
Comparative Analysis of the Characteristics, Phylogenetic Relationships of the Complete Chloroplast Genome, and Maternal Origin Track of White Poplar Interspecific Hybrid GM107

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant DNA Extraction and Sequencing
2.2. Plant RNA Extraction and Transcriptome Sequencing
2.3. Chloroplast Genome Assembly and Annotation
2.4. Comparative Genomic Analyses
2.5. Characterization of Repeat Sequences and SSRs
2.6. Phylogenetic Analyses
3. Results
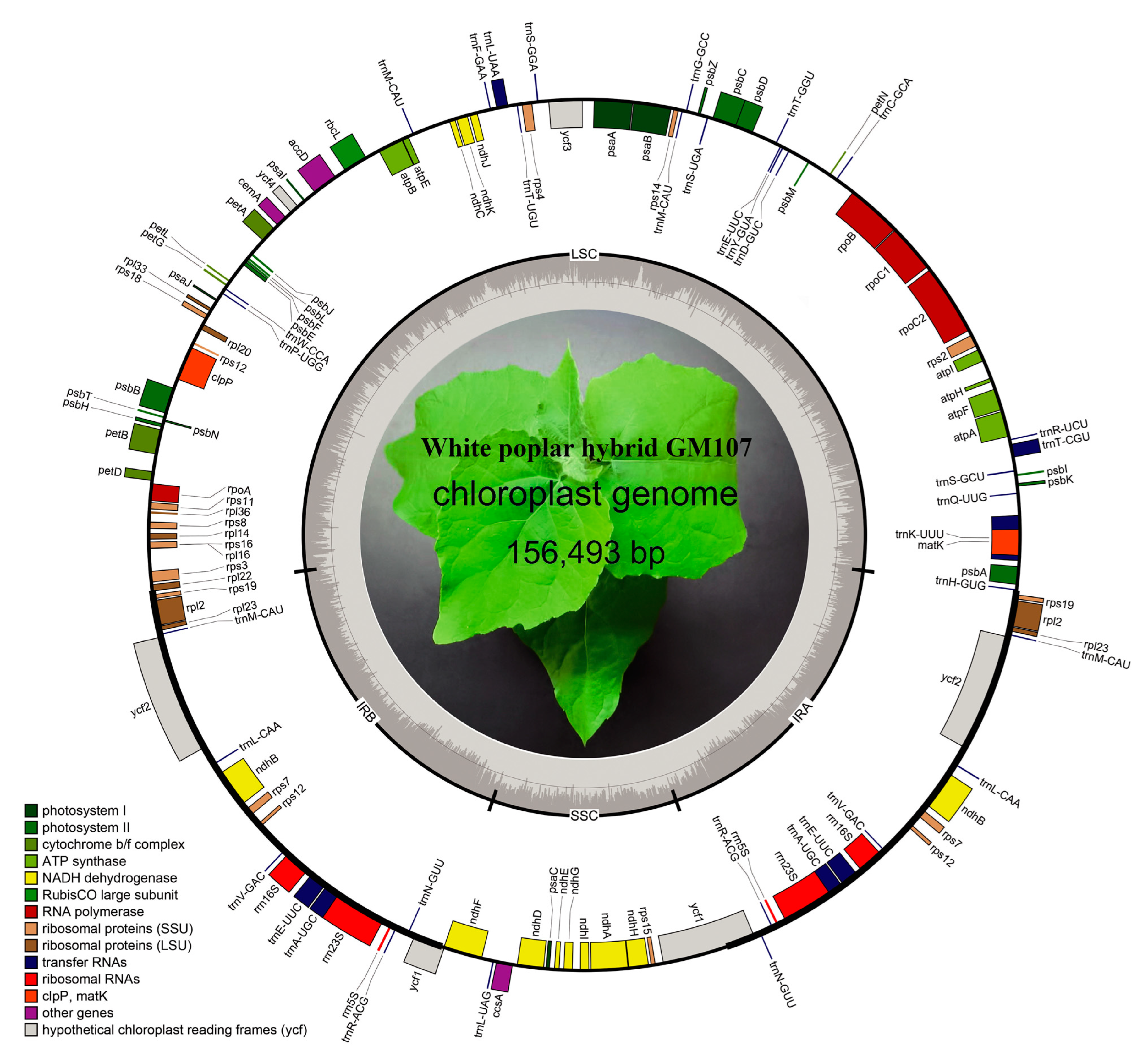
3.1. Cp Genome Structure
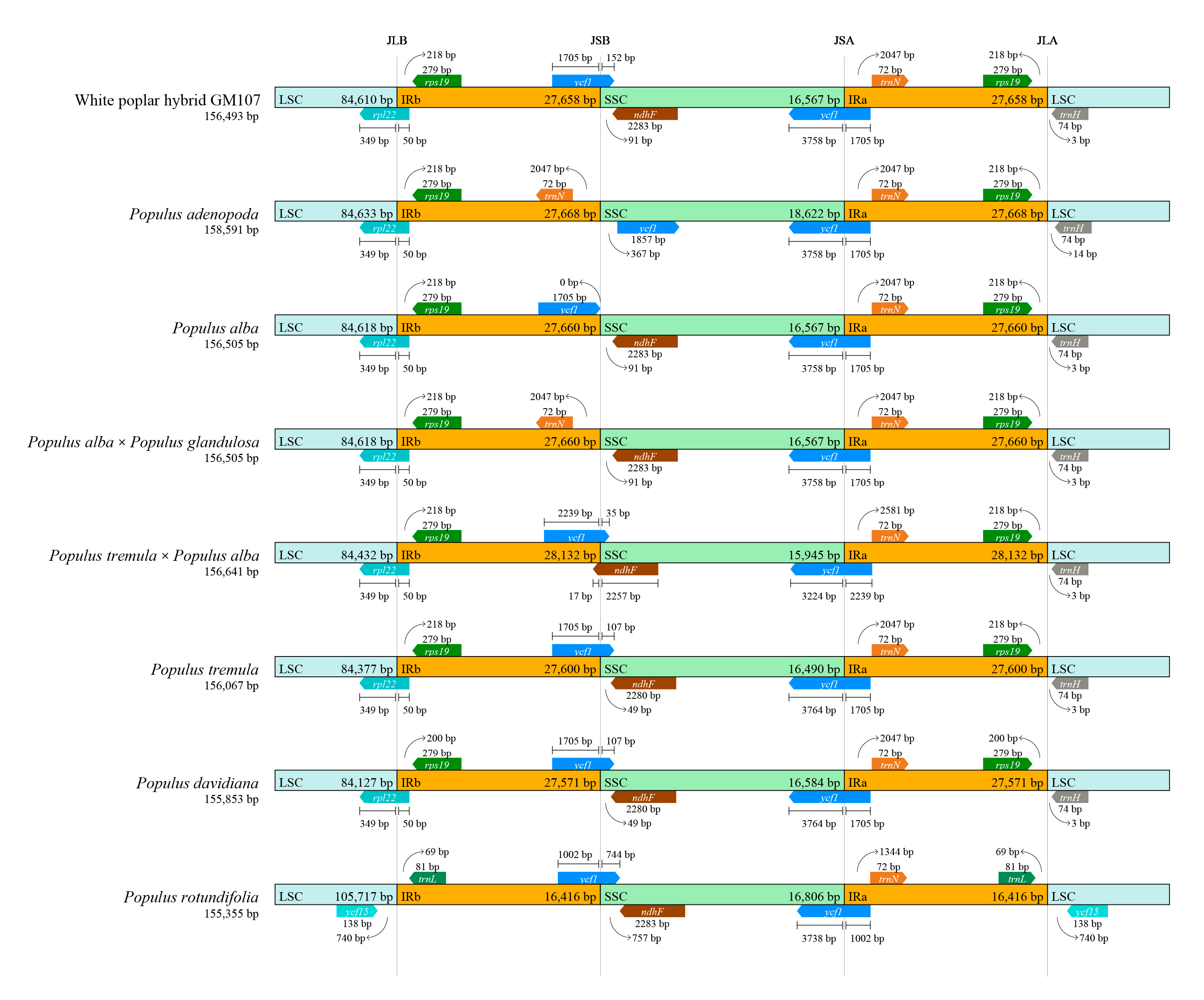
3.2. Expansion and Contraction of IR Regions
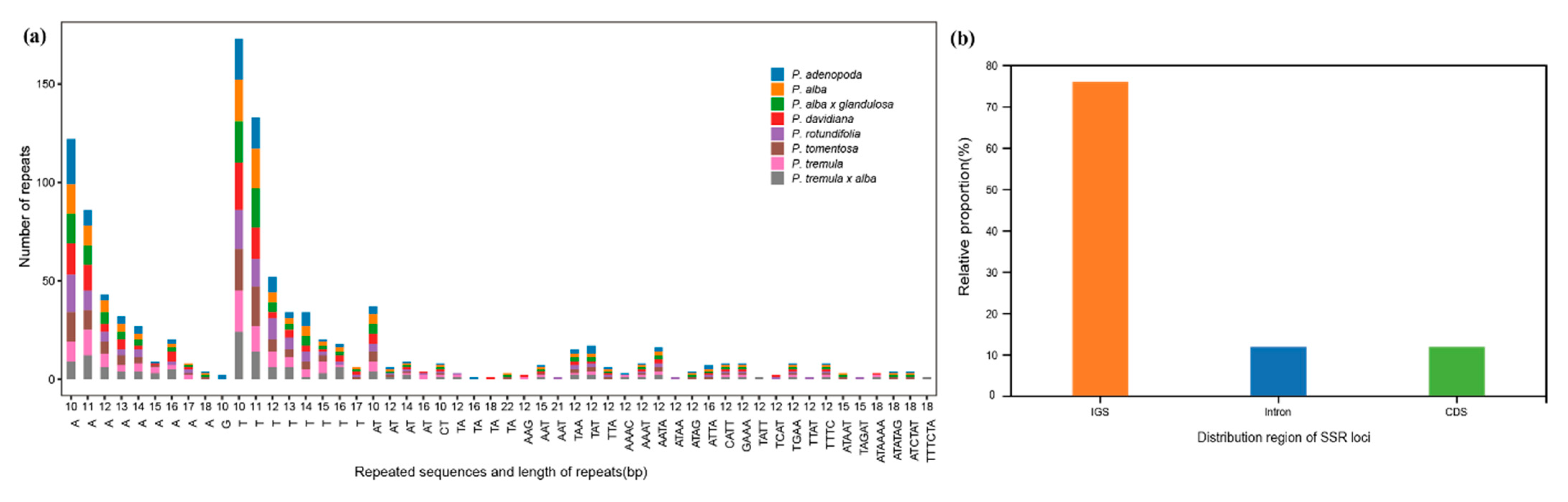
3.3. Repeat Structure and SSR Analyses
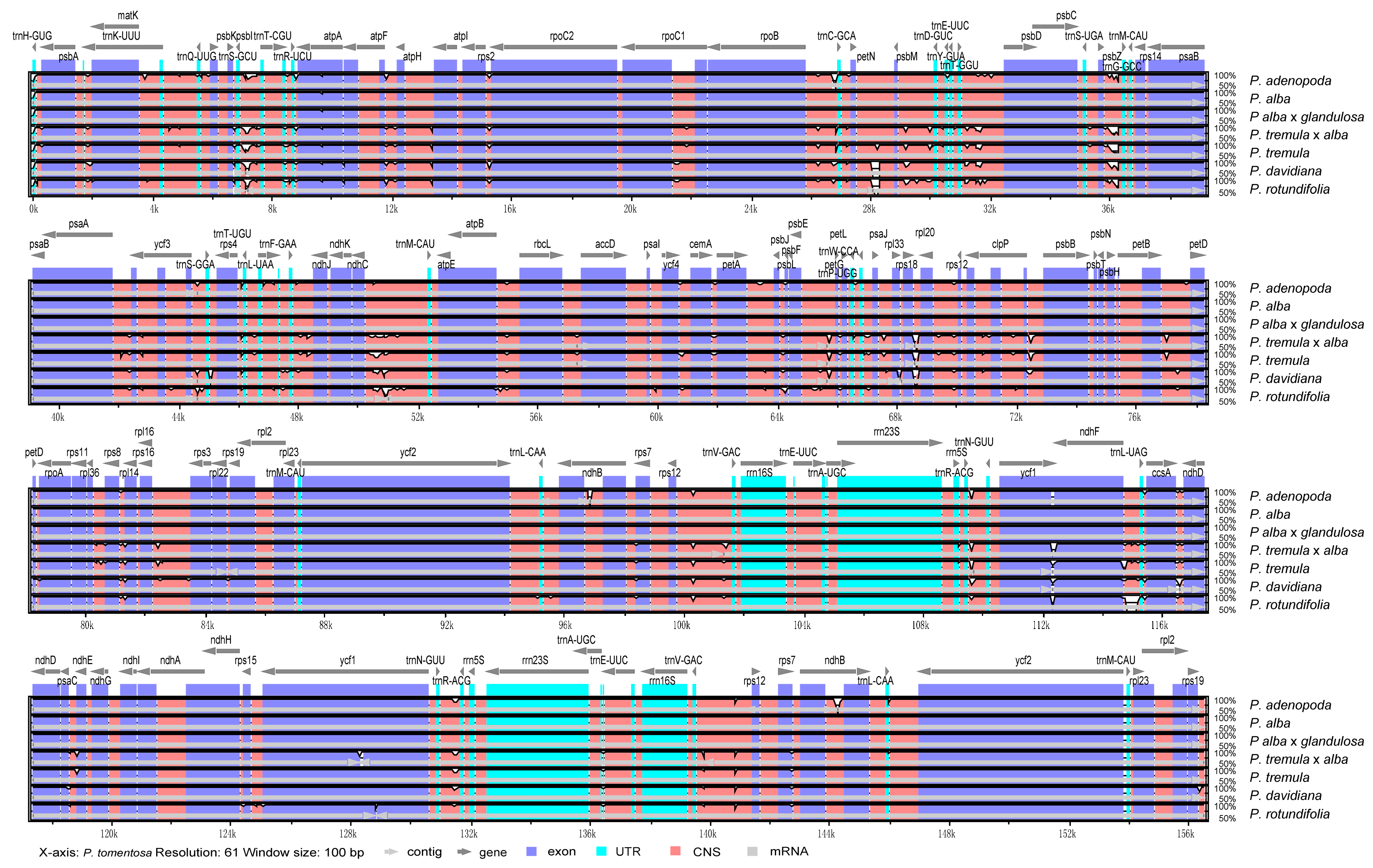
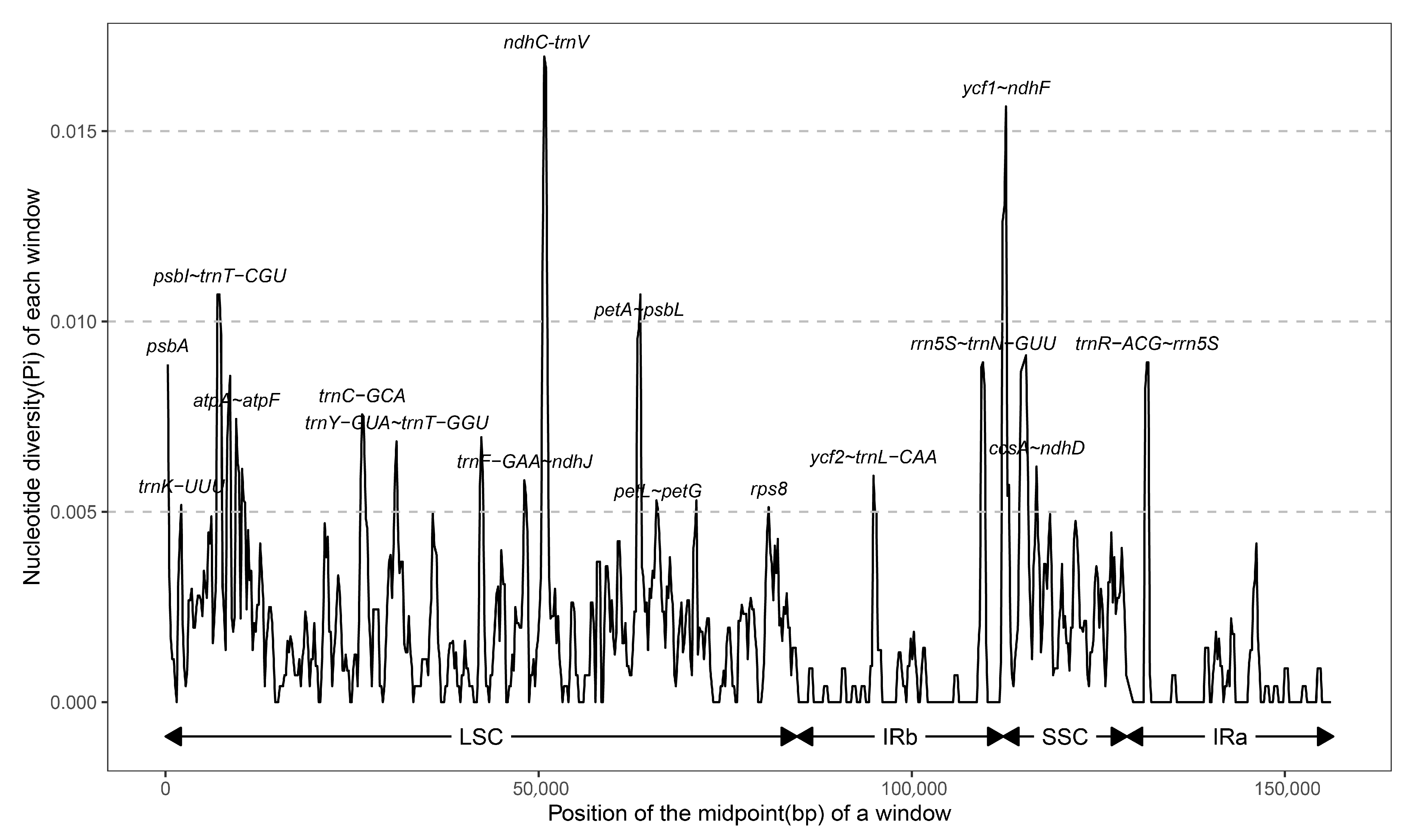
3.4. Sequence Divergence Analyses
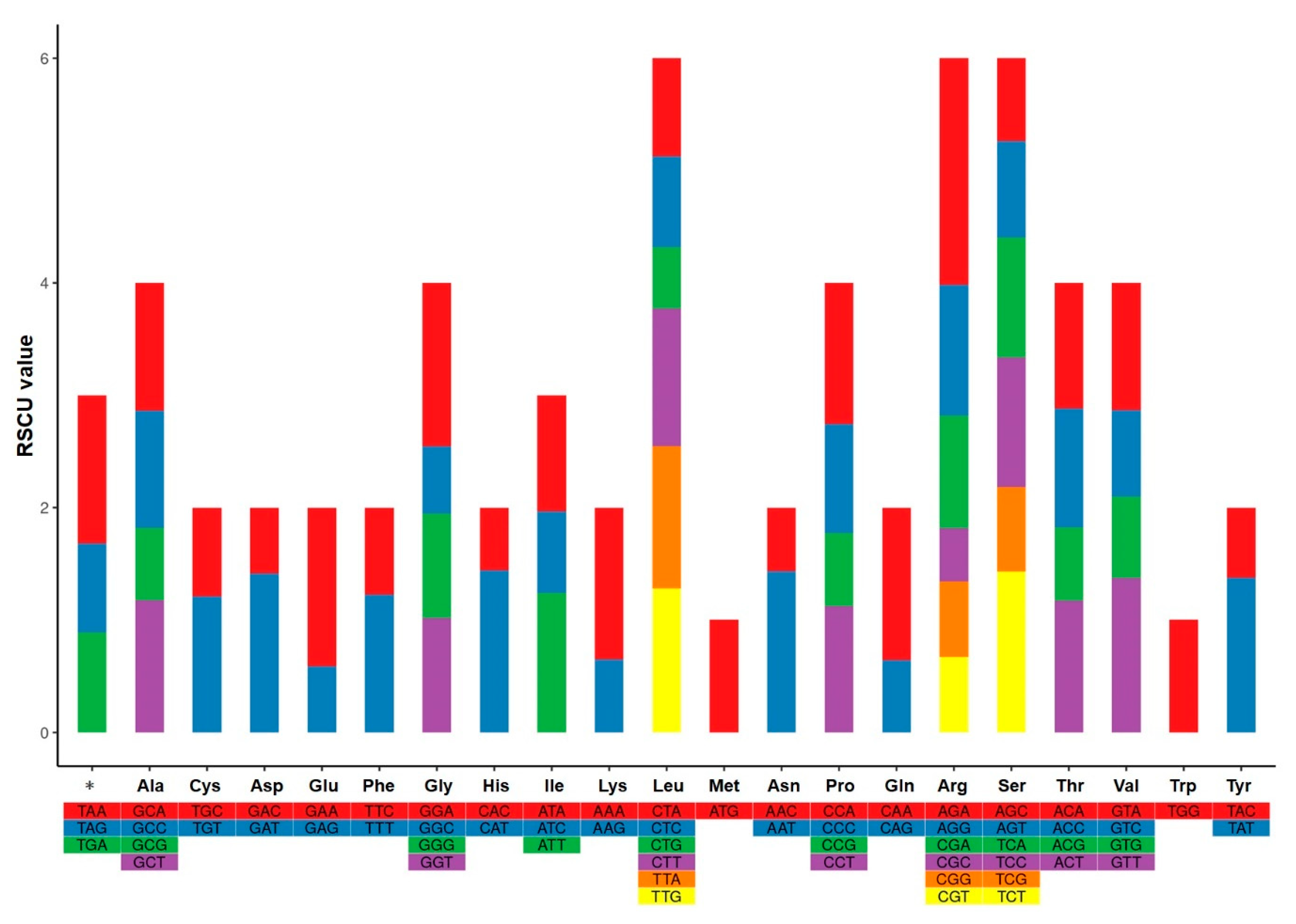
3.5. Codon Biased Usage Analysis
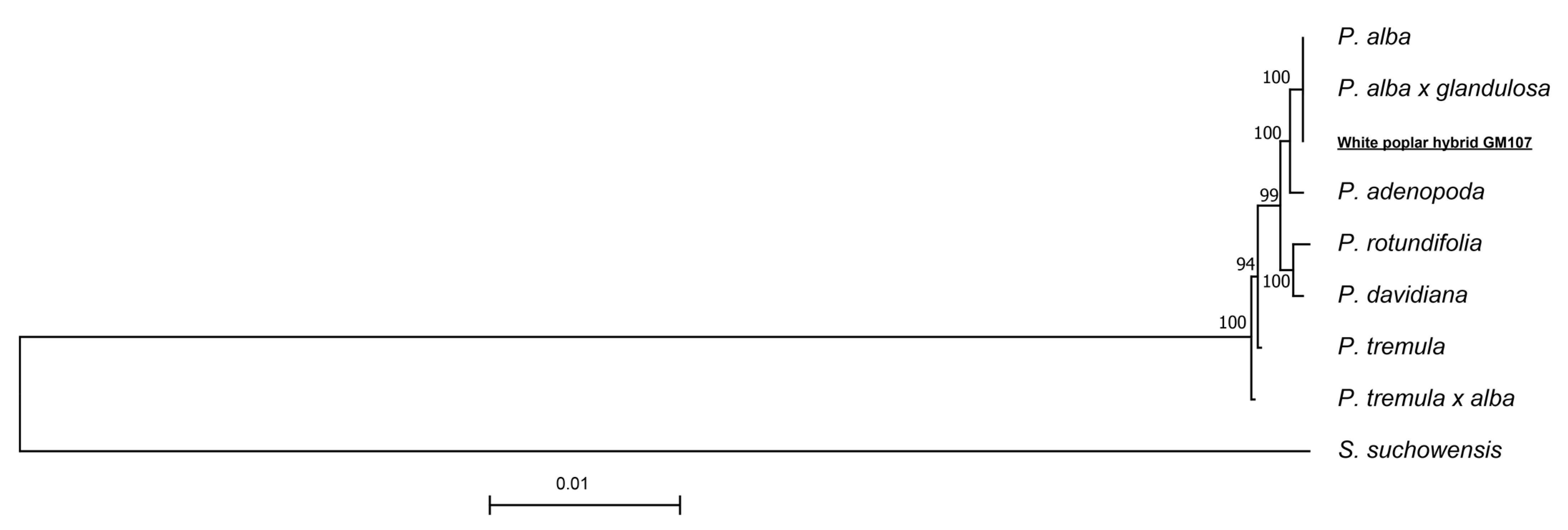
3.6. Phylogenetic Analyses
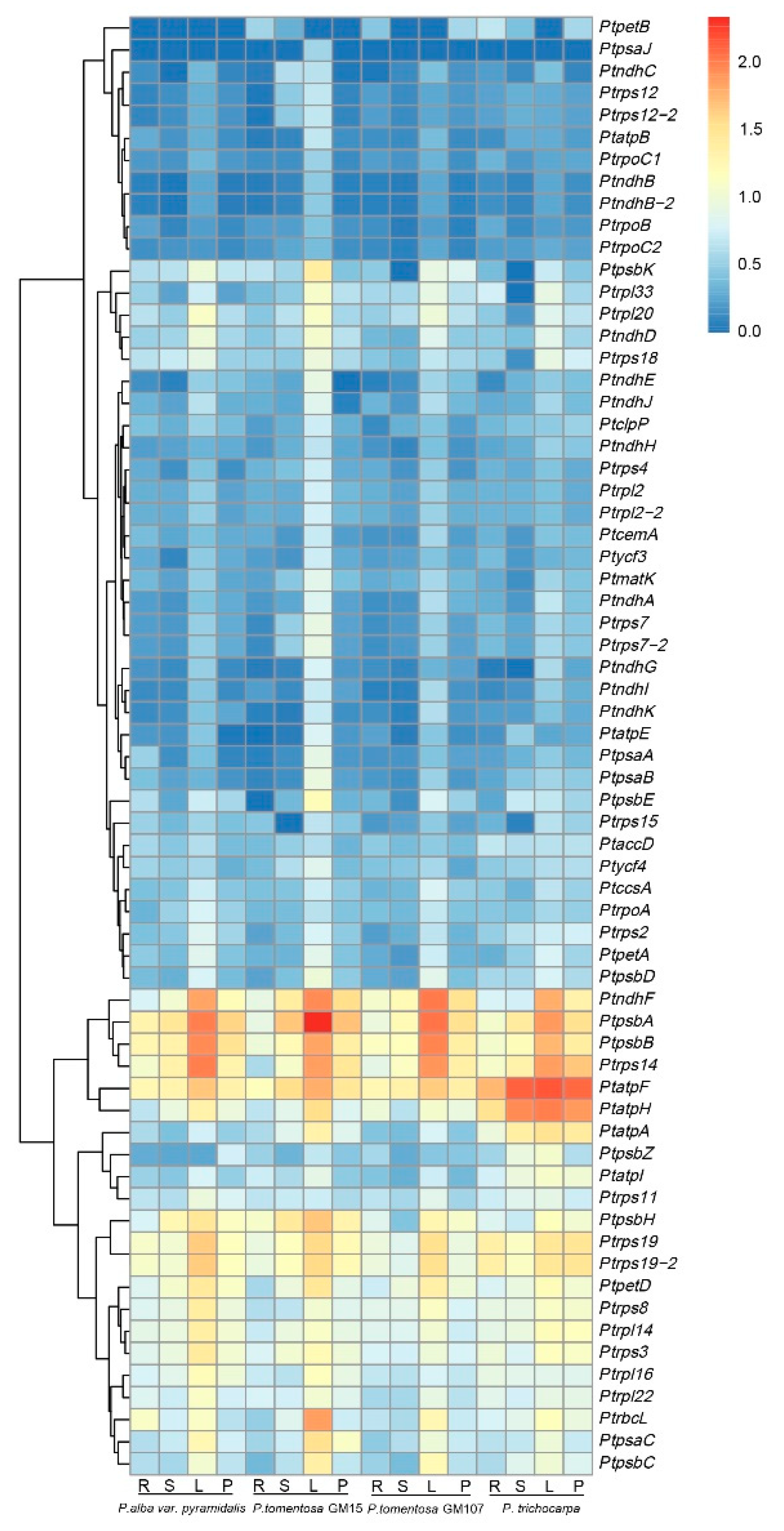
3.7. Transcription Profiles of cp Genes
4. Discussion
 ) and P. alba var. pyramidalis (
) and P. alba var. pyramidalis ( ) approximately 3.93 Mya [5]. Therefore, P. tomentosa may have multiple independent origins, suggesting a more complex evolutionary history.
) approximately 3.93 Mya [5]. Therefore, P. tomentosa may have multiple independent origins, suggesting a more complex evolutionary history.5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lexer, C.; Fay, M.F.; Joseph, J.A.; Nica, M.S.; Heinze, B. Barrier to gene flow between two ecologically divergent Populus species, P. alba (white poplar) and P. tremula (European aspen): The role of ecology and life history in gene introgression. Mol. Ecol. 2005, 14, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, D. Poplar Culture in North America; National Research Council Canada: Ottawa, ON, Canada, 2001. [Google Scholar]
- An, X.M.; Wang, D.M.; Wang, Z.L.; Li, B.; Bo, W.H.; Cao, G.L.; Zhang, Z.Y. Isolation of a LEAFY homolog from Populus tomentosa: Expression of PtLFY in P. tomentosa floral buds and PtLFY-IR-mediated gene silencing in tobacco (Nicotiana tabacum). Plant Cell Rep. 2011, 30, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, J.; Guo, T.; Guo, B.; Chen, Z.; An, X. Comprehensive analysis of the R2R3-MYB transcription factor gene family in Populus trichocarpa. Ind. Crops Prod. 2021, 168, 113614. [Google Scholar] [CrossRef]
- An, X.; Gao, K.; Chen, Z.; Li, J.; Yang, X.; Yang, X.; Zhou, J.; Guo, T.; Zhao, T.; Huang, S. High quality haplotype-resolved genome assemblies of Populus tomentosa Carr., a stabilized interspecific hybrid species widespread in Asia. Mol. Ecol. Resour. 2022, 22, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Li, J.; Khan, W.U.; Zhao, T.; Yang, X.; Yang, X.; Guo, B.; An, X. Comparative genomic and phylogenetic analyses of Populus section Leuce using complete chloroplast genome sequences. Tree Genet. Genomes. 2019, 15, 32. [Google Scholar] [CrossRef]
- He, C. Study on Genetic Diversity and Origin of Populus tomentosa Carr; Beijing Forestry University: Beijing, China, 2005. [Google Scholar]
- Wang, D.; Wang, Z.; Kang, X.; Zhang, J. Genetic analysis of admixture and hybrid patterns of Populus hopeiensis and P. tomentosa. Sci. Rep. 2019, 9, 4821. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Chen, Z.; He, L.; Feng, Q.; Zhang, H.; Du, G.; Shi, C.; Wang, S. Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications. Genes 2022, 13, 461. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.M.; Md Shah, M.U.; Makale, K.; Mohd-Yusuf, Y.; Khalid, N.; Othman, R.Y. Complete Chloroplast Genome Sequence of Corroborates Structural Heterogeneity of Inverted Repeats in Wild Progenitors of Cultivated Bananas and Plantains. Plant Genome 2016, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Lossl, A.G.; Waheed, M.T. Chloroplast-derived vaccines against human diseases: Achievements, challenges and scopes. Plant Biotechnol. J. 2011, 9, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I. Chloroplast genome sequencing: Some reflections. J. Next Gener. Seq. Appl. 2015, 2, 119. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craciunas, L.; Pickering, O.; Chu, J.; Choudhary, M.; Zurauskiene, J.; Coomarasamy, A. The transcriptomic profile of endometrial receptivity in recurrent miscarriage. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 261, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Sović, I.; Šikić, M.; Wilm, A.; Fenlon, S.N.; Chen, S.; Nagarajan, N. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 2016, 7, 11307. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Han, X.M.; Wang, Y.M.; Liu, Y.J. The complete chloroplast genome sequence of Populus wilsonii and its phylogenetic analysis. Mitochondrial DNA B Resour. 2017, 2, 932–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, B.; Faivre Rampant, P.; Mader, M.; Le Paslier, M.-C.; Bounon, R.; Berard, A.; Vettori, C.; Schroeder, H.; Leplé, J.-C.; Fladung, M. Genome sequences of Populus tremula chloroplast and mitochondrion: Implications for holistic poplar breeding. PLoS ONE 2016, 11, e0147209. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, T.; Bai, G.; Zhao, Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef] [Green Version]
- Asaf, S.; Khan, A.L.; Lubna; Khan, A.; Khan, A.; Khan, G.; Lee, I.J.; Al-Harrasi, A. Expanded inverted repeat region with large scale inversion in the first complete plastid genome sequence of Plantago ovata. Sci. Rep. 2020, 10, 3881. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.P.; Li, C.; Wang, J.; Wang, Y.; Ding, Z.T. Analysis of synonymous codon usage in FAD7 genes from different plant species. Genet. Mol. Res. 2015, 14, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mei, C.; Zhen, H.; Zhu, J. Codon preference optimization increases prokaryotic cystatin C expression. J. Biomed. Biotechnol. 2012, 732017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.I.; Hefer, C.A.; Kolosova, N.; Douglas, C.J.; Cronk, Q.C.B. Whole plastome sequencing reveals deep plastid divergence and cytonuclear discordance between closely related balsam poplars, Populus balsamifera and P. trichocarpa (Salicaceae). New Phytol. 2014, 204, 693–703. [Google Scholar] [CrossRef]
- Bausher, M.G.; Singh, N.D.; Lee, S.B.; Jansen, R.K.; Daniell, H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: Organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhou, T.; Han, K.; Yang, Y.; Zhao, J.; Liu, Z.L. The Complete Chloroplast Genome Sequences of Six Rehmannia Species. Genes 2017, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.; Baurens, F.C.; Cardi, C.; Aury, J.M.; D’Hont, A. The complete chloroplast genome of banana (Musa acuminata, Zingiberales): Insight into plastid monocotyledon evolution. PLoS ONE 2013, 8, e67350. [Google Scholar] [CrossRef] [Green Version]
- Melotto-Passarin, D.M.; Tambarussi, E.V.; Dressano, K.; De Martin, V.F.; Carrer, H. Characterization of chloroplast DNA microsatellites from Saccharum spp and related species. Genet. Mol. Res. 2011, 10, 2024–2033. [Google Scholar] [CrossRef]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; McLenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef]
- Cavender-Bares, J.; Gonzalez-Rodriguez, A.; Eaton, D.A.; Hipp, A.A.; Beulke, A.; Manos, P.S. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol. Ecol. 2015, 24, 3668–3687. [Google Scholar] [CrossRef]
- Hebert, P.D.; Ratnasingham, S.; deWaard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 2003, 270, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Little, D.M. A DNA barcode for land plants. Biol. Sci. 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef] [PubMed]
- Eckenwalder, J.E. Systematics and Evolution of Populus; National Research Council Canada: Ottawa, ON, Canada, 1996. [Google Scholar]
- Mejnartowicz, M. Inheritance of chloroplast DNA in Populus. Theor. Appl. Genet. 1991, 82, 477–480. [Google Scholar] [CrossRef]
- Palmer, J.D. Chloroplast DNA evolution and biosystematic uses of chloroplast DNA variation. Am. Nat. 1987, 130, 6–29. [Google Scholar] [CrossRef]
- Wang, Z.; Du, S.; Dayanandan, S.; Wang, D.; Zeng, Y.; Zhang, J. Phylogeny reconstruction and hybrid analysis of populus (Salicaceae) based on nucleotide sequences of multiple single-copy nuclear genes and plastid fragments. PLoS ONE 2014, 9, e103645. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Elite Individual GM107 |
|---|---|
| Length (bp) | 156,493 |
| GC content (%) | 36.74 |
| AT content (%) | 63.26 |
| LSC GC content (%) | 39.31 |
| SSC GC content (%) | 30.49 |
| IR GC content (%) | 41.95 |
| LSC length (bp) | 84,610 |
| SSC length (bp) | 16,567 |
| IR length (bp) | 27,658 |
| CDS length (bp) | 80,562 |
| tRNA length (bp) | 2648 |
| rRNA length (bp) | 8599 |
| Gene number | 127 |
| Gene number in IR regions | 34 |
| Protein-coding gene number | 85 |
| Protein-coding gene (%) | 66.93 |
| rRNA gene number | 6 |
| rRNA (%) | 4.72 |
| tRNA gene number | 36 |
| tRNA (%) | 28.35 |
| Category of Genes | Family Name List of Genes | List of Genes |
|---|---|---|
| Photosynthesis-related genes | Subunit of rubisco | rbcL |
| Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Assembly of photosystem I | ycf4, ycf3 ** | |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Subunits of cytochrome b/f complex | petA, petB *, petD, petG, petL, petN | |
| c-type cytochrom synthesis gene | ccsA | |
| Subunits of NADH-dehydrogenase | ndhA *, ndhB * (×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of protochlorophyllide reductase | - | |
| Transcription-related and translation-related genes | DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 |
| Small subunit of ribosome | rps11, rps12 * (×2), rps14, rps15, rps16, rps18, rps19 (×2), rps2, rps3, rps4, rps7 (×2), rps8 | |
| Large subunit of ribosome | rpl14, rpl16, rpl2 * (×2), rpl20, rpl22, rpl23(×2), rpl33, rpl36 | |
| Translational initiation factor | - | |
| Elongation factor | - | |
| RNA and its processing | rRNA genes | rrn16S (×2), rrn23S (×2), rrn5S (×2) |
| tRNA genes | trnH-GUG, trnK-UUU *, trnQ-UUG, trnS-GCU, trnT-CGU *, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC *, trnT-GGU, trnS-UGA, trnG-GCC, trnM-CAU, trnS-GGA, trnT-UGU, trnL-UAA *, trnF-GAA, trnM-CAU, trnW-CCA, trnP-UGG, trnM-CAU (×2), trnL-CAA (×2), trnV-GAC (×2), trnE-UUC (×2), trnA-UGC * (×2), trnR-ACG (×2), trnN-GUU (×2), trnL-UAG | |
| Other genes | RNA processing | matK |
| Subunit of Acetyl-CoA-carboxylase | accD | |
| Envelop membrane protein | cemA | |
| Protease | clpP ** | |
| Component of TIC complex | ycf1 (×2) | |
| Hypothetical proteins | ycf2 (×2) |
| Species | Number of Repeats | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 44 | 45 | 46 | 47 | 48 | 55 | 68 | 76 | |
| P. adenopoda | 13 | 4 | 4 | 1 | 3 | 1 | 1 | 0 | 0 | 6 | 0 | 1 | 6 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 |
| P. alba | 16 | 4 | 6 | 2 | 3 | 1 | 2 | 1 | 0 | 6 | 0 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| P. alba × P. glandulosa | 16 | 4 | 6 | 2 | 3 | 1 | 2 | 1 | 0 | 6 | 0 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| P. davidiana | 13 | 5 | 4 | 2 | 0 | 2 | 1 | 0 | 0 | 6 | 0 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 |
| P. rotundifolia | 12 | 8 | 11 | 3 | 0 | 2 | 1 | 1 | 1 | 6 | 2 | 1 | 4 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| Elite individual GM107 | 16 | 4 | 6 | 2 | 3 | 1 | 2 | 1 | 0 | 6 | 0 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| P. tremula | 13 | 3 | 3 | 1 | 1 | 2 | 1 | 1 | 0 | 7 | 0 | 2 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| P. tremula × P. alba | 16 | 4 | 6 | 2 | 3 | 1 | 2 | 1 | 0 | 6 | 0 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| Species | Number of Repeats | |||
|---|---|---|---|---|
| Forward | Palindromic | Reverse | Complement | |
| P. adenopoda | 23 | 19 | 3 | 0 |
| P. alba | 22 | 16 | 6 | 4 |
| P. alba × P. glandulosa | 22 | 16 | 6 | 4 |
| P. davidiana | 21 | 15 | 3 | 1 |
| P. rotundifolia | 25 | 20 | 10 | 2 |
| Elite individual GM107 | 22 | 16 | 6 | 4 |
| P. tremula | 19 | 17 | 4 | 0 |
| P. tremula × P. alba | 22 | 16 | 6 | 4 |
| Total | 176 | 135 | 44 | 19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, B.; Chen, T.; Li, Y.; Li, S.; Khan, W.U.; Zhang, R.-G.; Jia, K.-H.; An, X. Comparative Analysis of the Characteristics, Phylogenetic Relationships of the Complete Chloroplast Genome, and Maternal Origin Track of White Poplar Interspecific Hybrid GM107. Forests 2023, 14, 587. https://doi.org/10.3390/f14030587
Guo B, Chen T, Li Y, Li S, Khan WU, Zhang R-G, Jia K-H, An X. Comparative Analysis of the Characteristics, Phylogenetic Relationships of the Complete Chloroplast Genome, and Maternal Origin Track of White Poplar Interspecific Hybrid GM107. Forests. 2023; 14(3):587. https://doi.org/10.3390/f14030587
Chicago/Turabian StyleGuo, Bin, Tingting Chen, Ying Li, Shanwen Li, Wasif Ullah Khan, Ren-Gang Zhang, Kai-Hua Jia, and Xinmin An. 2023. "Comparative Analysis of the Characteristics, Phylogenetic Relationships of the Complete Chloroplast Genome, and Maternal Origin Track of White Poplar Interspecific Hybrid GM107" Forests 14, no. 3: 587. https://doi.org/10.3390/f14030587