CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
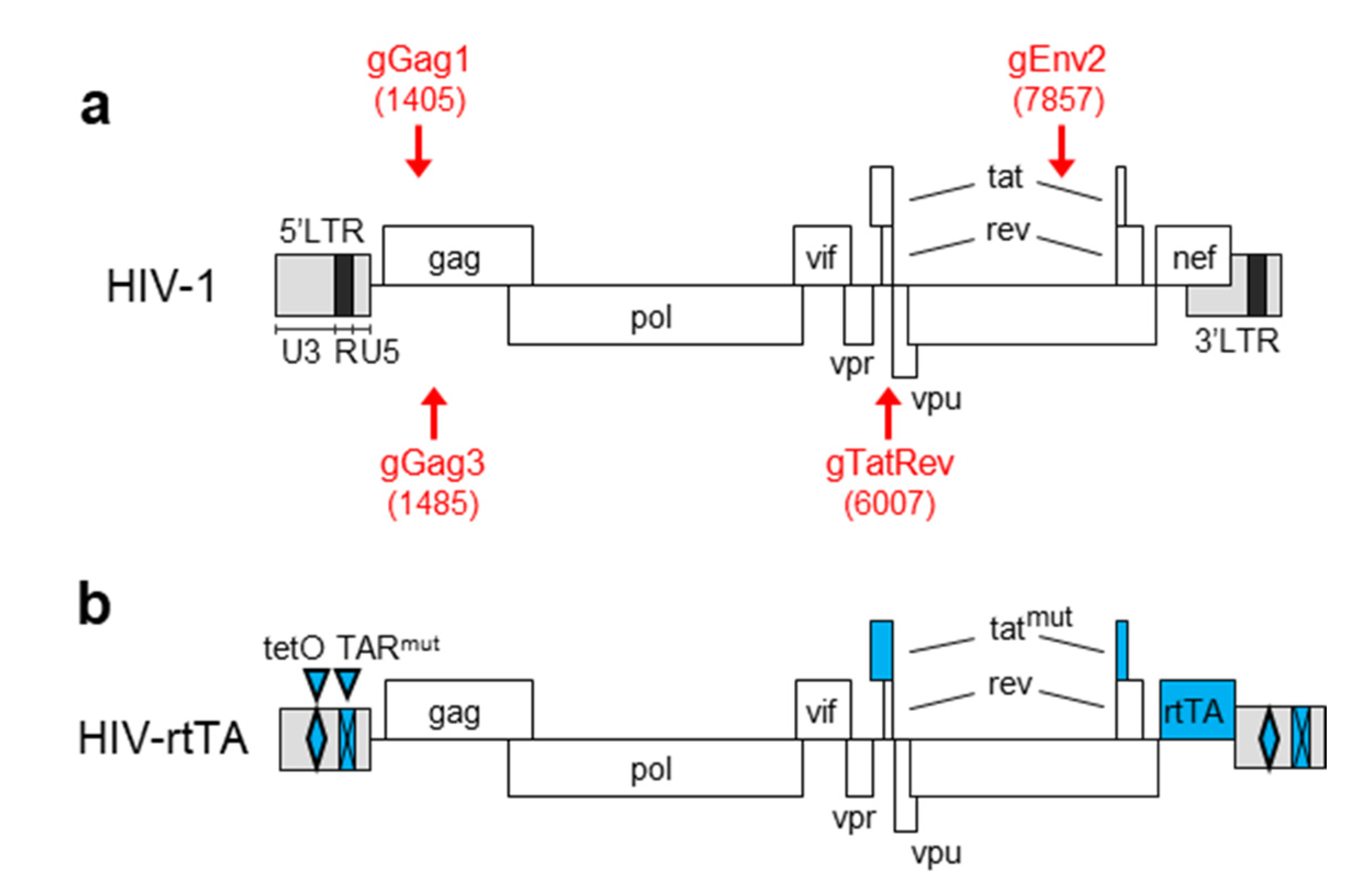
2.1. Plasmids
2.2. Cell Culture, Transfection and Transduction
2.3. Virus Stocks and Infection
2.4. PCR and qPCR Analysis
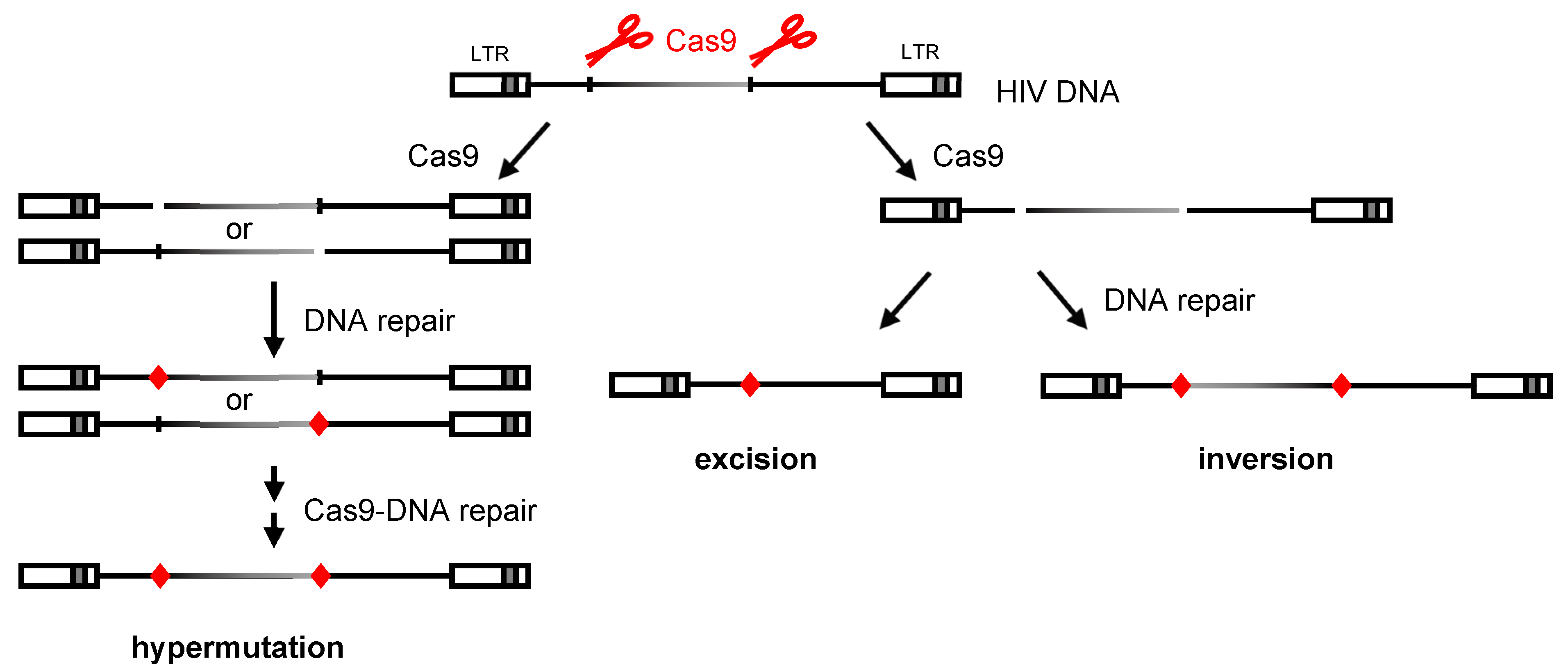
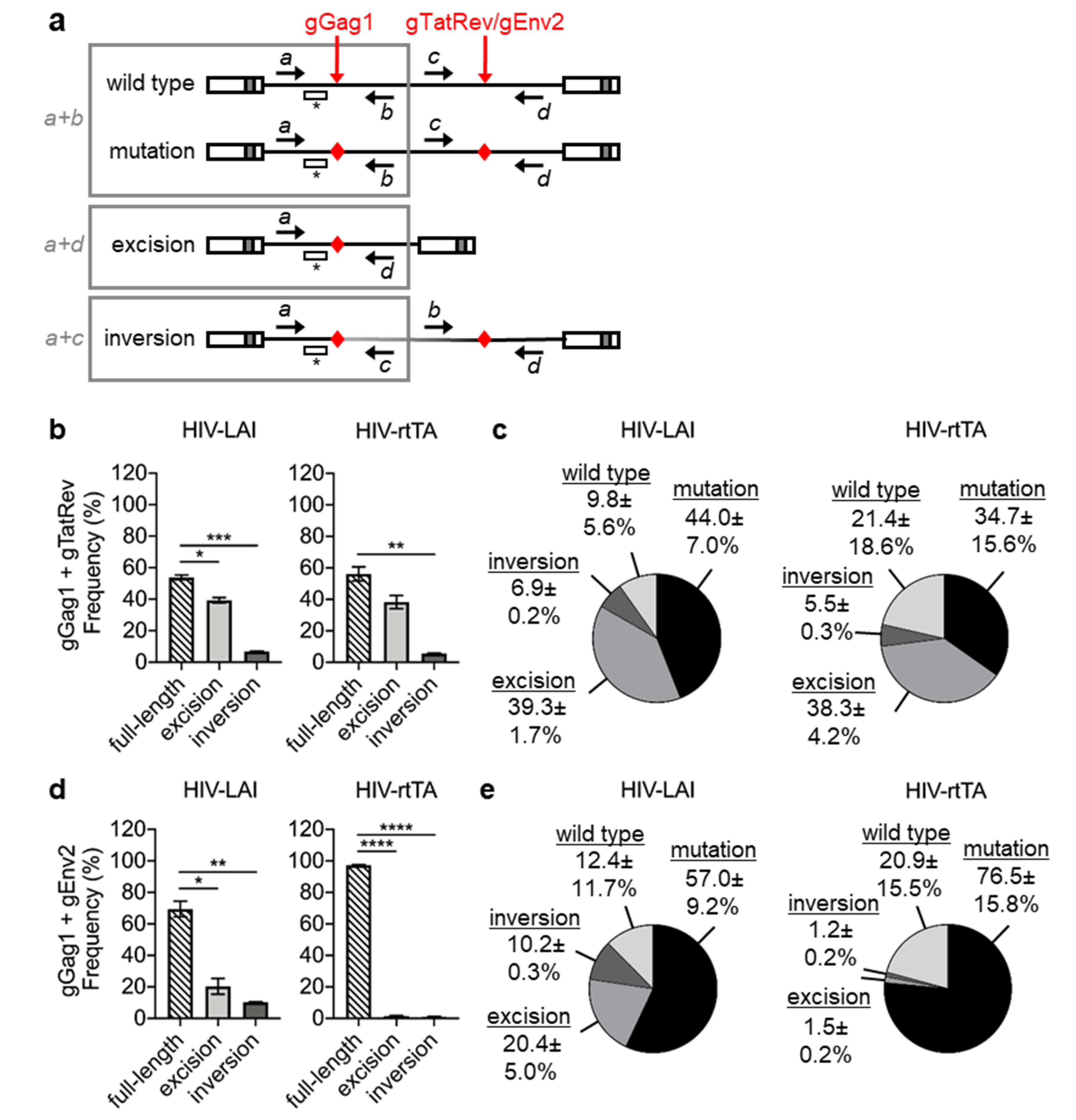
3. Results
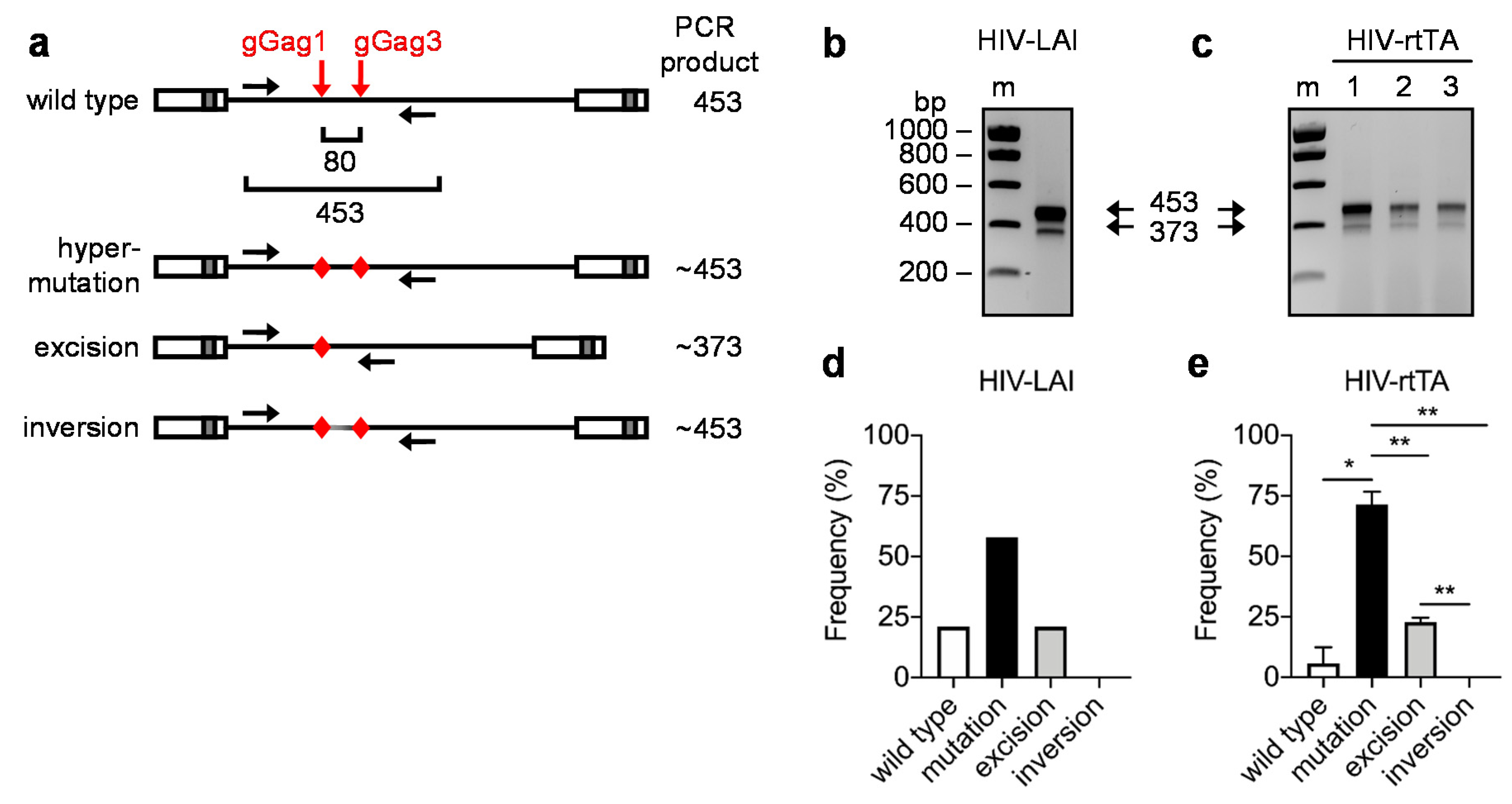
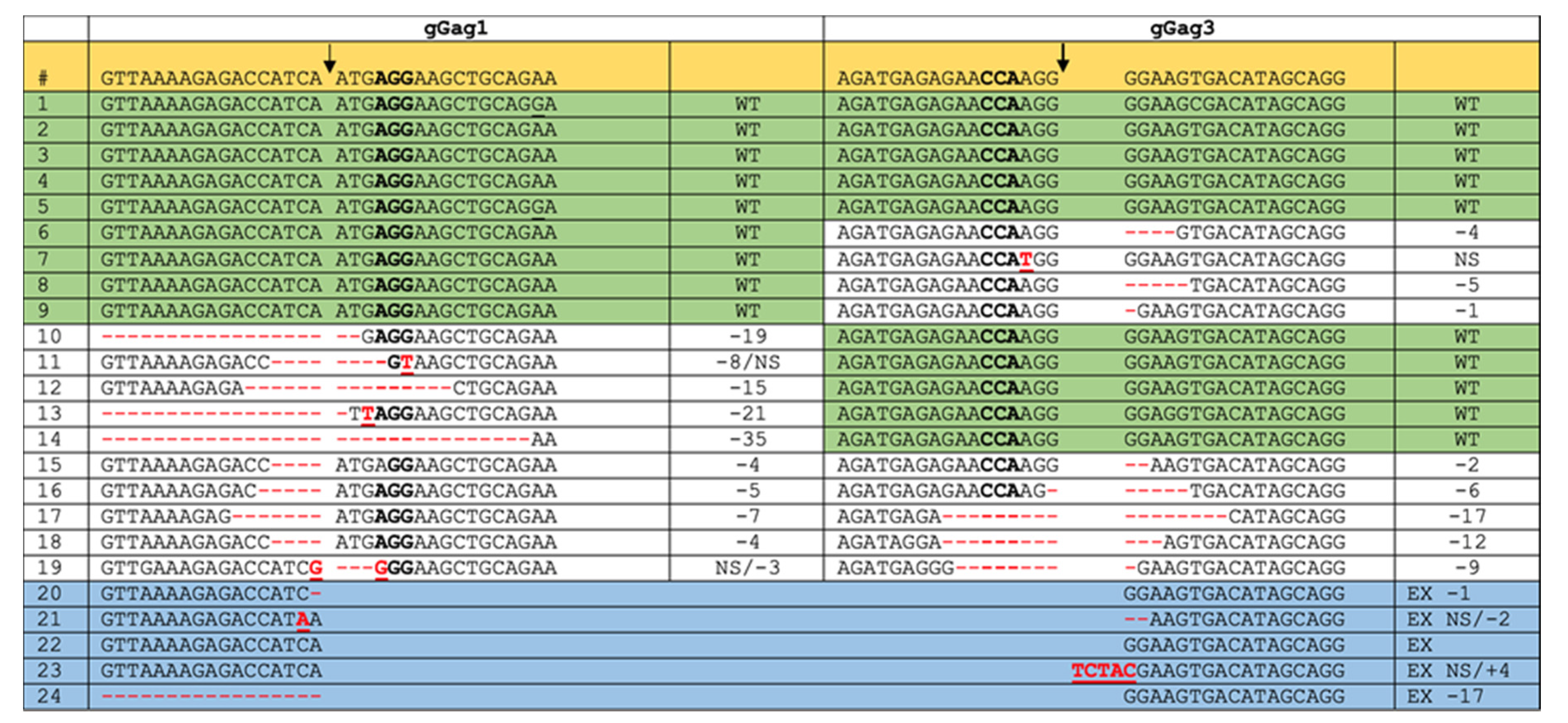
3.1. PCR and Sequence Analysis of HIV Proviral DNA upon Dual gRNA/Cas9 Attack
3.2. Quantitative PCR Analysis of HIV Proviral DNA upon Dual gRNA/Cas9 Attack
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Palenikova, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.Y.; Wang, C.; Lee, H.K.; Yoo, K.H.; Zeng, X.; Kuhns, T.; Yang, C.M.; Mohr, T.; Liu, C.; Hennighausen, L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017, 8, 15464. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, R.; Palange, N.J.; Satheka, A.C.; Togo, J.; An, Y.; Humphrey, M.; Ban, L.; Ji, Y.; Jin, H.; et al. Large Genomic Fragment Deletions and Insertions in Mouse Using CRISPR/Cas9. PLoS ONE 2015, 10, e0120396. [Google Scholar] [CrossRef] [Green Version]
- Boroviak, K.; Doe, B.; Banerjee, R.; Yang, F.; Bradley, A. Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 2016, 54, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Shou, J.; Guo, Y.; Tang, Y.; Wu, Y.; Jia, Z.; Zhai, Y.; Chen, Z.; Xu, Q.; Wu, Q. Efficient inversions and duplications of mammalian regulatory DNA elements and gene clusters by CRISPR/Cas9. J. Mol. Cell Biol. 2015, 7, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Li, F.; Yang, F.; Putatunda, R.; Young, W.B.; Khalili, K.; Hu, W.; Zhang, Y. Functional screening of guide RNAs targeting the regulatory and structural HIV-1 viral genome for a cure of AIDS. AIDS 2016, 30, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Dampier, W.; Nonnemacher, M.R.; Sullivan, N.T.; Jacobson, J.M.; Wigdahl, B. HIV excision utilizing CRISPR/Cas9 technology: Attacking the proviral quasispecies in reservoirs to achieve a cure. MOJ Immunol. 2014, 1. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, K.E.; Bundschuh, R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci. Rep. 2016, 6, 29530. [Google Scholar] [CrossRef]
- Ueda, S.; Ebina, H.; Kanemura, Y.; Misawa, N.; Koyanagi, Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol. Immunol. 2016, 60, 483–496. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Cui, Y.C.; Pan, Q.; Zhu, W.; Gendron, P.; Guo, F.; Cen, S.; Witcher, M.; Liang, C. HIV-1 employs multiple mechanisms to resist Cas9/single guide RNA targeting the viral primer binding site. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Wainberg, M.A.; Das, A.T.; Berkhout, B. CRISPR/Cas9: A double-edged sword when used to combat HIV infection. Retrovirology 2016, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A combinatorial CRISPR-Cas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [Green Version]
- Lebbink, R.J.; de Jong, D.C.; Wolters, F.; Kruse, E.M.; van Ham, P.M.; Wiertz, E.J.; Nijhuis, M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017, 7, 41968. [Google Scholar] [CrossRef]
- Pham, H.; Kearns, N.A.; Maehr, R. Transcriptional Regulation with CRISPR/Cas9 Effectors in Mammalian Cells. Methods Mol. Biol. 2016, 1358, 43–57. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peden, K.; Emerman, M.; Montagnier, L. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1 LAI, HIV-1 MAL, and HIV-1 ELI. Virology 1991, 185, 661–672. [Google Scholar] [CrossRef]
- Das, A.T.; Verhoef, K.; Berkhout, B. A conditionally replicating virus as a novel approach toward an HIV vaccine. Methods Enzymol. 2004, 388, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, K.; Marzio, G.; Hillen, W.; Bujard, H.; Berkhout, B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J. Virol. 2001, 75, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Vink, M.; Berkhout, B.; Das, A.T. Modification of the Tet-On regulatory system prevents the conditional-live HIV-1 variant from losing doxycycline-control. Retrovirology 2006, 3, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [Green Version]
- ter Brake, O.; Konstantinova, P.; Ceylan, M.; Berkhout, B. Silencing of HIV-1 with RNA interference: A multiple shRNA approach. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 883–892. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Jan, B.; van den Berg, H.; Berkhout, B. Construction of doxycyline-dependent mini-HIV-1 variants for the development of a virotherapy against leukemias. Retrovirology 2006, 3, 64. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Zhou, X.; Vink, M.; Klaver, B.; Verhoef, K.; Marzio, G.; Berkhout, B. Viral evolution as a tool to improve the tetracycline-regulated gene expression system. J. Biol. Chem. 2004, 279, 18776–18782. [Google Scholar] [CrossRef] [Green Version]
- Debode, F.; Marien, A.; Janssen, E.; Bragard, C.; Berben, G. Influence of the amplicon length on real-time PCR results. Biotechnol. Agron. Soc. Environ. 2017, 21, 3–11. [Google Scholar]
- Kalle, E.; Kubista, M.; Rensing, C. Multi-template polymerase chain reaction. Biomol. Detect. Quantif. 2014, 2, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.H.; Ren, Y.; Thomas, A.S.; Chan, D.; Mueller, S.; Ward, A.R.; Patel, S.; Bollard, C.M.; Cruz, C.R.; Karandish, S.; et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J. Clin. Investig. 2018, 128, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef]
- Montagna, C.; Petris, G.; Casini, A.; Maule, G.; Franceschini, G.M.; Zanella, I.; Conti, L.; Arnoldi, F.; Burrone, O.R.; Zentilin, L.; et al. VSV-G-enveloped vesicles for traceless delivery of CRISPR-Cas9. Mol. Nucleic Acids 2018, 12, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.A.; Coke, L.M.; Richie, C.T.; Fortuno, L.V.; Park, A.Y.; Harvey, B.K. Gesicle-mediated delivery of CRISPR/Cas9 ribonucleoprotein complex for inactivating the HIV provirus. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Bella, R.; Kaminski, R.; Mancuso, P.; Young, W.B.; Chen, C.; Sariyer, R.; Fischer, T.; Amini, S.; Ferrante, P.; Jacobson, J.M.; et al. Removal of HIV DNA by CRISPR from patient blood engrafts in humanized mice. Mol. Nucleic Acids 2018, 12, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcis, G.; Kootstra, N.A.; Hooibrink, B.; van Montfort, T.; Maurer, I.; Groen, K.; Jurriaans, S.; Bakker, M.; van Lint, C.; Berkhout, B.; et al. CD32(+)CD4(+) T cells are highly enriched for HIV DNA and can support transcriptional latency. Cell Rep. 2020, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descours, B.; Petitjean, G.; Lopez-Zaragoza, J.L.; Bruel, T.; Raffel, R.; Psomas, C.; Reynes, J.; Lacabaratz, C.; Levy, Y.; Schwartz, O.; et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature 2017, 543, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene 2015, 26, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Darcis, G.; Binda, C.S.; Klaver, B.; Herrera-Carrillo, E.; Berkhout, B.; Das, A.T. The impact of HIV-1 genetic diversity on CRISPR-Cas9 antiviral activity and viral escape. Viruses 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Dampier, W.; Sullivan, N.T.; Mell, J.C.; Pirrone, V.; Ehrlich, G.D.; Chung, C.H.; Allen, A.G.; DeSimone, M.; Zhong, W.; Kercher, K.; et al. Broad-Spectrum and Personalized Guide RNAs for CRISPR/Cas9 HIV-1 Therapeutics. Aids Res. Hum. Retrovir. 2018, 34, 950–960. [Google Scholar] [CrossRef]
- Sullivan, N.T.; Dampier, W.; Chung, C.H.; Allen, A.G.; Atkins, A.; Pirrone, V.; Homan, G.; Passic, S.; Williams, J.; Zhong, W.; et al. Novel gRNA design pipeline to develop broad-spectrum CRISPR/Cas9 gRNAs for safe targeting of the HIV-1 quasispecies in patients. Sci. Rep. 2019, 9, 17088. [Google Scholar] [CrossRef]
- Roychoudhury, P.; De Silva Feelixge, H.; Reeves, D.; Mayer, B.T.; Stone, D.; Schiffer, J.T.; Jerome, K.R. Viral diversity is an obligate consideration in CRISPR/Cas9 designs for targeting the HIV reservoir. BMC Biol. 2018, 16, 75. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binda, C.S.; Klaver, B.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA. Viruses 2020, 12, 330. https://doi.org/10.3390/v12030330
Binda CS, Klaver B, Berkhout B, Das AT. CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA. Viruses. 2020; 12(3):330. https://doi.org/10.3390/v12030330
Chicago/Turabian StyleBinda, Caroline S., Bep Klaver, Ben Berkhout, and Atze T. Das. 2020. "CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA" Viruses 12, no. 3: 330. https://doi.org/10.3390/v12030330