Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune Response against HCMV
2.1. Cellular Immune Response against HCMV
2.2. Humoral Immune Response against HCMV
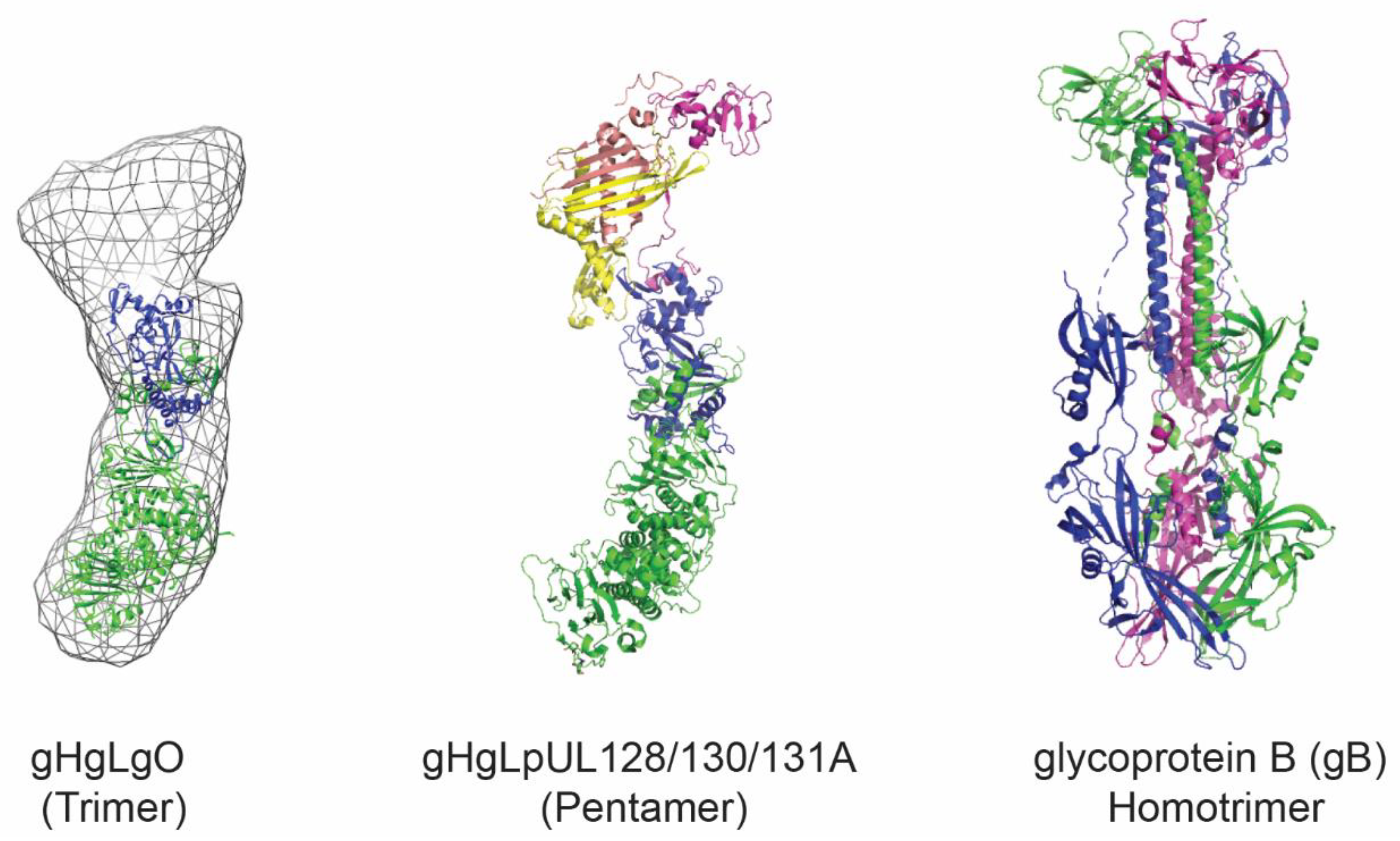
3. HCMV Antigens as Vaccine Candidates
4. Virus-Like Particles and Nanoparticles for Antigen Display
4.1. Developed HCMV VLP Vaccine
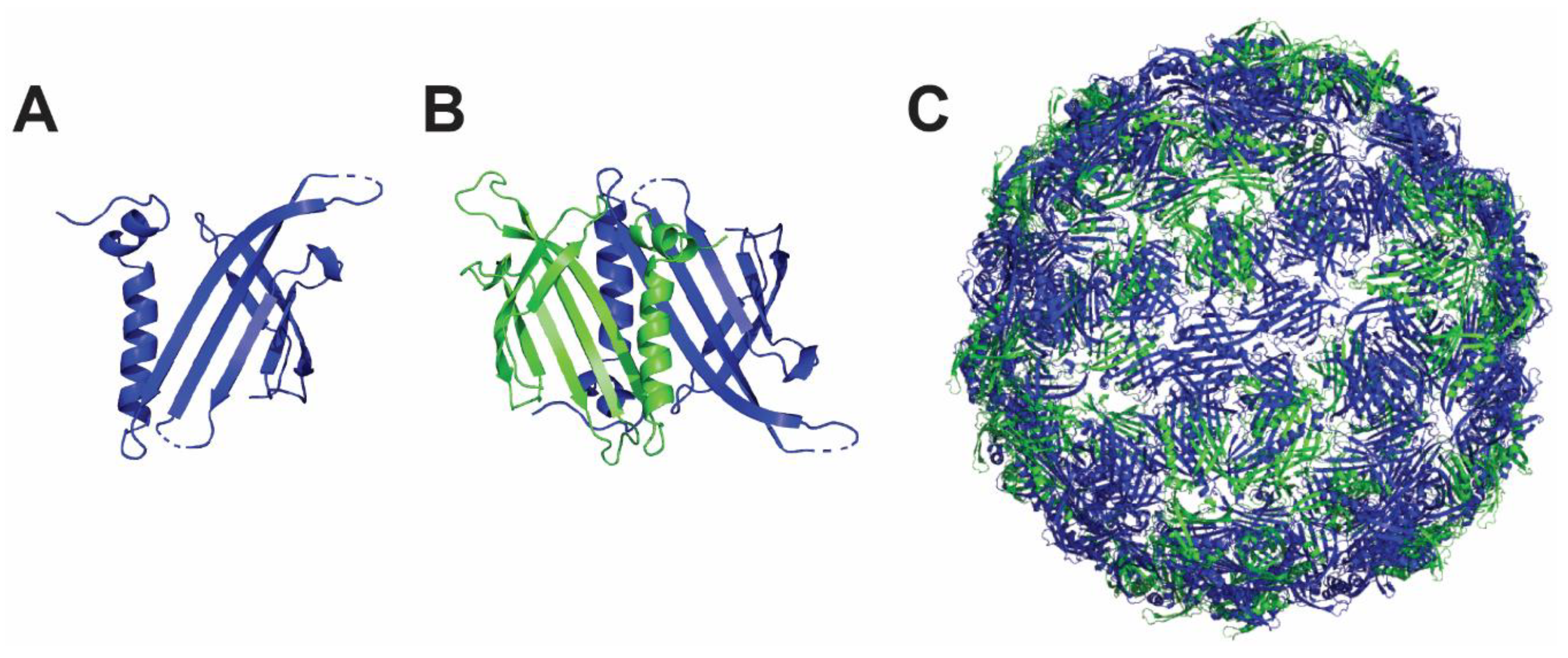
4.2. Hepatitis B Core Antigen Nanoparticle
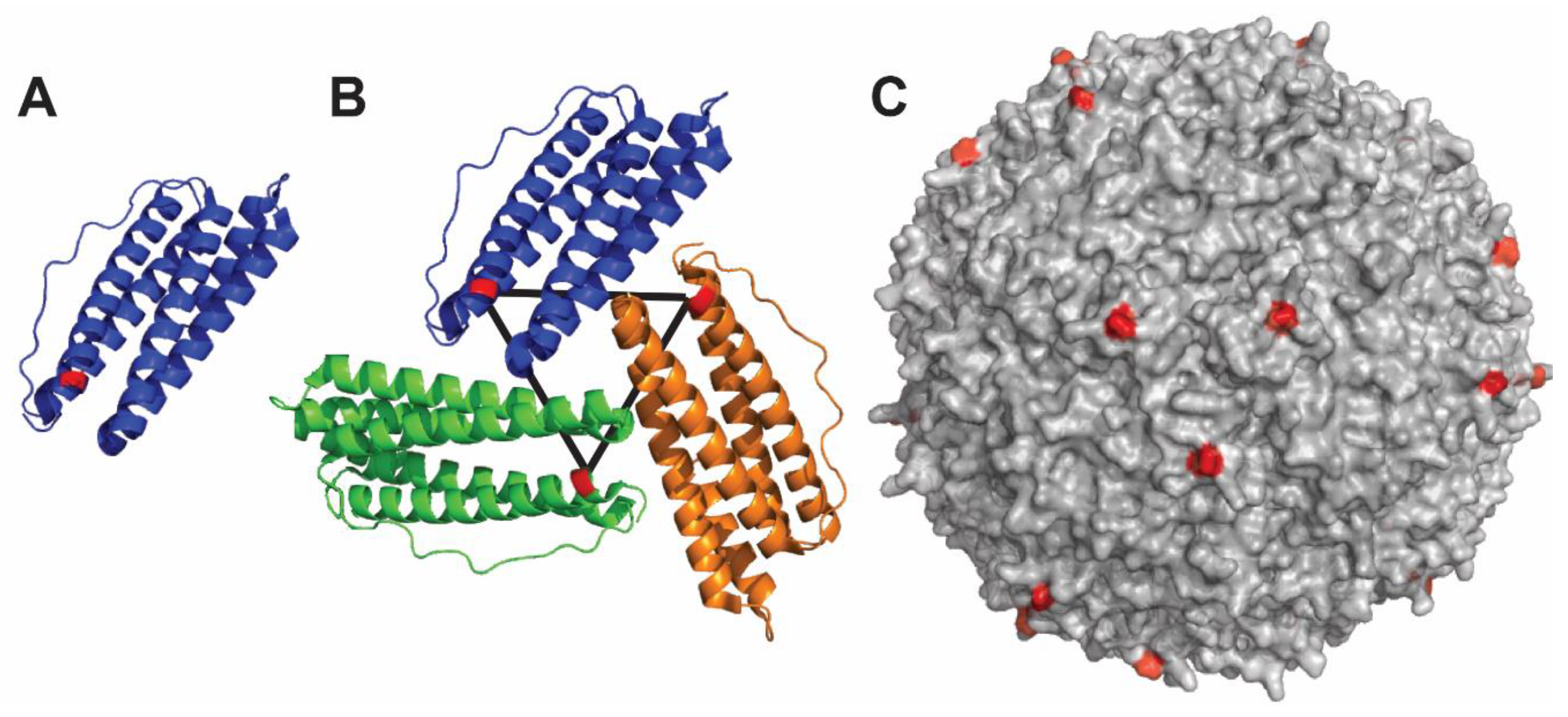
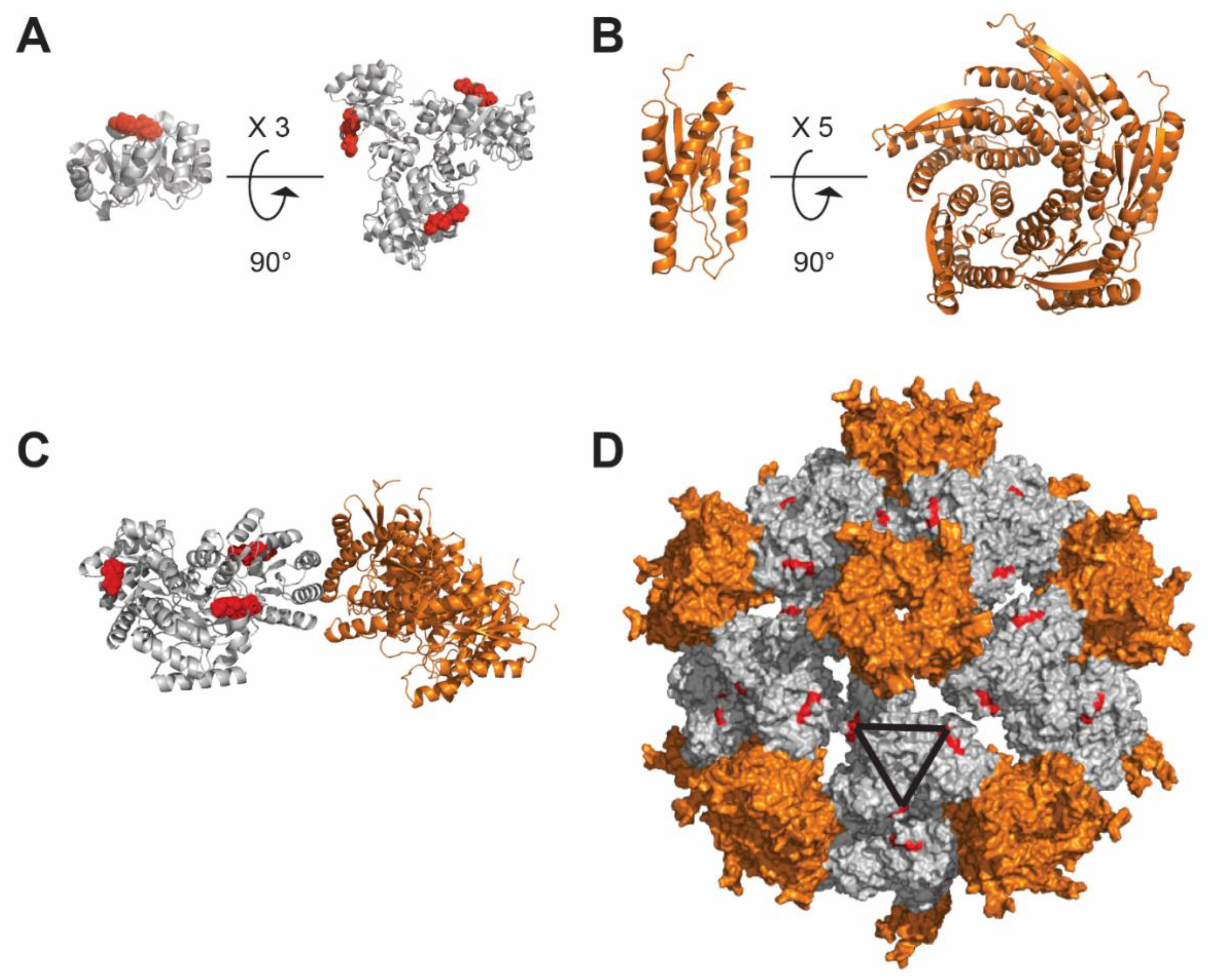
4.3. Ferritin-Based Nanoparticle
4.4. Qβ Nanoparticle
4.5. De Novo Design-Based Nanoparticle
4.6. SpyCatcher and SpyTag for Nanoparticle Formation
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2018, 37, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Zavattoni, M.; Baldanti, F.; Furione, M.; Chezzi, L.; Revello, M.G.; Percivalle, E. Circulating cytomegalic endothelial cells are associated with high human cytomegalovirus (HCMV) load in AIDS patients with late-stage disseminated HCMV disease. J. Med. Virol. 1998, 55, 64–74. [Google Scholar] [CrossRef]
- Lilleri, D.; Gerna, G. Strategies to control human cytomegalovirus infection in adult hematopoietic stem cell transplant recipients. Immunotherapy 2016, 8, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Britt, W.J. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J. Infect. Dis. 1995, 171, 1115–1121. [Google Scholar] [CrossRef]
- Spinillo, A.; Gerna, G. Hyperimmune globulin to prevent congenital CMV infection. N. Engl. J. Med. 2014, 370, 2544–2545. [Google Scholar] [CrossRef]
- James, S.H.; Kimberlin, D.W. Advances in the prevention and treatment of congenital cytomegalovirus infection. Curr. Opin. Pediatrics 2016, 28, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatrics 2017, 43, 38. [Google Scholar] [CrossRef]
- Gomes, A.C.; Griffiths, P.D.; Reeves, M.B. The Humoral Immune Response Against the gB Vaccine: Lessons Learnt from Protection in Solid Organ Transplantation. Vaccines 2019, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R. Recombinant cytomegalovirus glycoprotein B vaccine: Rethinking the immunological basis of protection. Proc. Natl. Acad. Sci. USA 2018, 115, 6110–6112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.S.; Herold, B.C.; Permar, S.R. A new era in cytomegalovirus vaccinology: Considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. Npj Vaccines 2018, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foglierini, M.; Marcandalli, J.; Perez, L. HCMV Envelope Glycoprotein Diversity Demystified. Front. Microbiol. 2019, 10, 1005. [Google Scholar] [CrossRef]
- Nelson, C.S.; Huffman, T.; Jenks, J.A.; Cisneros de la Rosa, E.; Xie, G.; Vandergrift, N.; Pass, R.F.; Pollara, J.; Permar, S.R. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc. Natl. Acad. Sci. USA 2018, 115, 6267–6272. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraniak, I.; Gomes, A.C.; Sodi, I.; Langstone, T.; Rothwell, E.; Atkinson, C.; Pichon, S.; Piras-Douce, F.; Griffiths, P.D.; Reeves, M.B. Seronegative patients vaccinated with cytomegalovirus gB-MF59 vaccine have evidence of neutralising antibody responses against gB early post-transplantation. EBioMedicine 2019, 50, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerna, G.; Lilleri, D.; Fornara, C.; Bruno, F.; Gabanti, E.; Cane, I.; Furione, M.; Revello, M.G. Differential kinetics of human cytomegalovirus load and antibody responses in primary infection of the immunocompetent and immunocompromised host. J. Gen. Virol. 2015, 96, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Quinn, M.; Snyder, C.M. CMV-Specific CD8 T Cell Differentiation and Localization: Implications for Adoptive Therapies. Front. Immunol. 2016, 7, 352. [Google Scholar] [CrossRef] [Green Version]
- Bialas, K.M.; Westreich, D.; Cisneros de la Rosa, E.; Nelson, C.S.; Kauvar, L.M.; Fu, T.M.; Permar, S.R. Maternal Antibody Responses and Nonprimary Congenital Cytomegalovirus Infection of HIV-1-Exposed Infants. J. Infect. Dis. 2016, 214, 1916–1923. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Nelson, C.S.; Martinez, D.R.; Permar, S.R. Maternal immune correlates of protection against placental transmission of cytomegalovirus. Placenta 2017, 60 (Suppl. 1), S73–S79. [Google Scholar] [CrossRef]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human Cytomegalovirus Latency and Reactivation in Allogeneic Hematopoietic Stem Cell Transplant Recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, B.A.; Wills, M.R.; Sinclair, J.H. Advances in the treatment of cytomegalovirus. Br. Med. Bull. 2019, 131, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.W.; Tomasec, P.; Stanton, R.J.; Armstrong, M.; Prod’homme, V.; Aicheler, R.; McSharry, B.P.; Rickards, C.R.; Cochrane, D.; Llewellyn-Lacey, S.; et al. Modulation of natural killer cells by human cytomegalovirus. J. Clin. Virol. 2008, 41, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Rist, M.; Cooper, L.; Elkington, R.; Walker, S.; Fazou, C.; Tellam, J.; Crough, T.; Khanna, R. Ex vivo expansion of human cytomegalovirus-specific cytotoxic T cells by recombinant polyepitope: Implications for HCMV immunotherapy. Eur. J. Immunol. 2005, 35, 996–1007. [Google Scholar] [CrossRef]
- Rohrlich, P.S.; Cardinaud, S.; Lule, J.; Montero-Julian, F.A.; Prodhomme, V.; Firat, H.; Davignon, J.L.; Perret, E.; Monseaux, S.; Necker, A.; et al. Use of a lentiviral vector encoding a HCMV-chimeric IE1-pp65 protein for epitope identification in HLA-Transgenic mice and for ex vivo stimulation and expansion of CD8(+) cytotoxic T cells from human peripheral blood cells. Hum. Immunol. 2004, 65, 514–522. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J. Recognition of human cytomegalovirus gene products by HCMV-specific cytotoxic T cells. Virology 1996, 222, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Hertoghs, K.M.; Moerland, P.D.; van Stijn, A.; Remmerswaal, E.B.; Yong, S.L.; van de Berg, P.J.; van Ham, S.M.; Baas, F.; ten Berge, I.J.; van Lier, R.A. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J. Clin. Invest. 2010, 120, 4077–4090. [Google Scholar] [CrossRef]
- Gillespie, G.M.; Wills, M.R.; Appay, V.; O’Callaghan, C.; Murphy, M.; Smith, N.; Sissons, P.; Rowland-Jones, S.; Bell, J.I.; Moss, P.A. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J. Virol. 2000, 74, 8140–8150. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, E.M.; Remmerswaal, E.B.; Vossen, M.T.; Rowshani, A.T.; Wertheim-van Dillen, P.M.; van Lier, R.A.; ten Berge, I.J. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J. Immunol. 2004, 173, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Kern, F.; Faulhaber, N.; Frommel, C.; Khatamzas, E.; Prosch, S.; Schonemann, C.; Kretzschmar, I.; Volkmer-Engert, R.; Volk, H.D.; Reinke, P. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur. J. Immunol. 2000, 30, 1676–1682. [Google Scholar] [CrossRef]
- Lilleri, D.; Zelini, P.; Fornara, C.; Zavaglio, F.; Rampino, T.; Perez, L.; Gabanti, E.; Gerna, G. Human cytomegalovirus (HCMV)-specific T cell but not neutralizing or IgG binding antibody responses to glycoprotein complexes gB, gHgLgO, and pUL128L correlate with protection against high HCMV viral load reactivation in solid-organ transplant recipients. J. Med. Virol. 2018, 90, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Reusser, P.; Gambertoglio, J.G.; Lilleby, K.; Meyers, J.D. Phase I-II trial of foscarnet for prevention of cytomegalovirus infection in autologous and allogeneic marrow transplant recipients. J. Infect. Dis. 1992, 166, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, P.D.; Reusser, P.; Goodrich, J.M.; Riddell, S.R. Development of a treatment regimen for human cytomegalovirus (CMV) infection in bone marrow transplantation recipients by adoptive transfer of donor-derived CMV-specific T cell clones expanded in vitro. Ann. N. Y. Acad. Sci. 1991, 636, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Fornara, C.; Cassaniti, I.; Zavattoni, M.; Furione, M.; Adzasehoun, K.M.G.; De Silvestri, A.; Comolli, G.; Baldanti, F. Human Cytomegalovirus-Specific Memory CD4+ T-Cell Response and Its Correlation With Virus Transmission to the Fetus in Pregnant Women With Primary Infection. Clin. Infect. Dis. 2017, 65, 1659–1665. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72 Pt 9, 2059–2064. [Google Scholar] [CrossRef]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef] [Green Version]
- Avdic, S.; Cao, J.Z.; Cheung, A.K.; Abendroth, A.; Slobedman, B. Viral interleukin-10 expressed by human cytomegalovirus during the latent phase of infection modulates latently infected myeloid cell differentiation. J. Virol. 2011, 85, 7465–7471. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef] [Green Version]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [Green Version]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [Green Version]
- Mason, G.M.; Jackson, S.; Okecha, G.; Poole, E.; Sissons, J.G.; Sinclair, J.; Wills, M.R. Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4(+) T cells. PLoS Pathog. 2013, 9, e1003635. [Google Scholar] [CrossRef]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS ONE 2013, 8, e59863. [Google Scholar] [CrossRef] [Green Version]
- Kabanova, A.; Perez, L.; Lilleri, D.; Marcandalli, J.; Agatic, G.; Becattini, S.; Preite, S.; Fuschillo, D.; Percivalle, E.; Sallusto, F.; et al. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 17965–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanarsdall, A.L.; Howard, P.W.; Wisner, T.W.; Johnson, D.C. Human Cytomegalovirus gH/gL Forms a Stable Complex with the Fusion Protein gB in Virions. PLoS Pathog. 2016, 12, e1005564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, W.; Joyce, M.G.; Nguyen, H.; Banh, D.V.; Aguilar, F.; Tariq, Z.; Yap, M.L.; Tsujimura, Y.; Gillespie, R.A.; Tsybovsky, Y.; et al. Immunization with Components of the Viral Fusion Apparatus Elicits Antibodies That Neutralize Epstein-Barr Virus in B Cells and Epithelial Cells. Immunity 2019. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e1119. [Google Scholar] [CrossRef] [Green Version]
- Malito, E.; Chandramouli, S.; Carfi, A. From recognition to execution-the HCMV Pentamer from receptor binding to fusion triggering. Curr. Opin. Virol. 2018, 31, 43–51. [Google Scholar] [CrossRef]
- Ciferri, C.; Chandramouli, S.; Leitner, A.; Donnarumma, D.; Cianfrocco, M.A.; Gerrein, R.; Friedrich, K.; Aggarwal, Y.; Palladino, G.; Aebersold, R.; et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11, e1005230. [Google Scholar] [CrossRef]
- Wiegers, A.K.; Sticht, H.; Winkler, T.H.; Britt, W.J.; Mach, M. Identification of a neutralizing epitope within antigenic domain 5 of glycoprotein B of human cytomegalovirus. J. Virol. 2015, 89, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Spindler, N.; Diestel, U.; Stump, J.D.; Wiegers, A.K.; Winkler, T.H.; Sticht, H.; Mach, M.; Muller, Y.A. Structural basis for the recognition of human cytomegalovirus glycoprotein B by a neutralizing human antibody. PLoS Pathog. 2014, 10, e1004377. [Google Scholar] [CrossRef]
- Potzsch, S.; Spindler, N.; Wiegers, A.K.; Fisch, T.; Rucker, P.; Sticht, H.; Grieb, N.; Baroti, T.; Weisel, F.; Stamminger, T.; et al. B cell repertoire analysis identifies new antigenic domains on glycoprotein B of human cytomegalovirus which are target of neutralizing antibodies. PLoS Pathog. 2011, 7, e1002172. [Google Scholar] [CrossRef] [Green Version]
- Baraniak, I.; Kropff, B.; McLean, G.R.; Pichon, S.; Piras-Douce, F.; Milne, R.S.B.; Smith, C.; Mach, M.; Griffiths, P.D.; Reeves, M.B. Epitope-Specific Humoral Responses to Human Cytomegalovirus Glycoprotein-B Vaccine With MF59: Anti-AD2 Levels Correlate With Protection From Viremia. J. Infect. Dis. 2018, 217, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, I.; Kern, F.; Holenya, P.; Griffiths, P.; Reeves, M. Original Antigenic Sin Shapes the Immunological Repertoire Evoked by Human Cytomegalovirus Glycoprotein B/MF59 Vaccine in Seropositive Recipients. J. Infect. Dis. 2019, 220, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Oberstein, A.; Wang, W.; Shenk, T. Role of PDGF receptor-alpha during human cytomegalovirus entry into fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, E9889–E9898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Prager, A.; Boos, S.; Resch, M.; Brizic, I.; Mach, M.; Wildner, S.; Scrivano, L.; Adler, B. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-alpha as a key for entry. PLoS Pathog. 2017, 13, e1006281. [Google Scholar] [CrossRef]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef]
- E, X.; Meraner, P.; Lu, P.; Perreira, J.M.; Aker, A.M.; McDougall, W.M.; Zhuge, R.; Chan, G.C.; Gerstein, R.M.; Caposio, P.; et al. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 7043–7052. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Gui, X.; Freed, D.C.; Ku, Z.; Li, L.; Chen, Y.; Xiong, W.; Fan, X.; Su, H.; He, X.; et al. Identification of adipocyte plasma membrane-associated protein as a novel modulator of human cytomegalovirus infection. PLoS Pathog. 2019, 15, e1007914. [Google Scholar] [CrossRef] [Green Version]
- Stein, K.R.; Gardner, T.J.; Hernandez, R.E.; Kraus, T.A.; Duty, J.A.; Ubarretxena-Belandia, I.; Moran, T.M.; Tortorella, D. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 2019, 10, 2699. [Google Scholar] [CrossRef] [PubMed]
- Vanarsdall, A.L.; Pritchard, S.R.; Wisner, T.W.; Liu, J.; Jardetzky, T.S.; Johnson, D.C. CD147 Promotes Entry of Pentamer-Expressing Human Cytomegalovirus into Epithelial and Endothelial Cells. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.T.; Compton, T. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 1998, 72, 8191–8197. [Google Scholar]
- Wille, P.T.; Knoche, A.J.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 2010, 84, 2585–2596. [Google Scholar] [CrossRef] [Green Version]
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P.A.; Cianfrocco, M.A.; Gerrein, R.; Feire, A.L.; Barnett, S.W.; Lilja, A.E.; Rappuoli, R.; et al. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lanchy, J.M.; Ryckman, B.J. Human Cytomegalovirus gH/gL/gO Promotes the Fusion Step of Entry into All Cell Types, whereas gH/gL/UL128-131 Broadens Virus Tropism through a Distinct Mechanism. J. Virol. 2015, 89, 8999–9009. [Google Scholar] [CrossRef] [Green Version]
- Bagdonaite, I.; Norden, R.; Joshi, H.J.; King, S.L.; Vakhrushev, S.Y.; Olofsson, S.; Wandall, H.H. Global Mapping of O-Glycosylation of Varicella Zoster Virus, Human Cytomegalovirus, and Epstein-Barr Virus. J. Biol. Chem. 2016, 291, 12014–12028. [Google Scholar] [CrossRef] [Green Version]
- Nogalski, M.T.; Chan, G.C.; Stevenson, E.V.; Collins-McMillen, D.K.; Yurochko, A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013, 9, e1003463. [Google Scholar] [CrossRef]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef] [Green Version]
- Zydek, M.; Petitt, M.; Fang-Hoover, J.; Adler, B.; Kauvar, L.M.; Pereira, L.; Tabata, T. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses 2014, 6, 1346–1364. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selinsky, C.; Luke, C.; Wloch, M.; Geall, A.; Hermanson, G.; Kaslow, D.; Evans, T. A DNA-based vaccine for the prevention of human cytomegalovirus-associated diseases. Hum. Vaccin. 2005, 1, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Y.; Kaur, A.; Zhou, S.S.; Barry, P.A. Characterization and immunological analysis of the rhesus cytomegalovirus homologue (Rh112) of the human cytomegalovirus UL83 lower matrix phosphoprotein (pp65). J. Gen. Virol. 2006, 87, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Reviews. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput Struct Biotechnol J. 2016, 14, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Duan, H.; Chen, X.; Boyington, J.C.; Cheng, C.; Zhang, Y.; Jafari, A.J.; Stephens, T.; Tsybovsky, Y.; Kalyuzhniy, O.; Zhao, P.; et al. Glycan Masking Focuses Immune Responses to the HIV-1 CD4-Binding Site and Enhances Elicitation of VRC01-Class Precursor Antibodies. Immunity 2018, 49, 301–311.e305. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Kirchmeier, M.; Fluckiger, A.C.; Soare, C.; Bozic, J.; Ontsouka, B.; Ahmed, T.; Diress, A.; Pereira, L.; Schodel, F.; Plotkin, S.; et al. Enveloped virus-like particle expression of human cytomegalovirus glycoprotein B antigen induces antibodies with potent and broad neutralizing activity. Clin. Vaccine Immunol. Cvi. 2014, 21, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Snapper, C.M. Development of novel vaccines against human cytomegalovirus. Hum. Vaccines Immunother. 2019, 15, 2673–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, T.; Burri, S.; Wellnitz, S.; Walsh, K.; Rothe, S.; Liderfelt, J. Fully aseptic single-use cross flow filtration system for clarification and concentration of cytomegalovirus-like particles. Eng. Life Sci. 2014, 14, 318–326. [Google Scholar] [CrossRef]
- Crowther, R.A.; Kiselev, N.A.; Bottcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [Google Scholar] [CrossRef]
- Roseman, A.M.; Berriman, J.A.; Wynne, S.A.; Butler, P.J.; Crowther, R.A. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. USA 2005, 102, 15821–15826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.; Skamel, C.; Vorreiter, J.; Nassal, M. Internal core protein cleavage leaves the hepatitis B virus capsid intact and enhances its capacity for surface display of heterologous whole chain proteins. J. Biol. Chem. 2008, 283, 33508–33515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, K.; Meijerink, E.; Speiser, D.E.; Tissot, A.C.; Cielens, I.; Renhof, R.; Dishlers, A.; Pumpens, P.; Bachmann, M.F. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur. J. Immunol. 2005, 35, 816–821. [Google Scholar] [CrossRef]
- Krishna, B.A.; Humby, M.S.; Miller, W.E.; O’Connor, C.M. Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc. Natl. Acad. Sci. USA 2019, 116, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Miller, W.E.; O’Connor, C.M. US28: HCMV’s Swiss Army Knife. Viruses 2018, 10, 445. [Google Scholar] [CrossRef] [Green Version]
- Watt, G.D.; Jacobs, D.; Frankel, R.B. Redox reactivity of bacterial and mammalian ferritin: Is reductant entry into the ferritin interior a necessary step for iron release? Proc. Natl. Acad. Sci. USA 1988, 85, 7457–7461. [Google Scholar] [CrossRef] [Green Version]
- Theil, E.C. Ferritin: Structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu. Rev. Biochem. 1987, 56, 289–315. [Google Scholar] [CrossRef]
- Cho, K.J.; Shin, H.J.; Lee, J.H.; Kim, K.J.; Park, S.S.; Lee, Y.; Lee, C.; Park, S.S.; Kim, K.H. The crystal structure of ferritin from Helicobacter pylori reveals unusual conformational changes for iron uptake. J. Mol. Biol. 2009, 390, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, K.W.; Villegas, J.A.; Roose, B.W.; Hicks, T.L.; Yoon, J.; Saven, J.G.; Dmochowski, I.J. Thermophilic Ferritin 24mer Assembly and Nanoparticle Encapsulation Modulated by Interdimer Electrostatic Repulsion. Biochemistry 2017, 56, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, I.S.; Joyce, M.G.; Chen, R.E.; Leung, K.; McKee, K.; Druz, A.; Van Galen, J.G.; Kanekiyo, M.; Tsybovsky, Y.; Yang, E.S.; et al. Two-Component Ferritin Nanoparticles for Multimerization of Diverse Trimeric Antigens. Acs. Infect. Dis. 2018, 4, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Joyce, M.G.; Gillespie, R.A.; Gallagher, J.R.; Andrews, S.F.; Yassine, H.M.; Wheatley, A.K.; Fisher, B.E.; Ambrozak, D.R.; Creanga, A.; et al. Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses. Nat. Immunol. 2019, 20, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Golmohammadi, R.; Fridborg, K.; Bundule, M.; Valegard, K.; Liljas, L. The crystal structure of bacteriophage Q beta at 3.5 A resolution. Structure 1996, 4, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Cornuz, J.; Zwahlen, S.; Jungi, W.F.; Osterwalder, J.; Klingler, K.; van Melle, G.; Bangala, Y.; Guessous, I.; Muller, P.; Willers, J.; et al. A vaccine against nicotine for smoking cessation: A randomized controlled trial. PLoS ONE 2008, 3, e2547. [Google Scholar] [CrossRef]
- Vasiljeva, I.; Kozlovska, T.; Cielens, I.; Strelnikova, A.; Kazaks, A.; Ose, V.; Pumpens, P. Mosaic Qbeta coats as a new presentation model. Febs. Lett. 1998, 431, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Hsia, Y.; Bale, J.B.; Gonen, S.; Shi, D.; Sheffler, W.; Fong, K.K.; Nattermann, U.; Xu, C.; Huang, P.S.; Ravichandran, R.; et al. Corrigendum: Design of a hyperstable 60-subunit protein icosahedron. Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- King, N.P.; Bale, J.B.; Sheffler, W.; McNamara, D.E.; Gonen, S.; Gonen, T.; Yeates, T.O.; Baker, D. Accurate design of co-assembling multi-component protein nanomaterials. Nature 2014, 510, 103–108. [Google Scholar] [CrossRef]
- Bale, J.B.; Gonen, S.; Liu, Y.; Sheffler, W.; Ellis, D.; Thomas, C.; Cascio, D.; Yeates, T.O.; Gonen, T.; King, N.P.; et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353, 389–394. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kumar, S.; Allen, J.D.; Huang, D.; Lin, X.; Mann, C.J.; Saye-Francisco, K.L.; Copps, J.; Sarkar, A.; Blizard, G.S.; et al. HIV-1 vaccine design through minimizing envelope metastability. Sci. Adv. 2018, 4, eaau6769. [Google Scholar] [CrossRef] [Green Version]
- Marcandalli, J.; Fiala, B.; Ols, S.; Perotti, M.; de van der Schueren, W.; Snijder, J.; Hodge, E.; Benhaim, M.; Ravichandran, R.; Carter, L.; et al. Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 2019, 176, 1420–1431.e1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, P.J.M.; Antanasijevic, A.; Berndsen, Z.; Yasmeen, A.; Fiala, B.; Bijl, T.P.L.; Bontjer, I.; Bale, J.B.; Sheffler, W.; Allen, J.D.; et al. Enhancing and shaping the immunogenicity of native-like HIV-1 envelope trimers with a two-component protein nanoparticle. Nat. Commun. 2019, 10, 4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyken, S.E.; Benhaim, M.A.; Busch, F.; Jia, M.; Bick, M.J.; Choi, H.; Klima, J.C.; Chen, Z.; Walkey, C.; Mileant, A.; et al. De novo design of tunable, pH-driven conformational changes. Science 2019, 364, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Ogohara, C.; Yi, S.; Hsia, Y.; Nattermann, U.; Belnap, D.M.; King, N.P.; Sundquist, W.I. Designed proteins induce the formation of nanocage-containing extracellular vesicles. Nature 2016, 540, 292–295. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [Green Version]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-Display: Decoration of Virus-Like Particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef] [Green Version]
- Palladini, A.; Thrane, S.; Janitzek, C.M.; Pihl, J.; Clemmensen, S.B.; de Jongh, W.A.; Clausen, T.M.; Nicoletti, G.; Landuzzi, L.; Penichet, M.L.; et al. Virus-like particle display of HER2 induces potent anti-cancer responses. Oncoimmunology 2018, 7, e1408749. [Google Scholar] [CrossRef] [Green Version]
- Thrane, S.; Janitzek, C.M.; Matondo, S.; Resende, M.; Gustavsson, T.; de Jongh, W.A.; Clemmensen, S.; Roeffen, W.; van de Vegte-Bolmer, M.; van Gemert, G.J.; et al. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J. Nanobiotechnology 2016, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.K.; Lee, J.H.; Menis, S.; Skog, P.; Rossi, M.; Ota, T.; Kulp, D.W.; Bhullar, D.; Kalyuzhniy, O.; Havenar-Daughton, C.; et al. Precursor Frequency and Affinity Determine B Cell Competitive Fitness in Germinal Centers, Tested with Germline-Targeting HIV Vaccine Immunogens. Immunity 2018, 48, 133–146.e6. [Google Scholar] [CrossRef] [Green Version]
- He, L.; de Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.T.; Gray, M.D.; Dosenovic, P.; Gitlin, A.D.; Freund, N.T.; Petersen, J.; Correnti, C.; Johnsen, W.; Kegel, R.; Stuart, A.B.; et al. Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat. Commun. 2016, 7, 10618. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; van Montfort, T.; Stunnenberg, M.; LaBranche, C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perotti, M.; Perez, L. Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV. Viruses 2020, 12, 35. https://doi.org/10.3390/v12010035
Perotti M, Perez L. Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV. Viruses. 2020; 12(1):35. https://doi.org/10.3390/v12010035
Chicago/Turabian StylePerotti, Michela, and Laurent Perez. 2020. "Virus-Like Particles and Nanoparticles for Vaccine Development against HCMV" Viruses 12, no. 1: 35. https://doi.org/10.3390/v12010035