J.; Garcia-Barreno, B.; Melero, J.A. Antigenic structure of human respiratory syncytial virus fusion glycoprotein. J. Virol. 1998, 72, 6922–6928. [Google Scholar] [CrossRef] [Green Version]
- Avanzato, V.A.; Oguntuyo, K.Y.; Escalera-Zamudio, M.; Gutierrez, B.; Golden, M.; Kosakovsky Pond, S.L.; Pryce, R.; Walter, T.S.; Seow, J.; Doores, K.J.; et al. A structural basis for antibody-mediated neutralization of Nipah virus reveals a site of vulnerability at the fusion glycoprotein apex. Proc. Natl. Acad. Sci. USA 2019, 116, 25057–25067. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.V.; Chan, Y.P.; Park, Y.J.; Snijder, J.; Da Silva, S.C.; Vu, B.; Yan, L.; Feng, Y.R.; Rockx, B.; Geisbert, T.W.; et al. An antibody against the F glycoprotein inhibits Nipah and Hendra virus infections. Nat. Struct. Mol. Biol. 2019, 26, 980–987. [Google Scholar] [CrossRef]
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Tse, C. Predicting protein druggability. Drug Discov. Today 2005, 10, 1675–1682. [Google Scholar] [CrossRef]
- Lowe, D. Replacing Antibodies With Small Molecules. In The Pipeline; Science Translational Medicine: Washington, DC, USA, 2018; Volume 2020. [Google Scholar]
- Wang, T.; Wu, X.; Guo, C.; Zhang, K.; Xu, J.; Li, Z.; Jiang, S. Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. 2019, 62, 1715–1730. [Google Scholar] [CrossRef]
- van Dongen, M.J.P.; Kadam, R.U.; Juraszek, J.; Lawson, E.; Brandenburg, B.; Schmitz, F.; Schepens, W.B.G.; Stoops, B.; van Diepen, H.A.; Jongeneelen, M.; et al. A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science 2019, 363. [Google Scholar] [CrossRef]
- Lambkin-Williams, R.; Noulin, N.; Mann, A.; Catchpole, A.; Gilbert, A.S. The human viral challenge model: Accelerating the evaluation of respiratory antivirals, vaccines and novel diagnostics. Respir. Res. 2018, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.P.; Whitley, R.J.; Mackman, R.L.; Scaglioni-Weinlich, C.; Harrison, L.; Farrell, E.; McBride, S.; Lambkin-Williams, R.; Jordan, R.; Xin, Y.; et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014, 371, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVincenzo, J.; Tait, D.; Efthimiou, J.; Mori, J.; Kim, Y.I.; Thomas, E.; Wilson, L.; Harland, R.; Mathews, N.; Cockerill, S.; et al. A Randomized, Placebo-Controlled, Respiratory Syncytial Virus Human Challenge Study of the Antiviral Efficacy, Safety, and Pharmacokinetics of RV521, an Inhibitor of the RSV-F Protein. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korell, J.; Green, B.; DeVincenzo, J.; Huntjens, D. A human challenge model for respiratory syncytial virus kinetics, the pharmacological effect of a novel fusion inhibitor, and the modelling of symptoms scores. Eur. J. Pharm. Sci. 2017, 109, S154–S160. [Google Scholar] [CrossRef]
- Hanfelt-Goade, D.; Maimon, N.; Nimer, A.; Riviere, F.; Catherinot, E.; Ison, M.; Jeong, S.; Walsh, E.; Gafter-Gvili, A.; Nama, S.; et al. A Phase 2b, Randomized, Double-Blind, Placebo-Controlled Trial of Presatovir (GS-5806), a Novel Oral RSV Fusion Inhibitor, for the Treatment of Respiratory Syncytial Virus (RSV) in Hospitalized Adults. Am. J. Respir. Crit. Care Med. 2018, 197, A4457. [Google Scholar]
- Gottlieb, J.; Torres, F.; Haddad, T.; Dhillon, G.; Dilling, D.F.; Knoop, C.; Rampolla, R.; Walia, R.; Ahya, V.; Kessler, R.; et al. A Phase 2b Randomized Controlled Trial of Presatovir, an Oral RSV Fusion Inhibitor, for the Treatment of Respiratory Syncytial Virus (RSV) in Lung Transplant (LT) Recipients. J. Heart Lung Transplant. 2018, 37, S155. [Google Scholar] [CrossRef]
- Marty, F.M.; Chemaly, R.F.; Mullane, K.M.; Lee, D.G.; Hirsch, H.H.; Small, C.B.; Bergeron, A.; Shoham, S.; Ljungman, P.; Waghmare, A.; et al. A Phase 2b, Randomized, Double-blind, Placebo-Controlled Multicenter Study Evaluating Antiviral Effects, Pharmacokinetics, Safety, and Tolerability of Presatovir in Hematopoietic Cell Transplant Recipients with Respiratory Syncytial Virus (RSV) Infection of the Lower Respiratory Tract. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Porter, D.P.; Guo, Y.; Perry, J.; Gossage, D.L.; Watkins, T.R.; Chien, J.W.; Jordan, R. Assessment of drug resistance during phase 2b clinical trials of presatovir in adults naturally infected with respiratory syncytial virus. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Noton, S.L.; Nagendra, K.; Dunn, E.F.; Mawhorter, M.E.; Yu, Q.; Fearns, R. Respiratory Syncytial Virus Inhibitor AZ-27 Differentially Inhibits Different Polymerase Activities at the Promoter. J. Virol. 2015, 89, 7786–7798. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.J.; Toots, M.; Lee, S.; Lee, M.E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob. Agents Chemother. 2018, 62, e00766-18. [Google Scholar] [CrossRef] [Green Version]
- DeVincenzo, J.P.; McClure, M.W.; Symons, J.A.; Fathi, H.; Westland, C.; Chanda, S.; Lambkin-Williams, R.; Smith, P.; Zhang, Q.; Beigelman, L.; et al. Activity of Oral ALS-008176 in a Respiratory Syncytial Virus Challenge Study. N. Engl. J. Med. 2015, 373, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, R.; Plemper, R.K. Structure-guided design of small-molecule therapeutics against RSV disease. Expert Opin. Drug Discov. 2016, 11, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Plemper, R.K. The paramyxovirus polymerase complex as a target for next-generation anti-paramyxovirus therapeutics. Front. Microbiol. 2015, 6, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plemper, R.K. Measles resurgence and drug development. Curr. Opin. Virol. 2020, in press. [Google Scholar]
- Svitek, N.; Gerhauser, I.; Goncalves, C.; Grabski, E.; Doring, M.; Kalinke, U.; Anderson, D.E.; Cattaneo, R.; von Messling, V. Morbillivirus control of the interferon response: Relevance of STAT2 and mda5 but not STAT1 for canine distemper virus virulence in ferrets. J. Virol. 2014, 88, 2941–2950. [Google Scholar] [CrossRef] [Green Version]
- Devaux, P.; Hudacek, A.W.; Hodge, G.; Reyes-Del Valle, J.; McChesney, M.B.; Cattaneo, R. A recombinant measles virus unable to antagonize STAT1 function cannot control inflammation and is attenuated in rhesus monkeys. J. Virol. 2011, 85, 348–356. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate ID | Sponsor | Phase | Outcome | Trial ID |
|---|---|---|---|---|
| GS-5806 (Presatovir) | Gilead Sciences | Phase IIb | well tolerated, did not achieve primary endpoints | NCT02254421 |
| GS-5806 (Presatovir) | Gilead Sciences | Phase IIb | well tolerated, did not achieve primary endpoints | NCT02534350 |
| GS-5806 (Presatovir) | Gilead Sciences | Phase IIb | well tolerated, did not achieve primary endpoints | NCT02135614 |
| GS-5806 (Presatovir) | Gilead Sciences | Phase IIa | reduction in RSV load | NCT01756482 |
| RV521 | ReViral Ltd. | Phase II | estimated completion October 2021 | NCT04225897 |
| MDT-637 | MicroDose Therapeutx, Inc | Phase I | well tolerated | NCT01556607 |
| BTA-C585 (Enzaplatovir) | Biota Pharmaceuticals, Inc. | Phase IIa | suspended | NCT02718937 |
| MK-1654 | Merck Sharp & Dohme Corp. | Phase IIa | estimated completion April 2020 | NCT04086472 |
| AK-0529 (Ziresovir) | Ark Biosciences Inc. | Phase II | completed | NCT02654171 |
| BTA9881 | Biota Scientific Management Pty Ltd. | Phase I | discontinued | NCT00504907 |
| JNJ-53718678 | Janssen Research & Development, LLC | Phase II | estimated completion November 2020 | NCT03656510 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggarwal, M.; Plemper, R.K. Structural Insight into Paramyxovirus and Pneumovirus Entry Inhibition. Viruses 2020, 12, 342. https://doi.org/10.3390/v12030342
Aggarwal M, Plemper RK. Structural Insight into Paramyxovirus and Pneumovirus Entry Inhibition. Viruses. 2020; 12(3):342. https://doi.org/10.3390/v12030342
Chicago/Turabian StyleAggarwal, Megha, and Richard K Plemper. 2020. "Structural Insight into Paramyxovirus and Pneumovirus Entry Inhibition" Viruses 12, no. 3: 342. https://doi.org/10.3390/v12030342