SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
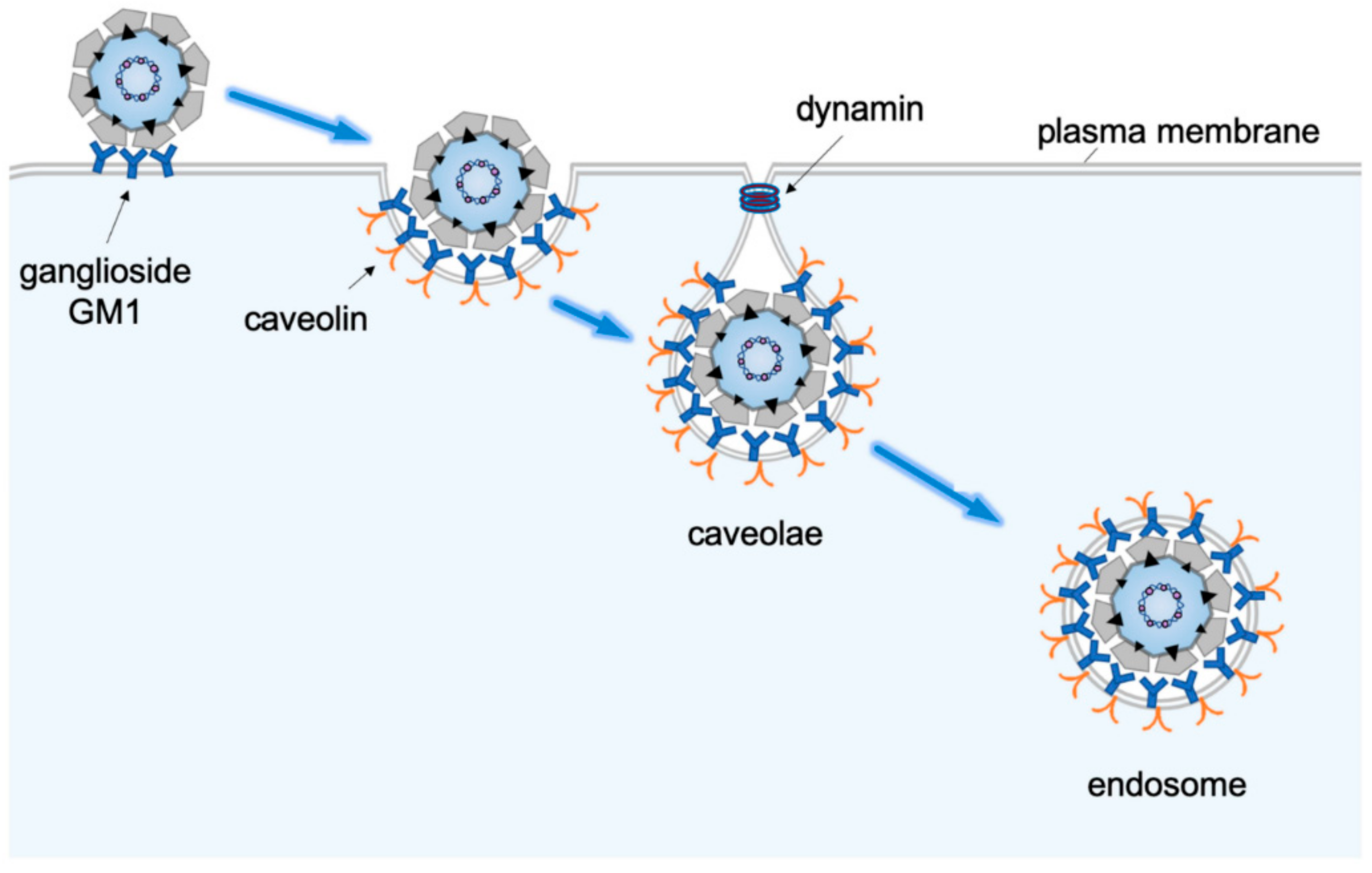
2. Receptor-Mediated Endocytosis to Endosomes
3. Targeting to the Endoplasmic Reticulum
4. ER Membrane Penetration to Enable Viral Escape into the Cytosol
5. Nuclear entry
6. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Moyer, C.L.; Nemerow, G.R. Viral weapons of membrane destruction: Variable modes of membrane penetration by non-enveloped viruses. Curr. Opin. Virol. 2011, 1, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Fay, N.; Pante, N. Old foes, new understandings: Nuclear entry of small non-enveloped DNA viruses. Curr. Opin. Virol. 2015, 12, 59–65. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Burd, C.G.; Goodner, K. Riding the R Train into the Cell. PLoS Pathog. 2015, 11, e1005036. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 2003, 22, 4346–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.E.; Lilie, H.; Helenius, A. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett. 2003, 555, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Low, J.A.; Magnuson, B.; Tsai, B.; Imperiale, M.J. Identification of gangliosides GD1b and GT1b as receptors for BK virus. J. Virol. 2006, 80, 1361–1366. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef]
- Lewis, M.J.; Pelham, H.R. SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell 1996, 85, 205–215. [Google Scholar] [CrossRef]
- Johannes, L.; Popoff, V. Tracing the retrograde route in protein trafficking. Cell 2008, 135, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Kartenbeck, J.; Stukenbrok, H.; Helenius, A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell. Biol. 1989, 109, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cai, D.; Verhey, K.J.; Tsai, B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Chang, Y.; Moore, P.S. MCV and Merkel cell carcinoma: A molecular success story. Curr. Opin. Virol. 2012, 2, 489–498. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Abend, J.R.; Johnson, S.F.; Imperiale, M.J. The role of polyomaviruses in human disease. Virology 2009, 384, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.S.; Stehle, T.; Harrison, S.C. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef]
- Stehle, T.; Gamblin, S.J.; Yan, Y.; Harrison, S.C. The structure of simian virus 40 refined at 3.1 A resolution. Structure 1996, 4, 165–182. [Google Scholar] [CrossRef]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus 40 at 3.8-A resolution. Nature 1991, 354, 278–284. [Google Scholar] [CrossRef]
- White, M.K.; Gordon, J.; Khalili, K. The rapidly expanding family of human polyomaviruses: Recent developments in understanding their life cycle and role in human pathology. PLoS Pathog. 2013, 9, e1003206. [Google Scholar] [CrossRef] [PubMed]
- Campanero-Rhodes, M.A.; Smith, A.; Chai, W.; Sonnino, S.; Mauri, L.; Childs, R.A.; Zhang, Y.; Ewers, H.; Helenius, A.; Imberty, A.; et al. N-glycolyl GM1 ganglioside as a receptor for simian virus 40. J. Virol. 2007, 81, 12846–12858. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Benjamin, T. Uptake pathway of polyomavirus via ganglioside GD1a. J. Virol. 2004, 78, 12259–12267. [Google Scholar] [CrossRef] [PubMed]
- Chinnapen, D.J.; Hsieh, W.T.; te Welscher, Y.M.; Saslowsky, D.E.; Kaoutzani, L.; Brandsma, E.; D’Auria, L.; Park, H.; Wagner, J.S.; Drake, K.R.; et al. Lipid sorting by ceramide structure from plasma membrane to ER for the cholera toxin receptor ganglioside GM1. Dev. Cell 2012, 23, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar] [PubMed]
- Fujinaga, Y.; Wolf, A.A.; Rodighiero, C.; Wheeler, H.; Tsai, B.; Allen, L.; Jobling, M.G.; Rapoport, T.; Holmes, R.K.; Lencer, W.I. Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol. Biol. Cell 2003, 14, 4783–4793. [Google Scholar] [CrossRef] [PubMed]
- Ewers, H.; Romer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell. Biol. 2010, 12, 11–18; sup pp. 10–12. [Google Scholar] [CrossRef]
- Engel, S.; Heger, T.; Mancini, R.; Herzog, F.; Kartenbeck, J.; Hayer, A.; Helenius, A. Role of endosomes in simian virus 40 entry and infection. J. Virol. 2011, 85, 4198–4211. [Google Scholar] [CrossRef]
- Szklarczyk, O.M.; Gonzalez-Segredo, N.; Kukura, P.; Oppenheim, A.; Choquet, D.; Sandoghdar, V.; Helenius, A.; Sbalzarini, I.F.; Ewers, H. Receptor concentration and diffusivity control multivalent binding of Sv40 to membrane bilayers. PLoS Comput. Biol. 2013, 9, e1003310. [Google Scholar] [CrossRef]
- Damm, E.M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzchalia, T.; Helenius, A. Clathrin- and caveolin-1-independent endocytosis: Entry of simian virus 40 into cells devoid of caveolae. J. Cell. Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef]
- Stang, E.; Kartenbeck, J.; Parton, R.G. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell. 1997, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell. Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.A.; Chen, Y.; Norkin, L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 1996, 7, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Norkin, L.C.; Anderson, H.A.; Wolfrom, S.A.; Oppenheim, A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 2002, 76, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.D.; Holicky, E.L.; Cheng, Z.J.; Kim, S.Y.; Wheatley, C.L.; Marks, D.L.; Bittman, R.; Pagano, R.E. Inhibition of caveolar uptake, SV40 infection, and beta1-integrin signaling by a nonnatural glycosphingolipid stereoisomer. J. Cell. Biol. 2007, 176, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Puntener, D.; Helenius, A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef]
- Eash, S.; Querbes, W.; Atwood, W.J. Infection of vero cells by BK virus is dependent on caveolae. J. Virol. 2004, 78, 11583–11590. [Google Scholar] [CrossRef]
- Bouley, S.J.; Maginnis, M.S.; Derdowski, A.; Gee, G.V.; O’Hara, B.A.; Nelson, C.D.; Bara, A.M.; Atwood, W.J.; Dugan, A.S. Host cell autophagy promotes BK virus infection. Virology 2014, 456–457, 87–95. [Google Scholar] [CrossRef]
- Ashok, A.; Atwood, W.J. Contrasting roles of endosomal pH and the cytoskeleton in infection of human glial cells by JC virus and simian virus 40. J. Virol. 2003, 77, 1347–1356. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Zhao, L.; Imperiale, M.J. Identification of Rab18 as an Essential Host Factor for BK Polyomavirus Infection Using a Whole-Genome RNA Interference Screen. mSphere 2017, 2, e00291-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.D.; Derdowski, A.; Maginnis, M.S.; O’Hara, B.A.; Atwood, W.J. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 2012, 428, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelhaas, M.; Malmstrom, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grunewald, K.; Helenius, A. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef]
- Walczak, C.P.; Tsai, B. A PDI family network acts distinctly and coordinately with ERp29 to facilitate polyomavirus infection. J. Virol. 2011, 85, 2386–2396. [Google Scholar] [CrossRef]
- Gilbert, J.; Ou, W.; Silver, J.; Benjamin, T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 2006, 80, 10868–10870. [Google Scholar] [CrossRef]
- Daniels, R.; Rusan, N.M.; Wadsworth, P.; Hebert, D.N. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: Implications for DNA translocation out of the ER. Mol. Cell 2006, 24, 955–966. [Google Scholar] [CrossRef]
- Kuksin, D.; Norkin, L.C. Disassembly of simian virus 40 during passage through the endoplasmic reticulum and in the cytoplasm. J. Virol. 2012, 86, 1555–1562. [Google Scholar] [CrossRef]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog. 2014, 10, e1004007. [Google Scholar] [CrossRef]
- Ravindran, M.S.; Bagchi, P.; Inoue, T.; Tsai, B. A Non-enveloped Virus Hijacks Host Disaggregation Machinery to Translocate across the Endoplasmic Reticulum Membrane. PLoS Pathog. 2015, 11, e1005086. [Google Scholar] [CrossRef]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Walczak, C.P.; Tsai, B. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J. Virol. 2015, 89, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Inoue, T.; Tsai, B. EMC1-dependent stabilization drives membrane penetration of a partially destabilized non-enveloped virus. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Dupzyk, A.; Williams, J.M.; Bagchi, P.; Inoue, T.; Tsai, B. SGTA-Dependent Regulation of Hsc70 Promotes Cytosol Entry of Simian Virus 40 from the Endoplasmic Reticulum. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Dupzyk, A.; Tsai, B. Bag2 Is a Component of a Cytosolic Extraction Machinery That Promotes Membrane Penetration of a Nonenveloped Virus. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.; Van Goor, K.E.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio 2011, 2, e00101–e00111. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog 2011, 7, e1002037. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Engelke, M.F.; Verhey, K.J.; Tsai, B. Exploiting the kinesin-1 molecular motor to generate a virus membrane penetration site. Nat. Commun. 2017, 8, 15496. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Tsai, B. Regulated Erlin-dependent release of the B12 transmembrane J-protein promotes ER membrane penetration of a non-enveloped virus. PLoS Pathog. 2017, 13, e1006439. [Google Scholar] [CrossRef]
- Fahrenkrog, B.; Aebi, U. The nuclear pore complex: Nucleocytoplasmic transport and beyond. Nat. Rev. Mol. Cell Biol. 2003, 4, 757–766. [Google Scholar] [CrossRef]
- Nakanishi, A.; Shum, D.; Morioka, H.; Otsuka, E.; Kasamatsu, H. Interaction of the Vp3 nuclear localization signal with the importin alpha 2/beta heterodimer directs nuclear entry of infecting simian virus 40. J. Virol. 2002, 76, 9368–9377. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, D.S.; Corbett, A.H.; Mason, D.A.; Harreman, M.T.; Adam, S.A. Importin alpha: A multipurpose nuclear-transport receptor. Trends Cell. Biol. 2004, 14, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell. Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Ben-nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011, 2, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Toscano, M.G.; de Haan, P. How Simian Virus 40 Hijacks the Intracellular Protein Trafficking Pathway to Its Own Benefit and Ours. Front. Immunol. 2018, 9, 1160. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Spriggs, C.C.; Verhey, K.J.; Tsai, B. Dynein Engages and Disassembles Cytosol-Localized Simian Virus 40 To Promote Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Schlager, M.A.; Hoang, H.T.; Urnavicius, L.; Bullock, S.L.; Carter, A.P. In vitro reconstitution of a highly processive recombinant human dynein complex. EMBO J. 2014, 33, 1855–1868. [Google Scholar] [CrossRef]
- Zhang, K.; Foster, H.E.; Rondelet, A.; Lacey, S.E.; Bahi-Buisson, N.; Bird, A.W.; Carter, A.P. Cryo-EM Reveals How Human Cytoplasmic Dynein Is Auto-inhibited and Activated. Cell 2017, 169, 1303–1314. [Google Scholar] [CrossRef]
- Strunze, S.; Engelke, M.F.; Wang, I.H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-mediated capsid disassembly and disruption of the nuclear pore complex promote virus infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef]
- Chromy, L.R.; Oltman, A.; Estes, P.A.; Garcea, R.L. Chaperone-mediated in vitro disassembly of polyoma- and papillomaviruses. J. Virol. 2006, 80, 5086–5091. [Google Scholar] [CrossRef]
- Gharakhanian, E.; Takahashi, J.; Kasamatsu, H. The carboxyl 35 amino acids of SV40 Vp3 are essential for its nuclear accumulation. Virology 1987, 157, 440–448. [Google Scholar] [CrossRef]
- Bennett, S.M.; Zhao, L.; Bosard, C.; Imperiale, M.J. Role of a nuclear localization signal on the minor capsid proteins VP2 and VP3 in BKPyV nuclear entry. Virology 2015, 474, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Sawa, H.; Suzuki, T.; Semba, S.; Henmi, C.; Okada, Y.; Tsuda, M.; Tanaka, S.; Atwood, W.J.; Nagashima, K. Nuclear entry mechanism of the human polyomavirus JC virus-like particle: Role of importins and the nuclear pore complex. J. Biol. Chem. 2004, 279, 27735–27742. [Google Scholar] [CrossRef]
- Soldatova, I.; Prilepskaja, T.; Abrahamyan, L.; Forstova, J.; Huerfano, S. Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus. Viruses 2018, 10, 165. [Google Scholar] [CrossRef]
- Rainey-Barger, E.K.; Magnuson, B.; Tsai, B. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J. Virol. 2007, 81, 12996–13004. [Google Scholar] [CrossRef]
- Yamada, M.; Kasamatsu, H. Role of nuclear pore complex in simian virus 40 nuclear targeting. J. Virol. 1993, 67, 119–130. [Google Scholar] [Green Version]
- Clever, J.; Yamada, M.; Kasamatsu, H. Import of simian virus 40 virions through nuclear pore complexes. Proc. Natl. Acad. Sci. USA 1991, 88, 7333–7337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. MBio 2014, 5, e01777-14. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; Liu, X.; Tsai, B. SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection. Viruses 2019, 11, 917. https://doi.org/10.3390/v11100917
Chen Y-J, Liu X, Tsai B. SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection. Viruses. 2019; 11(10):917. https://doi.org/10.3390/v11100917
Chicago/Turabian StyleChen, Yu-Jie, Xiaofang Liu, and Billy Tsai. 2019. "SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection" Viruses 11, no. 10: 917. https://doi.org/10.3390/v11100917