

High-Throughput DNA Metabarcoding of Stem Sections from Trees with Cavities Describes Fungal Communities Associated with Variable Wood Decay, Position on Stem and Tree Species

Abstract
:1. Introduction
2. Methods
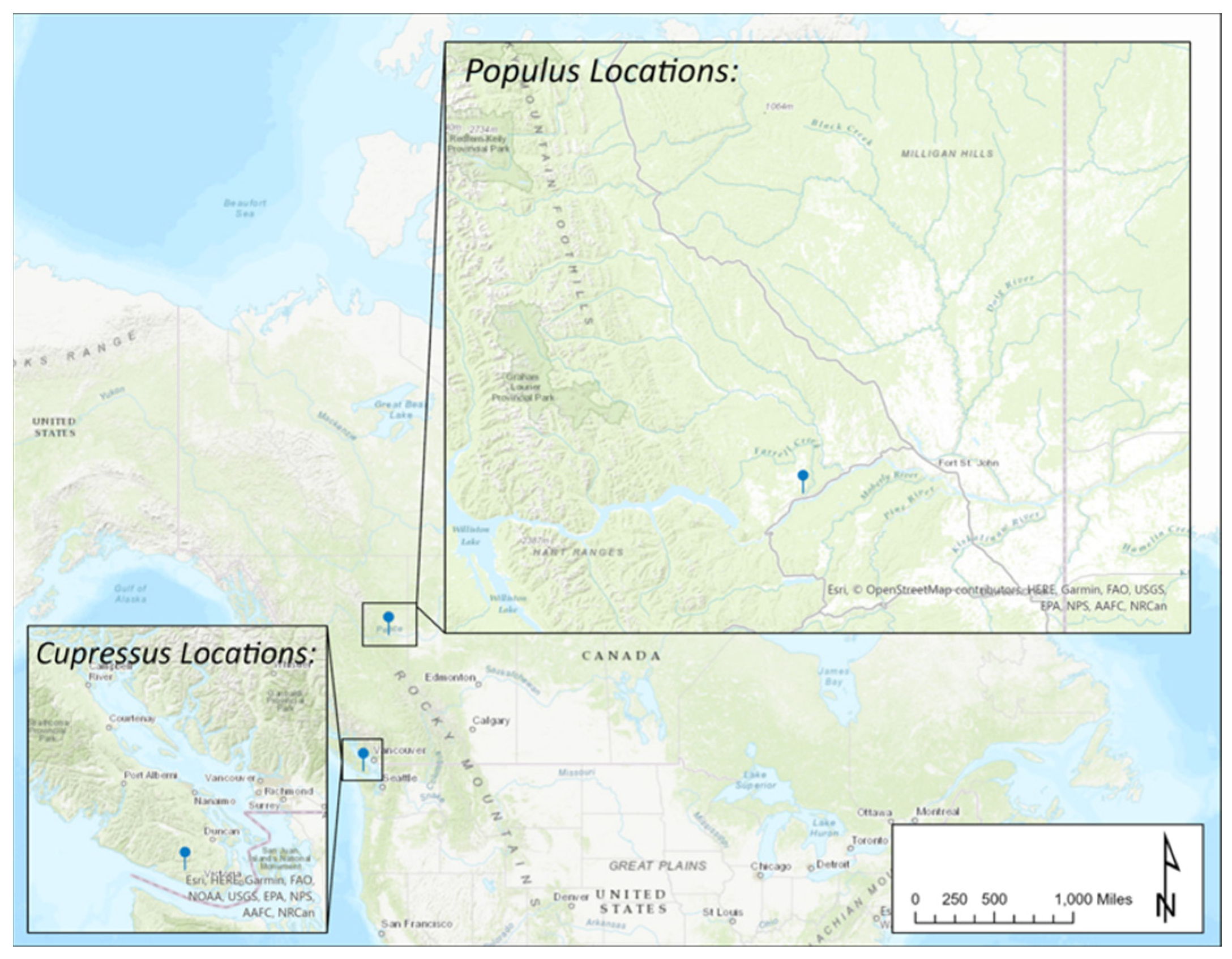
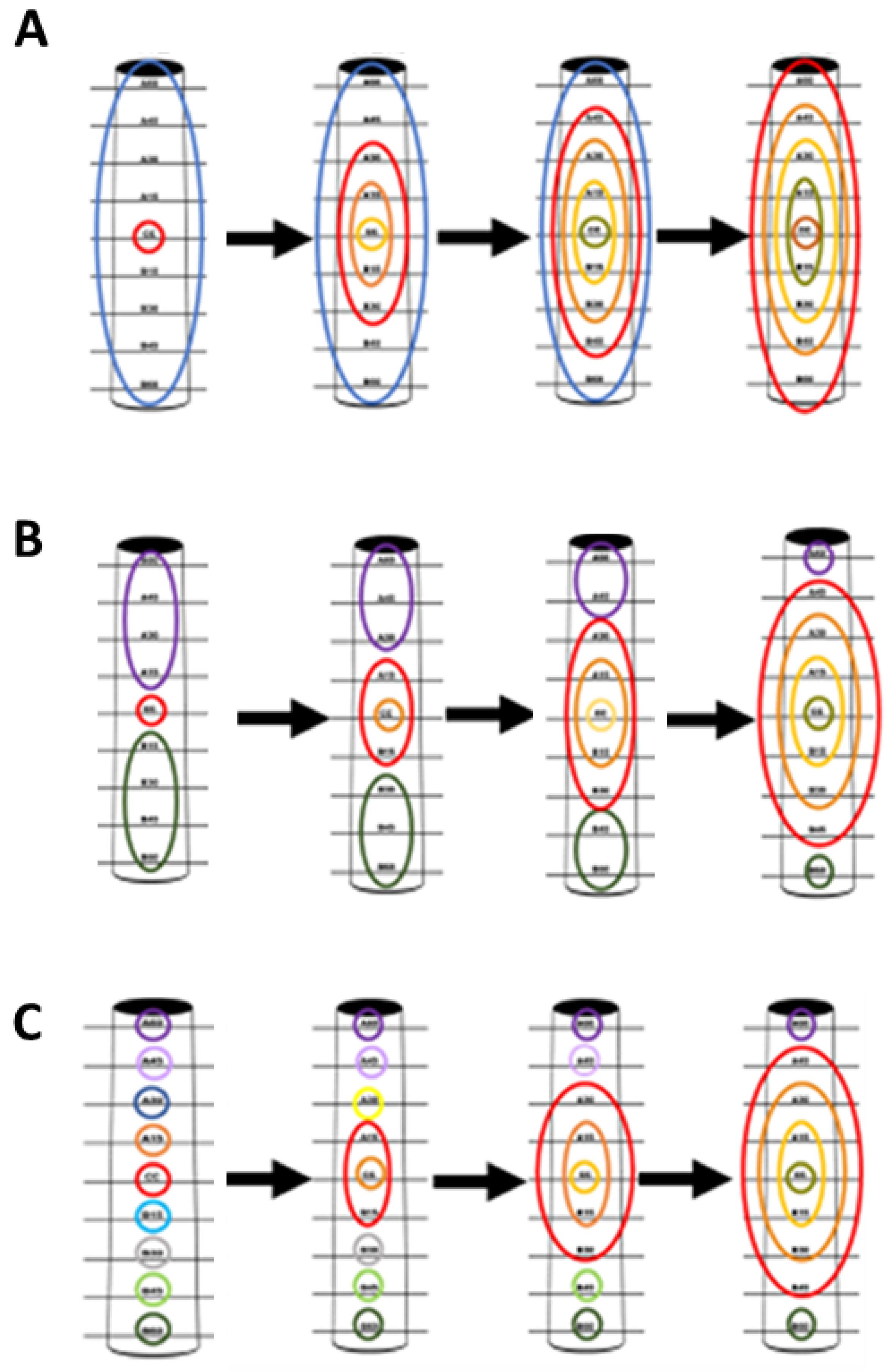
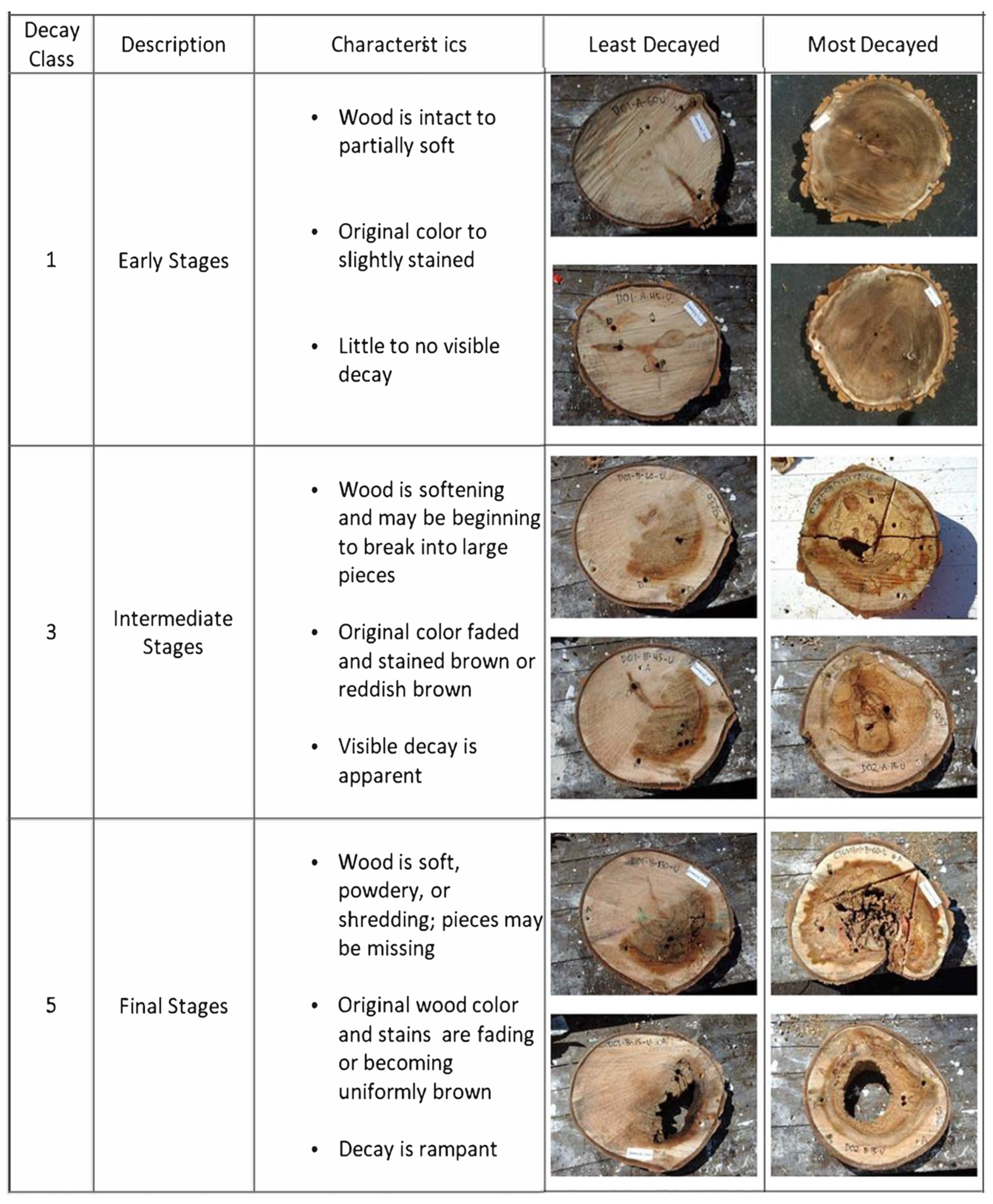
2.1. Tree Selection and Sampling Approach
2.2. Metabarcoding Analysis
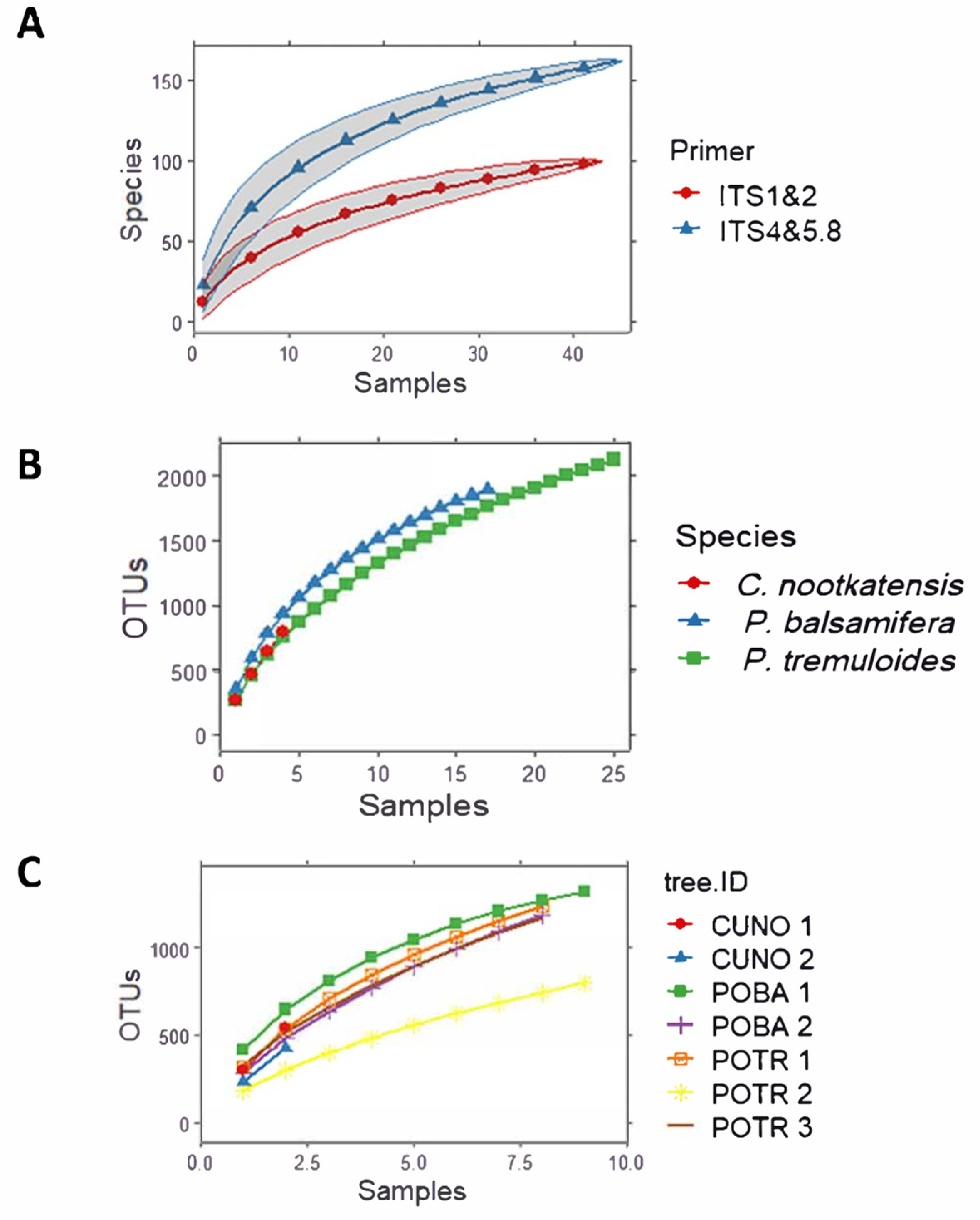
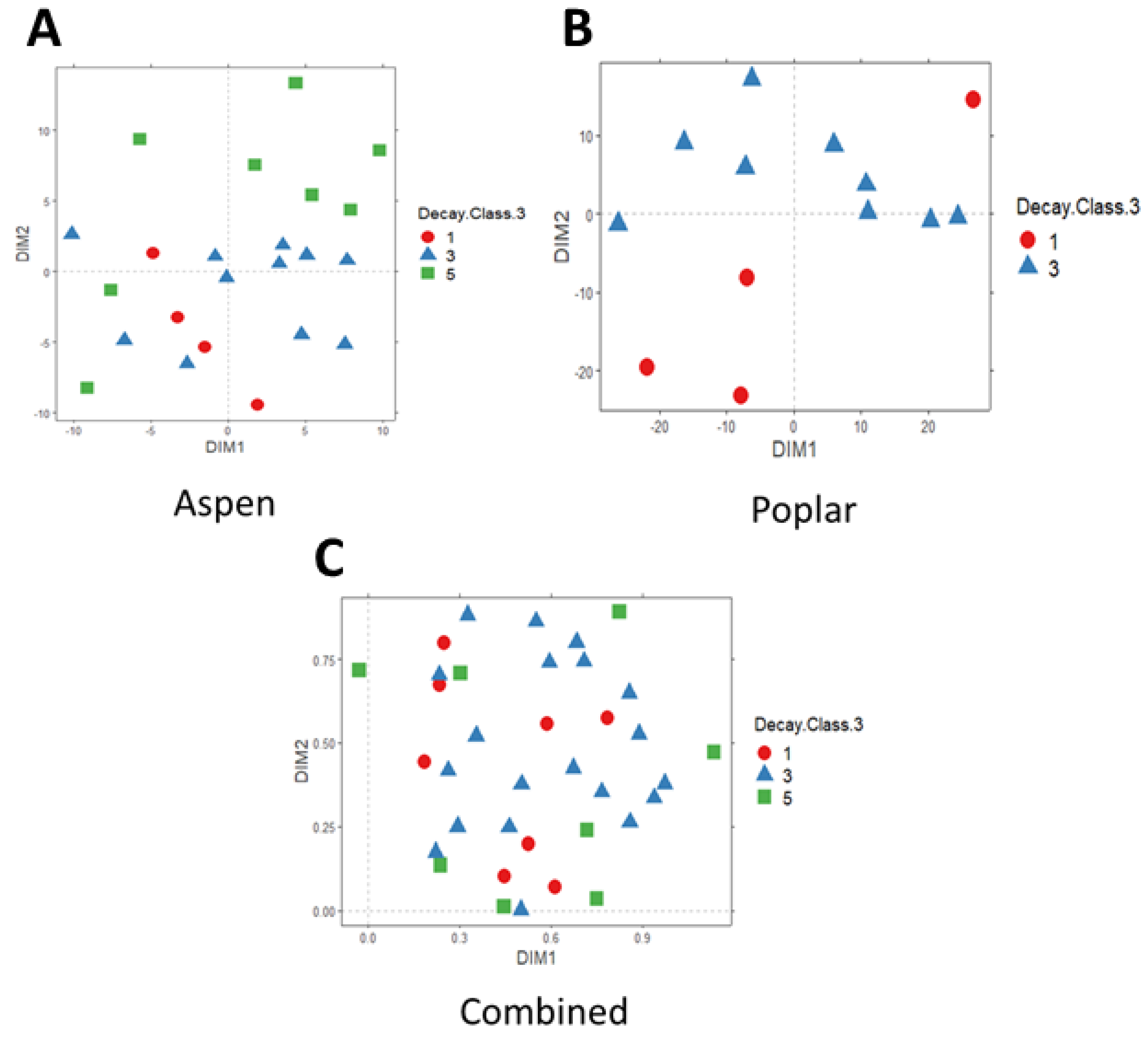
3. Results
4. Discussion
4.1. A Hyper-Rich Fungal Community Is Found in the Wood of Two Populus Species
4.2. Wood Fungal Communities Vary Depending on PCR Primers Used in High-Throughput DNA Metabarcoding
4.3. Wood Fungal Communities Vary Depending on Tree Species
4.4. The Composition of Wood Fungal Communities Varies Depending on Wood Decay Stage
4.5. Fungal Communities Change Progressively as Wood Transitions from Healthy to Decayed, with Some Fungi Being Present in Different Wood Decay Stages
4.6. Decay and Location on Stem Interact to Drive Composition of Fungal Communities in the Wood of Tree Stems
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boddy, L.; Rayner, A.D.M. Origins of decay in living deciduous trees: The role of moisture content and a re-appraisal of the expanded concept of tree decay. New Phytol. 1983, 94, 623–641. [Google Scholar] [CrossRef]
- Rayner, A.D.M.; Boddy, L. Fungal Decomposition of Wood: Its Biology and Ecology; John Wiley Sons Ltd.: Hoboken, NJ, USA, 1988. [Google Scholar] [CrossRef]
- Watkinson, S.; Bebber, D.P.; Darrah, P.E.; Fricker, M.A.; Tlalka, M.; Boddy, L. The role of wood decay fungi in the carbon and nitrogen dynamics of the forest floor Related papers. In Fungi Biogeochem Cycles; Gadd, G.M., Ed.; Cambridge University Press: Cambridge, UK, 2006; Volume 24, p. 151. [Google Scholar]
- van der Wal, A.; Ottosson, E.; De Boer, W. Neglected role of fungal community composition in explaining variation in wood decay rates. Ecology 2015, 96, 124–133. [Google Scholar] [CrossRef]
- Hoppe, B.; Purahong, W.; Wubet, T.; Kahl, T.; Bauhus, J.; Arnstadt, T.; Hofrichter, M.; Buscot, F.; Krüger, D. Linking molecular deadwood-inhabiting fungal diversity and community dynamics to ecosystem functions and processes in Central European forests. Fungal Divers. 2016, 77, 367–379. [Google Scholar] [CrossRef]
- Parfitt, D.; Hunt, J.; Dockrell, D.; Rogers, H.J.; Boddy, L. Do all trees carry the seeds of their own destruction? PCR reveals numerous wood decay fungi latently present in sapwood of a wide range of angiosperm trees. Fungal Ecol. 2010, 3, 338–346. [Google Scholar] [CrossRef]
- Ulyshen, M.D. Wood decomposition as influenced by invertebrates. Biol. Rev. 2016, 91, 70–85. [Google Scholar] [CrossRef]
- Jusino, M.A.; Lindner, D.L.; Banik, M.T.; Walters, J.R.; Davey, M.L. Heart rot hotel: Fungal communities in red-cockaded woodpecker excavations. Fungal Ecol. 2015, 14, 33–43. [Google Scholar] [CrossRef]
- Elliott, T.F.; Jusino, M.A.; Trappe, J.M.; Lepp, H.; Ballard, G.A.; Bruhl, J.J.; Vernes, K. A global review of the ecological significance of symbiotic associations between birds and fungi. Fungal Divers. 2019, 98, 161–194. [Google Scholar] [CrossRef]
- Coates, D.; Rayner, A.D.M. Fungal population and community development in cut beech logs: I. Establishment via the aerial cut surface. New Phytol. 1985, 101, 153–171. [Google Scholar] [CrossRef]
- Coates, D.; Rayner, A.D.M. Fungal population and community development in cut beech logs: III. Spatial dynamics, interactions and strategies. New Phytol. 1985, 101, 183–198. [Google Scholar] [CrossRef]
- Chapela, I.H.; Boddy, L. Fungal Colonization of Attached Beech Branches II. Spatial and temporal organization of communities arising from latent invaders in bark and functional sapwood, under different moisture regimes. New Phytol. 1988, 110, 47–57. [Google Scholar] [CrossRef]
- Boddy, L.; Griffith, G.S. Role of endophytes and latent invasion in the development of decay communities in sapwood of angiospermous trees. Sydowia 1989, 41, 41–73. [Google Scholar]
- Griffith, G.S.; Boddy, L. Fungal decomposition of attached angiosperm twigs I. Decay community development in ash, beech and oak. New Phytol. 1990, 116, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Guglielmo, F.; Gonthier, P.; Garbelotto, M.; Nicolotti, G. Optimization of sampling procedures for DNA-based diagnosis of wood decay fungi in standing trees. Lett. Appl. Microbiol. 2010, 51, 90–97. [Google Scholar] [CrossRef]
- Martin, R.; Gazis, R.; Skaltsas, D.; Chaverri, P.; Hibbett, D. Unexpected diversity of basidiomycetous endophytes in sapwood and leaves of Hevea. Mycologia 2017, 107, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, E.C.; Jusino, M.A.; Pyne, E.J.; Banik, M.T.; Lindner, D.L.; Boddy, L. Fungal endophytes and origins of decay in beech (Fagus sylvatica) sapwood. Fungal Ecol. 2022, 59, 101–161. [Google Scholar] [CrossRef]
- Boddy, L.; Watkinson, S.C. Wood decomposition, higher fungi, and their role in nutrient redistribution. Can. J. Bot. 1994, 73, 1377–1383. [Google Scholar] [CrossRef]
- Kebli, H.; Drouin, P.; Brais, S.; Kernaghan, G. Species composition of saproxylic fungal communities on decaying logs in the boreal forest. Microb. Ecol. 2011, 61, 898–910. [Google Scholar] [CrossRef]
- Fischer, A.L.; Moncalvo, J.M.; Klironomos, J.N.; Malcolm, J.R. Fruiting Body and Molecular rDNA Sampling of fungi in Woody Debris from Logged and Unlogged Boreal Forests in Northeastern Ontario. Ecoscience 2012, 19, 374–390. [Google Scholar] [CrossRef]
- Kubartová, A.; Ottosson, E.; Dahlberg, A.; Stenlid, J. Patterns of fungal communities among and within decaying logs, revealed by 454 sequencing. Mol. Ecol. 2012, 21, 4514–4532. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Osono, T.; Takeda, H. Dynamics of physicochemical properties and occurrence of fungal fruit bodies during decomposition of coarse woody debris of Fagus crenata. J. For. Res. 2008, 14, 20–29. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Osono, T.; Takeda, H. Fungal decomposition of woody debris of Castanopsis sieboldii in a subtropical old-growth forest. Ecol. Res. 2011, 27, 211–218. [Google Scholar] [CrossRef]
- Heilmann-Clausen, J.; Christensen, M. Does size matter? On the importance of various dead wood fractions for fungal diversity in Danish beech forests. For. Ecol. Manag. 2004, 201, 105–117. [Google Scholar] [CrossRef]
- Olsson, J.; Jonsson, B.G.; Hjältén, J.; Ericson, L. Addition of coarse woody debris—The early fungal succession on Picea abies logs in managed forests and reserves. Biol. Conserv. 2011, 144, 1100–1110. [Google Scholar] [CrossRef]
- Lim, Y.W.; Kim, J.J.; Chedgy, R.; Morris, P.I.; Breuil, C. Fungal diversity from western redcedar fences and their resistance to beta-thujaplicin. Antonie Van Leeuwenhoek 2005, 87, 109–117. [Google Scholar] [CrossRef]
- Kurth, V.J.; Fransioli, N.; Fulé, P.Z.; Hart, S.C.; Gehring, C.A. Stand-replacing wildfires alter the community structure of wood-inhabiting fungi in southwestern ponderosa pine forests of the USA. Fungal Ecol. 2013, 6, 192–204. [Google Scholar] [CrossRef]
- Hiscox, J.; Savoury, M.; Vaughan, I.P.; Müller, C.T.; Boddy, L. Antagonistic fungal interactions influence carbon dioxide evolution from decomposing wood. Fungal Ecol. 2015, 14, 24–32. [Google Scholar] [CrossRef]
- Ding, S.; Hu, H.; Gu, J.D. Diversity, abundance, and distribution of wood-decay fungi in major Parks of Hong Kong. Forests 2020, 1, 1030. [Google Scholar] [CrossRef]
- Bari, E.; Karimi, K.; Aghajani, H.; Schmidt, O.; Zahari, S.; Tajick-Ghanbary, M.A.; Juybari, H.Z. Characterizations of tree-decay fungi by molecular and morphological investigations in the Iranian Alamerdadeh forest. Maderas Cienc. Tecnol. 2021, 23, 1–24. [Google Scholar] [CrossRef]
- Ovaskainen, O.; Schigel, D.; Ali-Kovero, H.; Auvinen, P.; Paulin, L.; Nordén, B.; Nordén, J. Combining high-throughput sequencing with fruit body surveys reveals contrasting life-history strategies in fungi. ISME J. 2013, 7, 1696–1709. [Google Scholar] [CrossRef]
- Boddy, L.; Büntgen, U.; Egli, S.; Gange, A.C.; Heegaard, E.; Kirk, P.M.; Mohammad, A.; Kauserud, H. Climate variation effects on fungal fruiting. Fungal Ecol. 2014, 10, 20–33. [Google Scholar] [CrossRef]
- Wu, B.; Hussain, M.; Zhang, W.; Stadler, M.; Liu, X.; Xiang, M. Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 2019, 10, 127–140. [Google Scholar] [CrossRef]
- Peay, K.G.; Kennedy, P.G.; Talbot, J.M. Dimensions of biodiversity in the Earth mycobiome. Nat. Rev. Microbiol. 2016, 14, 434–447. [Google Scholar] [CrossRef]
- Rajala, T.; Peltoniemi, M.; Pennanen, T.; Mäkipää, R. Fungal community dynamics in relation to substrate quality of decaying Norway spruce (Picea abies [L.] Karst.) logs in boreal forests. FEMS Microbiol. Ecol. 2012, 81, 494–505. [Google Scholar] [CrossRef]
- Vaz, A.B.; Fonseca, P.L.; Leite, L.R.; Badotti, F.; Salim, A.C.; Araujo, F.M.; Cuadros-Orellana, S.; Duarte, A.A.; Rosa, C.A.; Oliveira, G.; et al. Using next-generation sequencing (NGS) to uncover diversity of wood-decaying fungi in neotropical Atlantic forests. Phytotaxa 2017, 295, 1–21. [Google Scholar] [CrossRef]
- Heilmann-Clausen, J. A gradient analysis of communities of macrofungi and slime moulds on decaying beech logs. Mycol. Res. 2001, 105, 575–596. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Osono, T.; Takeda, H. Microfungus communities of Japanese beech logs at different stages of decay in a cool temperate deciduous forest. Can. J. For. Res. 2009, 39, 1606–1614. [Google Scholar] [CrossRef]
- Runnel, K.; Drenkhan, R.; Adamson, K.; Lõhmus, P.; Rosenvald, K.; Rosenvald, R.; Rähn, E.; Tedersoo, L. The factors and scales shaping fungal assemblages in fallen spruce trunks: A DNA metabarcoding study. For. Ecol. Manag. 2021, 495, 119381. [Google Scholar] [CrossRef]
- Gilmartin, E.C. Fungal Communities of Beech (Fagus sylvatica) Trees: Heart Rot and Origins of Decay. Ph.D. Thesis, Cardiff University, Wales, UK, 2020. [Google Scholar]
- Tedersoo, L.; Bahram, M.; Zinger, L.; Nilsson, R.H.; Kennedy, P.G.; Yang, T.; Anslan, S.; Mikryukov, V. Best practices in metabarcoding of fungi: From experimental design to results. Mol. Ecol. 2022, 31, 2769–2795. [Google Scholar] [CrossRef]
- Weslien, J.; Djupström, L.B.; Schroeder, M.; Widenfalk, O. Long-term priority effects among insects and fungi colonizing decaying wood. J. Anim. Ecol. 2011, 80, 1155–1162. [Google Scholar] [CrossRef]
- Janisch, J.E.; Harmon, M.E.; Chen, H.; Fasth, B.; Sexton, J. Decomposition of coarse woody debris originating by clearcutting of an old-growth conifer forest. Ecoscience 2005, 12, 151–160. [Google Scholar] [CrossRef]
- Boddy, L. Fungal community ecology and wood decomposition processes in angiosperms: From standing tree to complete decay of coarse woody debris. Ecol. Bull. 2001, 49, 43–56. [Google Scholar]
- Oses, R.; Valenzuela, S.; Freer, J.; Baeza, J.; Rodríguez, J. Evaluation of fungal endophytes for lignocellulolytic enzyme production and wood biodegradation. Int. Biodeterior. Biodegrad. 2006, 57, 129–135. [Google Scholar] [CrossRef]
- Cline, L.C.; Schilling, J.S.; Menke, J.; Groenhof, E.; Kennedy, P.G.; Schilling, S. Ecological and functional effects of fungal endophytes on wood decomposition. Funct. Ecol. 2018, 32, 181–191. [Google Scholar] [CrossRef]
- Maser, C.; Cline, S.P.; Cromack, K.; Trappe, J.M.; Hansen, E. Chapter 2. What we know about large trees that fall to the forest floor. In From the Forest to the Sea: A Story of Fallen Trees; USDA Forest Survey General Technical Report, PNWGTR-22; U.S. Department of Agriculture, Forest Service, Pacific Northwest Research Station: Portland, OR, USA, 1979; p. 153. [Google Scholar]
- Boddy, L.; Hiscox, J.; Gilmartin, E.C.; Johnston, S.R.; Heilmann-Clausen, J. Wood decay communities in angiosperm wood. In The Fungal Community: Its Organization and Role in the Ecosystem, 4th ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 169–190. [Google Scholar]
- Weir, R.D.; Phinney, M.; Lofroth, E.C. Big, sick, and rotting: Why tree size, damage, and decay are important to fisher reproductive habitat. For. Ecol. Manag. 2012, 265, 230–240. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl. Environ. Microb. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wen, C.; Qin, Y.; Yin, H.; Tu, Q.; Van Nostrand, J.D.; Yuan, T.; Yuan, M.; Deng, Y.; Zhou, J. Phasing amplicon sequencing on Illumina Miseq for robust environmental microbial community analysis. BMC Microbiol. 2015, 15, 125. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 081257:1–081257:21. [Google Scholar] [CrossRef]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv 2016, 074161:1–074161:20. [Google Scholar] [CrossRef]
- Kindt, R.; Coe, R. Tree Diversity Analysis. A Manual and Software for Common Statistical Methods for Ecological and Biodiversity Studies; World Agroforestry Centre (ICRAF): Nairobi, Kenya, 2005; ISBN 92-9059-179-X. Available online: https://www.worldagroforestry.org/output/tree-diversity-analysis (accessed on 12 April 2023).
- Dufrêne, M.; Legendre, P. Species assemblages and Indicator Species:the need for a flexible asymmetrical approach. Ecol. Monogr. 1997, 67, 345–366. [Google Scholar] [CrossRef]
- Trochine, A.; Turchetti, B.; Vaz, A.B.M.; Brandao, L.; Rosa, L.H.; Buzzini, P.; Rosa, C.; Libkind, D. Description of Dioszegia patagonica sp. nov., a novel carotenogenic yeast isolated from cold environments. Int. J. Syst. Evol. Microbiol. 2007, 67, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Amend, A. From dandruff to deep-sea vents: Malassezia-like fungi are ecologically hyper-diverse. PLoS Pathog. 2014, 10, e1004277. [Google Scholar] [CrossRef] [PubMed]
- Glassman, S.I.; Martiny, J.B. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. MSphere 2018, 3, e00148-18. [Google Scholar] [CrossRef]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS variability in the Kingdom Fungi as expressed in the International Sequence Databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, ebo.s653:1–ebo.s653:5. [Google Scholar] [CrossRef] [PubMed]
- Estensmo, E.L.F.; Maurice, S.; Morgado, L.; Martin-Sanchez, P.M.; Skrede, I.; Kauserud, H. The influence of intraspecific sequence variation during DNA metabarcoding: A case study of eleven fungal species. Mol. Ecol. Resour. 2021, 21, 1141–1148. [Google Scholar] [CrossRef]
- Osmundson, T.W.; Robert, V.A.; Schoch, C.L.; Baker, L.J.; Smith, A.; Robich, G.; Mizzan, L.; Garbelotto, M.M. Filling gaps in biodiversity knowledge for macrofungi: Contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS ONE 2013, 8, e62419. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Põlme, S.; Riit, T.; Liiv, I.; Kõljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.A.; Bálint, M.; Greshake, B.; Bandow, C.; Römbke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 2013, 65, 128–132. [Google Scholar] [CrossRef]
- Zhou, D.; Hyde, K.D. Host-specificity, host-exclusivity, and host-recurrence in saprobic fungi. Mycol. Res. 2001, 105, 1449–1457. [Google Scholar] [CrossRef]
- Johnston, P.R.; Park, D.; Manning, M.A. Neobulgaria alba sp. nov. and its Phialophora-like anamorph in native forests and kiwifruit orchards in New Zealand. Mycotaxon 2010, 113, 385–396. [Google Scholar] [CrossRef]
- Vazquez, F.; Heluane, H.; Spencer, J.F.T.; Spencer, D.M.; De Figueroa, L.I.C. Fusion between protoplasts of Pichia stipitis and isolated filamentous fungi nuclei. Enzym. Microb. Technol. 1997, 21, 32–38. [Google Scholar] [CrossRef]
- Cadete, R.M.; Lopes, M.R.; Rosa, C.A. Yeasts associated with decomposing plant material and rotting wood. In Yeasts in Natural Ecosystems: Diversity; Buzzini, P., Lachance, M.A., Yurkov, A., Eds.; Springer: Cham, Switzerland, 2017; pp. 265–292. [Google Scholar] [CrossRef]
- Nagy, L.G.; Riley, R.; Tritt, A.; Adam, C.; Daum, C.; Floudas, D.; Sun, H.; Yadav, J.S.; Pangilinan, J.; Larsson, K.-H.; et al. Comparative genomics of early-diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol. Biol. Evol. 2016, 33, 959–970. [Google Scholar] [CrossRef]
- Untereiner, W.A.; Malloch, D. Patterns of substrate utilization in species of Capronia and allied black yeasts: Ecological and taxonomic implications. Mycologia 1999, 91, 417–427. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total OTUs (n = 3467) | ||||
| OTUs | Family | Genus | Species | |
| Ascomycota | 2093 | 121 (684) | 297 (929) | 222 (1654) |
| Basidiomycota | 929 | 74 (186) | 159 (308) | 160 (593) |
| Chytridiomycota | 46 | 2 (34) | 3 (36) | 1 (45) |
| Glomeromycota | 18 | 3 (5) | 3 (13) | 2 (15) |
| Neocallimastigomycota | 2 | 1 (0) | 2 (0) | 0 (2) |
| Rozellomycota | 5 | 0 (5) | 0 (5) | 0 (5) |
| Mucoromycota/Zoopagomycota | 79 | 5 (4) | 8 (22) | 14 (51) |
| UNKN | 295 | NA | NA | NA |
| Total | 3467 | 206 (918) | 472 (1313) | 397 (2365) |
| Only OTUs at species level (n = 397) | ||||
| OTUs | Family | Genus | ||
| Ascomycota | 185 | 39 | 69 | 98 |
| Basidiomycota | 196 | 38 | 71 | 94 |
| Mucoromycota/Zoopagomycota | 16 | 4 | 5 | 9 |
| Total | 397 | 81 | 145 | 201 |
| Unknown Taxa | Unconfident Taxa | Confident Taxa | |
|---|---|---|---|
| Phylum | 295 | 338 | 2834 |
| Class | 525 | 780 | 2162 |
| Order | 751 | 1160 | 1556 |
| Family | 1213 | 930 | 1324 |
| Genus | 1595 | 867 | 1005 |
| Species | 2660 | 410 | 397 |
| Phylum | Family | # of OTUs | Phylum | Family | # of OTUs |
|---|---|---|---|---|---|
| Ascomycota | Herpotrichiellaceae | 96 | Basidiomycota | Tremellales fam Incertae sedis | 44 |
| Trichocomaceae | 71 | Malasseziaceae | 35 | ||
| Saccharomycetales fam Incertae sedis | 23 | Meruliaceae | 27 | ||
| Helotiales fam Incertae sedis | 22 | Polyporaceae | 24 | ||
| Hypocreales fam Incertae sedis | 21 | Sporidiobolales fam Incertae sedis | 22 | ||
| Microascaceae | 18 | Schizoporaceae | 21 | ||
| Xylariaceae | 17 | Hymenochaetaceae | 20 | ||
| Hypocreaceae | 16 | Strophariaceae | 20 | ||
| Pleosporaceae | 14 | Psathyrellaceae | 19 | ||
| Amphisphaeriaceae | 13 | Phanerochaetaceae | 15 | ||
| Dothioraceae | 13 | Hydnodontaceae | 14 | ||
| Orbiliaceae | 13 | Fomitopsidaceae | 13 | ||
| Erysiphaceae | 12 | Marasmiaceae | 13 | ||
| Helotiaceae | 12 | Peniophoraceae | 11 | ||
| Pezizomycotina fam Incertae sedis | 12 | Botryobasidiaceae | 10 | ||
| Mycosphaerellaceae | 11 | Ceratobasidiaceae | 10 | ||
| Phaeosphaeriaceae | 11 | Cystofilobasidiaceae | 9 | ||
| Diatrypaceae | 10 | Hydnaceae | 9 | ||
| Parmeliaceae | 10 | Tricholomataceae | 9 | ||
| Pleosporales fam Incertae sedis | 10 | Pluteaceae | 8 | ||
| Total | 425 | Total | 353 |
| Reads | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|
| 478,059 | Ascomycota | Sordariomycetes | Xylariales | Xylariaceae | Hypoxylon | macrocarpum |
| 403,522 | Ascomycota | Sordariomycetes | Xylariales | Xylariaceae | Hypoxylon | macrocarpum |
| 164,872 | Ascomycota | Sordariomycetes | Xylariales | Xylariaceae | Xylaria | UNKN |
| 156,995 | Ascomycota | Sordariomycetes | Xylariales | UNKN | UNKN | UNKN |
| 156,069 | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales fam Incertae sedis | Candida | UNKN |
| 141,112 | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales fam Incertae sedis | Candida | UNKN |
| 115,889 | Ascomycota | Eurotiomycetes | Chaetothyriales | Herpotrichiellaceae | Capronia | pilosella |
| 96,463 | Ascomycota | Leotiomycetes | Helotiales | Helotiaceae | Neobulgaria | UNKN |
| 96,356 | Ascomycota | Leotiomycetes | Helotiales | Helotiaceae | Neobulgaria | UNKN |
| 94,026 | Ascomycota | Eurotiomycetes | Chaetothyriales | Herpotrichiellaceae | Capronia | pilosella |
| 87,025 | Ascomycota | Sordariomycetes | Xylariales | Diatrypaceae | UNKN | UNKN |
| 83,331 | Ascomycota | Sordariomycetes | Xylariales | Diatrypaceae | UNKN | UNKN |
| 76,299 | Ascomycota | UNKN | UNKN | UNKN | UNKN | UNKN |
| 68,459 | Ascomycota | UNKN | UNKN | UNKN | UNKN | UNKN |
| 67,929 | Ascomycota | Saccharomycetes | Saccharomycetales | UNKN | UNKN | UNKN |
| 67,722 | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales fam Incertae sedis | Cyberlindnera | sargentensis |
| 66,523 | Ascomycota | Saccharomycetes | Saccharomycetales | Pichiaceae | Pichia | norvegensis |
| 61,158 | Ascomycota | Eurotiomycetes | Chaetothyriales | Herpotrichiellaceae | Rhinocladiella | UNKN |
| 60,451 | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetacea e | Williopsis | UNKN |
| 58,157 | Ascomycota | Sordariomycetes | Xylariales | Diatrypaceae | UNKN | UNKN |
| 306,359 | Basidiomycota | Agaricomycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 288,893 | Basidiomycota | Agaricomycetes | Agaricales | Pleurotaceae | Pleurotus | dryinus |
| 270,948 | Basidiomycota | Agaricomycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 146,897 | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 124,892 | Basidiomycota | Agaricomycetes | UNKN | UNKN | UNKN | UNKN |
| 105,172 | Basidiomycota | Agaricomycetes | UNKN | UNKN | UNKN | UNKN |
| 91,483 | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 90,592 | Basidiomycota | Agaricomycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 87,551 | Basidiomycota | Agaricomycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 85,156 | Basidiomycota | Microbotryomycetes | Sporidiobolales | Sporidiobolales fam Incertae sedis | Rhodotorula | UNKN |
| 82,402 | Basidiomycota | Microbotryomycetes | Sporidiobolales | Sporidiobolales fam Incertae sedis | Rhodotorula | UNKN |
| 71,796 | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 63,190 | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 53,508 | Basidiomycota | Agaricomycetes | Russulales | Auriscalpiaceae | Lentinellus | UNKN |
| 47,017 | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 46,379 | Basidiomycota | Agaricomycetes | Russulales | Auriscalpiaceae | Lentinellus | UNKN |
| 37,400 | Basidiomycota | Ustilaginomycotina cls Incertae sedis | Malasseziales | Malasseziaceae | Malassezia | restricta |
| 35,845 | Basidiomycota | Ustilaginomycotina cls Incertae sedis | Malasseziales | Malasseziaceae | Malassezia | restricta |
| 34,408 | Basidiomycota | Agaricomycetes | Sebacinales | Sebacinales Group B | UNKN | UNKN |
| 31,389 | Basidiomycota | Tremellomycetes | Tremellales | Tremellales fam Incertae sedis | Derxomyces | qinlingensis |
| DF | Sum Of Sqs | Mean Sqs | F. Model | R2 | p-Value | |
|---|---|---|---|---|---|---|
| Tree Sp. | 1 | 2.120 | 2.120 | 6.3420 | 0.05732 | 0.000999 |
| Tree ID | 3 | 3.469 | 1.156 | 3.4593 | 0.09380 | 0.000999 |
| Primer | 1 | 3.015 | 3.015 | 9.0195 | 0.08152 | 0.000999 |
| Tree Sp. * Primer | 1 | 1.679 | 1.679 | 5.0228 | 0.04540 | 0.000999 |
| Tree ID * Primer | 3 | 2.969 | 0.999 | 2.9608 | 0.08028 | 0.000999 |
| Residuals | 71 | 23.734 | 0.334 | 0.64169 | ||
| Total | 89 | 41.599 | 1 |
| (a) Y = Species * Tree * Distance * Ab vs. Be | ||||||
| Degrees of Freedom | Sum of Sqrs | Mean Squares | F Statistic | R2 | p-Value | |
| Species | 1 | 1.9584 | 1.95845 | 8.2916 | 0.11208 | 0.000999 |
| Tree | 3 | 3.2307 | 1.07689 | 4.5593 | 0.18488 | 0.000999 |
| Distance | 1 | 0.3976 | 0.39764 | 1.6835 | 0.02276 | 0.018981 |
| Ab-Be | 2 | 1.0926 | 0.54629 | 2.3129 | 0.06252 | 0.000999 |
| Species: Distance | 1 | 0.4425 | 0.44251 | 1.8735 | 0.02532 | 0.008991 |
| Tree: Distance | 3 | 1.1604 | 0.38681 | 1.6377 | 0.06641 | 0.000999 |
| Species: Ab vs. Be | 2 | 0.9569 | 0.47845 | 2.0257 | 0.05476 | 0.000999 |
| Tree: Ab vs.Be | 5 | 2.2786 | 0.45572 | 1.9294 | 0.1304 | 0.000999 |
| Distance: Ab vs. Be | 1 | 0.4097 | 0.40972 | 1.7347 | 0.02345 | 0.015984 |
| Species: Distance:Ab vs. Be | 1 | 0.3675 | 0.36755 | 1.5561 | 0.02103 | 0.040959 |
| Tree: Distance: Ab vs. Be | 3 | 0.9278 | 0.30926 | 1.3094 | 0.05309 | 0.034965 |
| Residuals | 18 | 4.2515 | 0.2362 | 0.2433 | ||
| Total | 41 | 17.4744 | 1 | |||
| (b) Y = Species * Tree * Model B * Ab vs. Be | ||||||
| Degrees of Freedom | Sum of Sqrs | Mean Squares | F Statistic | R2 | p-Value | |
| Species | 1 | 1.9584 | 1.95845 | 7.9251 | 0.11208 | 0.000999 |
| Tree | 3 | 3.2307 | 1.07689 | 4.3578 | 0.18488 | 0.000999 |
| Model B | 4 | 1.5811 | 0.39527 | 1.5995 | 0.09048 | 0.005994 |
| Ab vs. Be | 1 | 0.5363 | 0.53632 | 2.1703 | 0.03069 | 0.001998 |
| Species: Model B | 4 | 1.5262 | 0.38155 | 1.544 | 0.08734 | 0.008991 |
| Tree: Model B | 11 | 3.6997 | 0.33634 | 1.361 | 0.21172 | 0.010989 |
| Species: Ab vs. Be | 1 | 0.4658 | 0.46577 | 1.8848 | 0.02665 | 0.006993 |
| Tree: Ab vs. Be | 3 | 1.2417 | 0.4139 | 1.6749 | 0.07106 | 0.001998 |
| Residuals | 8 | 1.977 | 0.24712 | 0.18509 | ||
| Total | 41 | 17.4744 | 1 | |||
| ITS1&2 (358 OTUs) | ITS4&5.8 (381 OTUs) | ||
|---|---|---|---|
| Ascomycota | 237 | Ascomycota | 278 |
| Basidiomycota | 81 | Basidiomycota | 81 |
| Chytridiomycota | 2 | UNKN | 14 |
| Glomeromycota | 1 | Mucoromycota/Zoopagomycota | 8 |
| UNKN | 32 | ||
| Mucoromycota/Zoopagomycota | 5 | ||
| Aspen | ||||
| Decay Class Pair | F. Model | R2 | p-Value | Adjusted p-Value |
| 3 vs. 5 | 2.222836 | 0.1156352 | 0.005 | 0.01 |
| 3 vs. 1 | 2.100934 | 0.1391261 | 0.009 | 0.01 |
| 5 vs. 1 | 2.064238 | 0.171139 | 0.003 | 0.009 |
| Poplar | ||||
| Decay Class Pair | F. Model | R2 | p-Value | Adjusted p-Value |
| 3 vs. 1 | 1.930536 | 0.1493005 | 0.032 | 0.032 |
| Aspen and Poplar Combined | ||||
| Decay Class Pair | F. Model | R2 | p-Value | Adjusted p-Value |
| 3 vs. 5 | 2.355585 | 0.08307 | 0.002 | 0.004 |
| 3 vs. 1 | 1.803715 | 0.06487 | 0.2 | 0.02 |
| 5 vs. 1 | 2.59398 | 0.15632 | 0.001 | 0.003 |
| Aspen Y = Tree * Decay Class | ||||||
| Degrees of Freedom | Sum of Squares | Mean Squares | F Statistic | R2 | p-Value | |
| Tree | 2 | 2.0153 | 1.00765 | 3.2834 | 0.2153 | 0.001 |
| Decay Class | 2 | 1.4809 | 0.74045 | 2.4128 | 0.1582 | 0.001 |
| Tree:Decay Class | 2 | 0.9535 | 0.47677 | 1.5535 | 0.1019 | 0.01798 |
| Residuals | 16 | 4.9102 | 0.30689 | 0.5246 | ||
| Total | 22 | 9.36 | 1 | |||
| Poplar Y = Tree * Decay Class | ||||||
| Degrees of Freedom | Sum of Squares | Mean Squares | F Statistic | R2 | p-Value | |
| Tree | 1 | 0.8983 | 0.89829 | 3.501 | 0.2239 | 0.001 |
| Decay Class | 1 | 0.5485 | 0.54848 | 2.1377 | 0.1367 | 0.01898 |
| Residuals | 10 | 2.5658 | 0.25658 | 0.6394 | ||
| Total | 12 | 4.0126 | 1 | |||
| Aspen and Poplar Combined Y = Tree * Decay Class | ||||||
| Degrees of Freedom | Sum of Squares | Mean Squares | F Statistic | R2 | p-Value | |
| Species | 1 | 1.5766 | 1.57656 | 5.4829 | 0.1055 | 0.001 |
| Tree | 3 | 2.9136 | 0.97119 | 3.3776 | 0.1949 | 0.001 |
| Decay Class | 2 | 1.5003 | 0.75017 | 2.6089 | 0.1004 | 0.001 |
| Species: Decay Class | 1 | 0.529 | 0.52903 | 1.8399 | 0.0354 | 0.01299 |
| Tree: Decay Class | 2 | 0.9535 | 0.47677 | 1.6581 | 0.0638 | 0.005 |
| Residuals | 26 | 7.476 | 0.28754 | 0.5001 | ||
| Total | 35 | 14.9491 | 1 | |||
| Aspen Decay Class 1 (n = 2) | |||||||
| Reads | Domain | Phylum | Class | Order | Family | Genus | Species |
| 14,523 | Fungi | Ascomycota | UNKN | UNKN | UNKN | UNKN | UNKN |
| 13,894 | Fungi | Ascomycota | UNKN | UNKN | UNKN | UNKN | UNKN |
| 12,090 | Fungi | Basidiomycota | Ustilaginomycotina cls Incertae sedis | Malasseziales | Malaseziaceae | Malassezia | restricta |
| 10,967 | Fungi | Basidiomycota | Ustilaginomycotina cls Incertae sedis | Malasseziales | Malaseziaceae | Malassezia | restricta |
| 6623 | Fungi | Ascomycota | Leotiomycetes | Helotiales | UNKN | UNKN | UNKN |
| 5302 | Fungi | Ascomycota | Leotiomycetes | Helotiales | UNKN | UNKN | UNKN |
| 4720 | Fungi | Basidiomycota | Agaricomycetes | UNKN | UNKN | UNKN | UNKN |
| 3972 | Fungi | Basidiomycota | Agaricomycetes | UNKN | UNKN | UNKN | UNKN |
| 3814 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | UNKN | UNKN | UNKN |
| 3531 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales_fam Incertae sedis | Cyberlindnera | sargentesis |
| Pleurotus_dryinus is three reads short of tied for 10th | |||||||
| Aspen Class 3 (n = 14) | |||||||
| Reads | Domain | Phylum | Class | Order | Family | Genus | Species |
| 305,240 | Fungi | Basid io mycota | Agaricom ycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 269,980 | Fungi | Basidiomycota | Agaricomycetes | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 102,147 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales_fam Incertae sedis | Candida | UNKN |
| 91,852 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetales_fam Incertae sedis | Candida | UNKN |
| 86,392 | Fungi | Basidiomycota | Agarico-myces | Hymenochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 83,504 | Fungi | Basidiomycota | Agaricomycetes | Hym enochaetales | Hymenochaetaceae | Phellinus | UNKN |
| 62,174 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | Pichiaceae | Pichia | norvegen-sis |
| 54,224 | Fungi | Ascomycota | Saccharomycetes | Saccharomycetales | UNKN | UNKN | UNKN |
| 40,411 | Fungi | Ascomycota | Leotiomycetes | Helotiales | Helotiaceae | Neobulgaria | UNKN |
| 39,793 | Fungi | Ascomycota | Leotiomycetes | Helotiales | Helotiaceae | Neobulgaria | UNKN |
| Aspen Decay Class 5 (n = 7) | |||||||
| Reads | Domain | Phylum | Class | Order | Family | Genus | Species |
| 478,031 | Fungi | Ascomycota | Sordariomycetes | Xylariales | Xylariaceae | Hypoxylon | macrocarpum |
| 403,497 | Fungi | Ascomycota | Sordariomycetes | Xylariales | Xylariaceae | Hypoxylon | macrocarpum |
| 164,510 | Fungi | Ascomycota | Sord ar iomycetes | Xylariales | Xylariaceae | Xylaria | UNKN |
| 156,660 | Fungi | Ascomycota | Sordariomycetes | Xylariales | UNKN | UNKN | UNKN |
| 135,434 | Fungi | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 115,881 | Fungi | Ascomycota | Eurotiomycetes | Chaetothyriales | Herpotrichiellaceae | Capronia | pilosella |
| 94,020 | Fungi | Ascomycota | Eurotiomycetes | Chaetothyriales | Herpotrichiellaceae | Capronia | pilosella |
| 82,341 | Fungi | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 62,060 | Fungi | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
| 54,882 | Fungi | Basidiomycota | Agaricomycetes | Cantharellales | Hydnaceae | Sistotrema | raduloides |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garbelotto, M.; Johnson, M.G. High-Throughput DNA Metabarcoding of Stem Sections from Trees with Cavities Describes Fungal Communities Associated with Variable Wood Decay, Position on Stem and Tree Species. Forests 2023, 14, 1070. https://doi.org/10.3390/f14061070
Garbelotto M, Johnson MG. High-Throughput DNA Metabarcoding of Stem Sections from Trees with Cavities Describes Fungal Communities Associated with Variable Wood Decay, Position on Stem and Tree Species. Forests. 2023; 14(6):1070. https://doi.org/10.3390/f14061070
Chicago/Turabian StyleGarbelotto, Matteo, and Michael G. Johnson. 2023. "High-Throughput DNA Metabarcoding of Stem Sections from Trees with Cavities Describes Fungal Communities Associated with Variable Wood Decay, Position on Stem and Tree Species" Forests 14, no. 6: 1070. https://doi.org/10.3390/f14061070