Potential of Genome-Wide Association Studies and Genomic Selection to Improve Productivity and Quality of Commercial Timber Species in Tropical Rainforest, a Case Study of Shorea platyclados

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
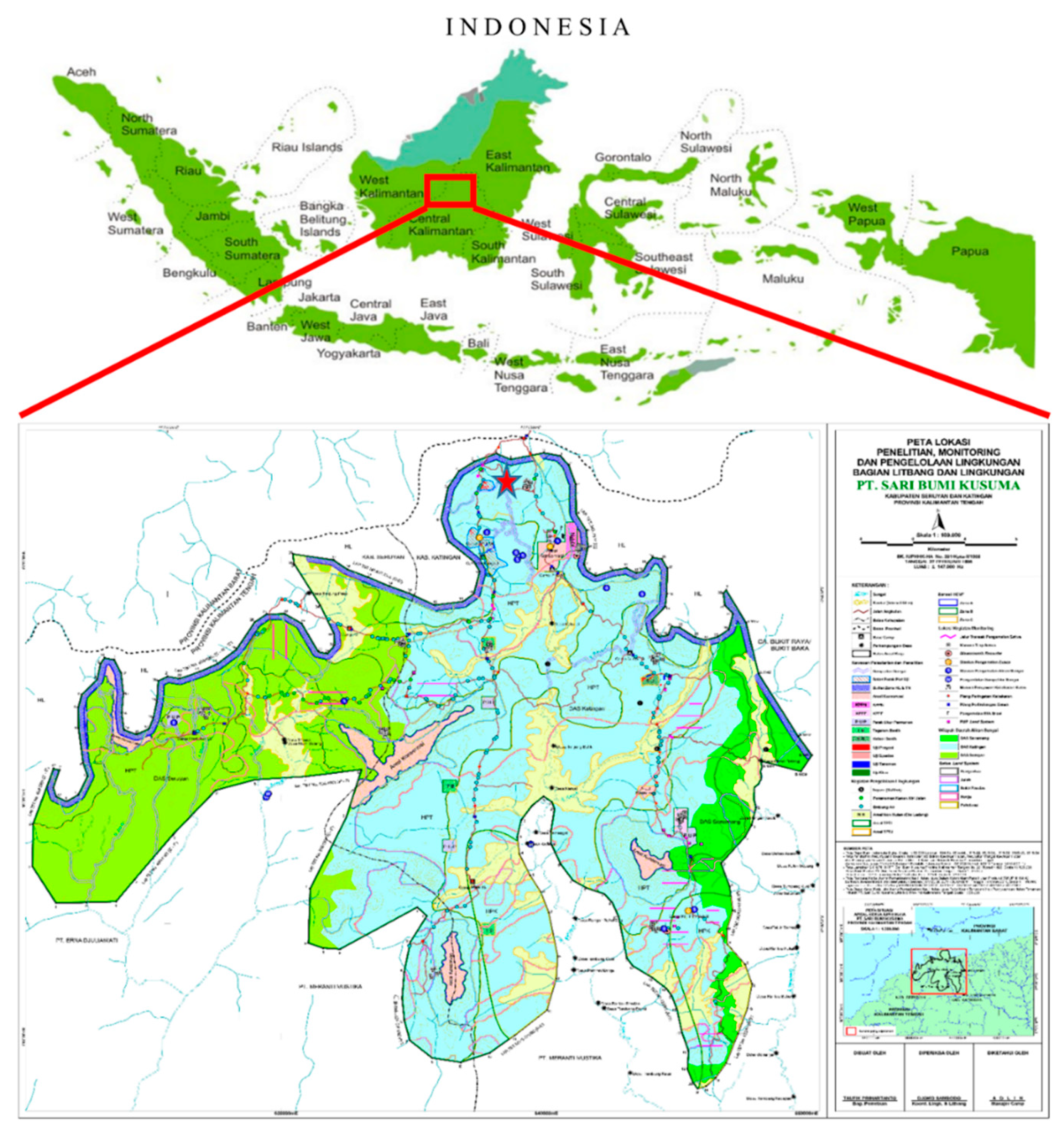
2.1. Study Site
2.2. Phenotypic Data
2.3. Genotypic Data and SNP Discovery
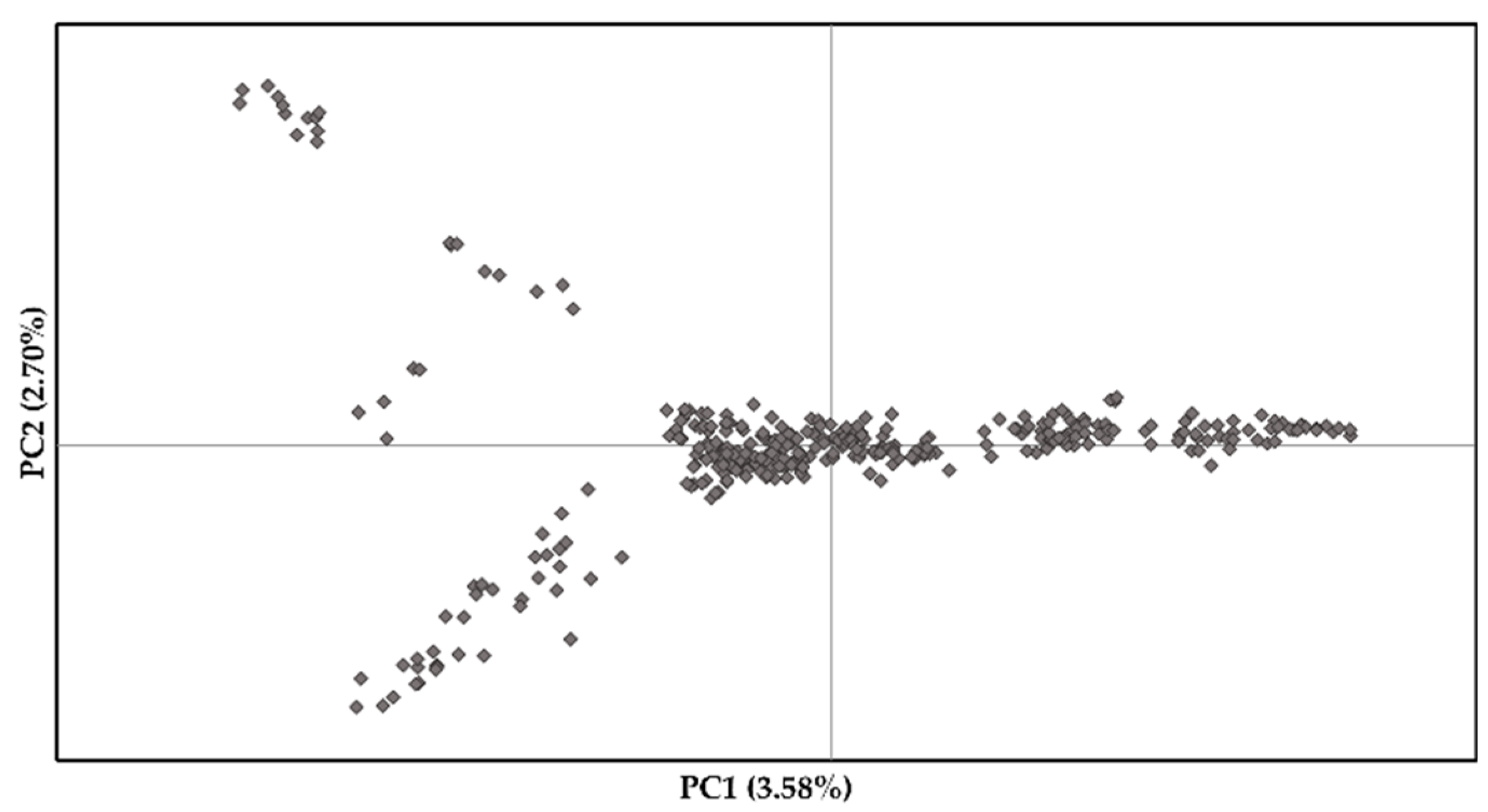
2.4. Population Structure and Genetic Diversity
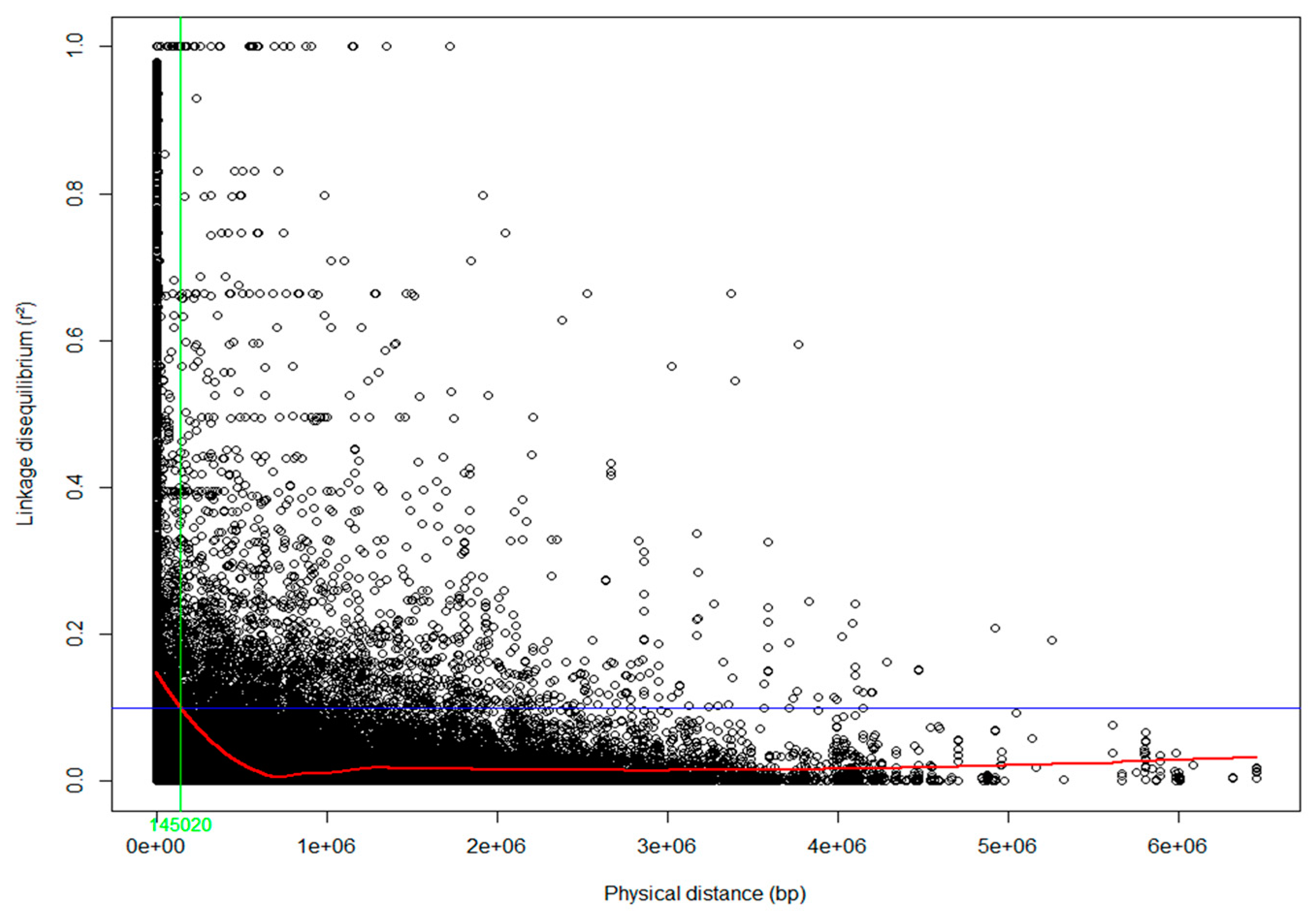
2.5. Linkage Disequilibrium
2.6. Spatial Analysis and Genotype Imputation
2.7. GWAS Using All Individuals and Markers
2.8. Four-Fold Cross-Validation of GWAS-Based Genomic Prediction
- (a)
- all SNP markers in the whole genome (5900 SNPs);
- (b)
- selected SNP markers with high −log10(P) values according to the GWAS analysis (applying five GWAS-based thresholds: −log10(P) > 0.5, −log10(P) > 0.75, −log10(P) > 1, −log10(P) > 1.25, and −log10(P) > 1.5.
2.9. Genomic Heritability
3. Results
3.1. Population Structure and Linkage Disequilibrium
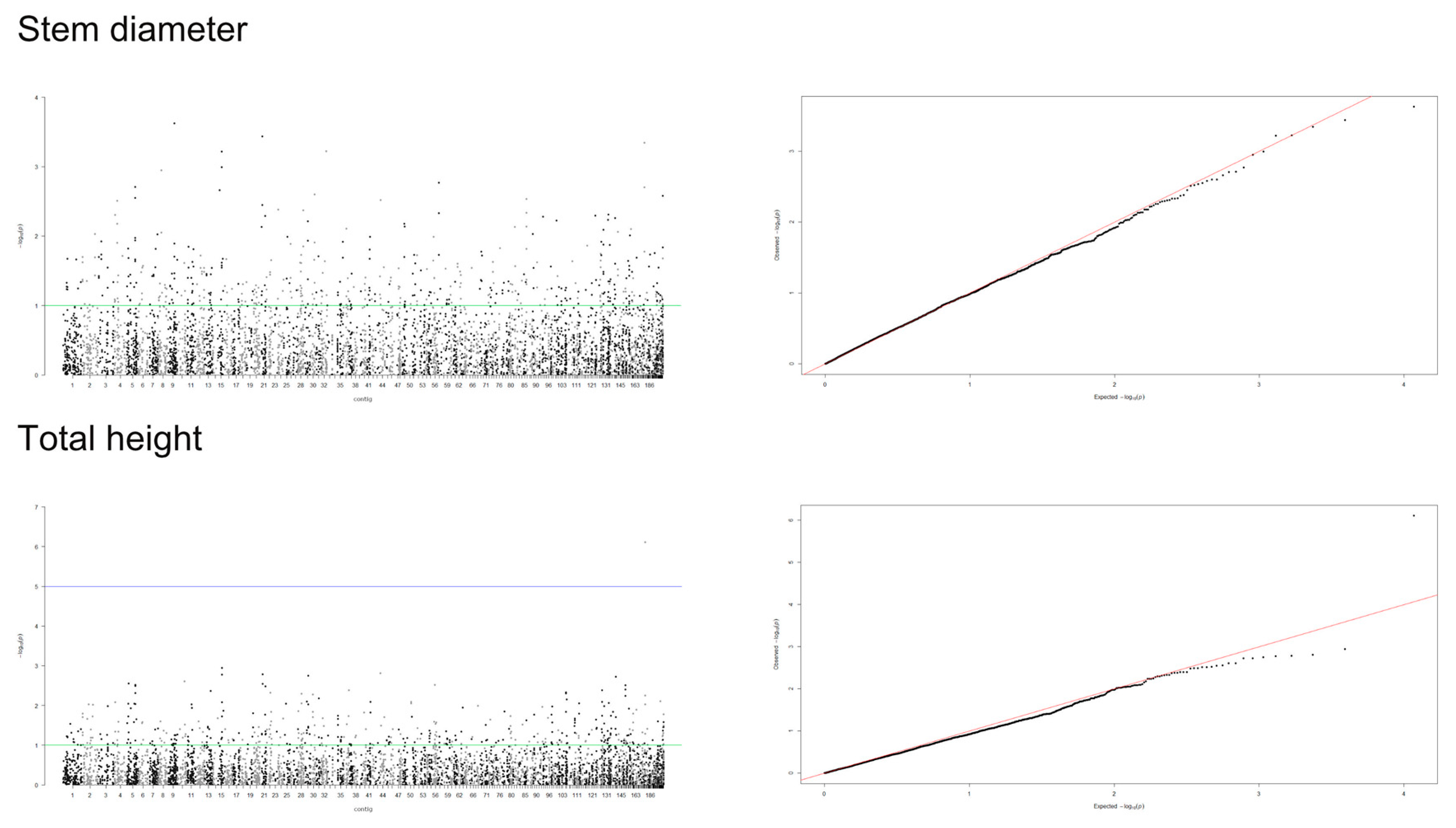
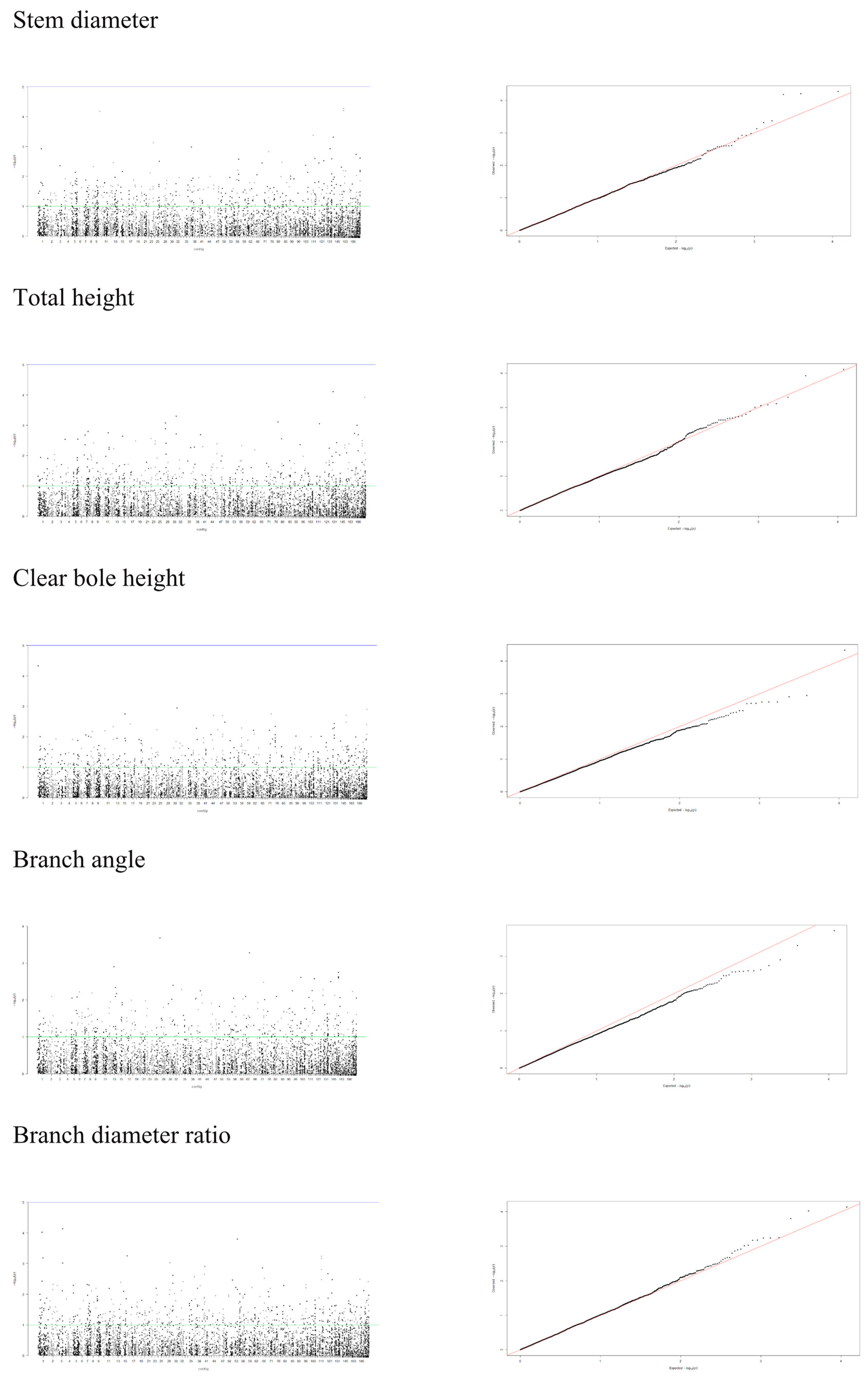
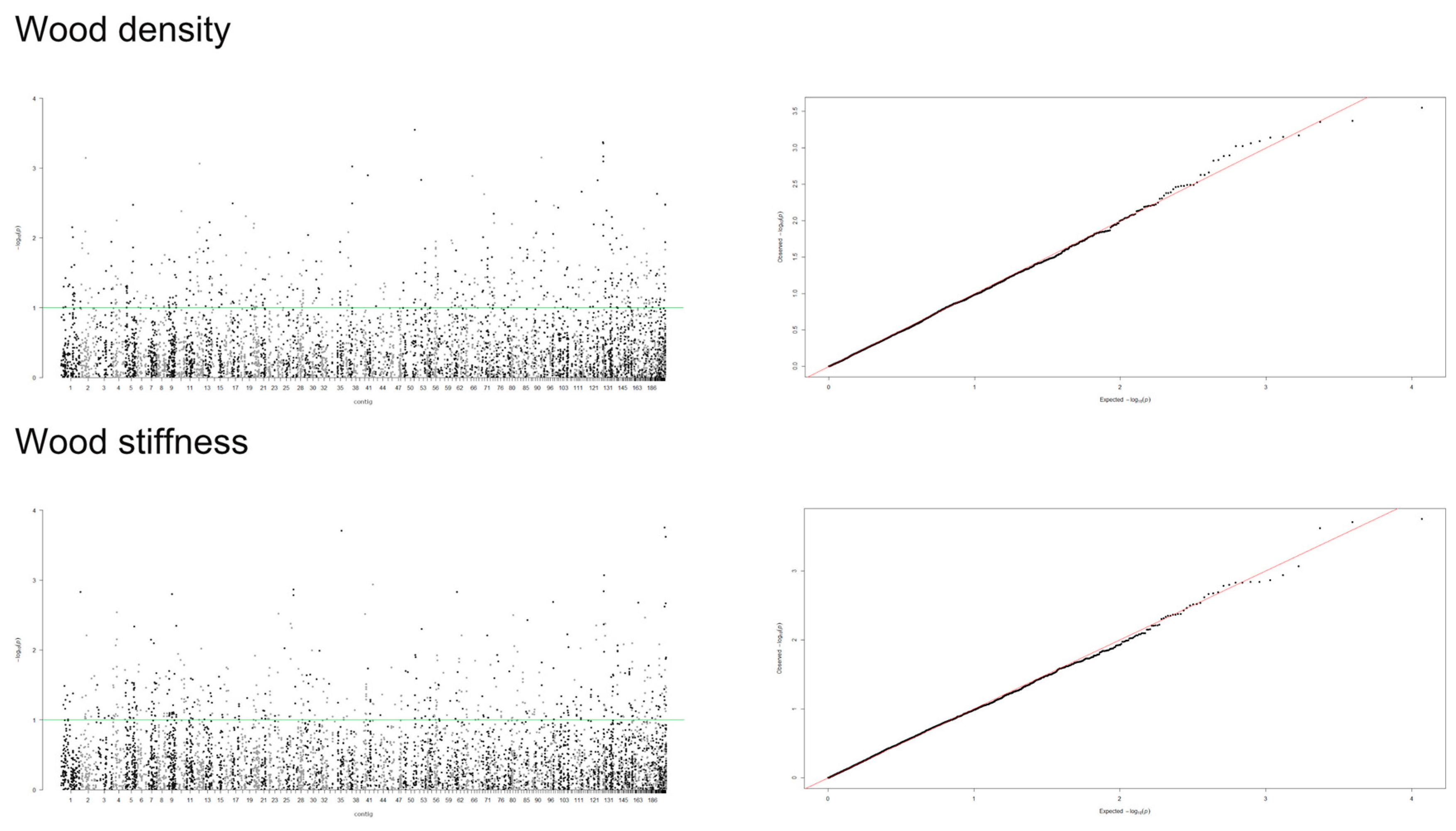
3.2. Genome-Wide Association Study
3.3. Prediction Accuracies Based on the Bayesian Models
3.4. Prediction Accuracies Based on the Machine Learning Methods
3.5. Genomic Heritability
4. Discussion
4.1. Population Structure
4.2. Detection of Significant Markers by GWAS
4.3. Genomic Predictions Based on All SNPs and GWAS-Based Thresholds
4.4. Genomic Prediction Accuracies of Bayesian and Machine Learning Methods
4.5. Genomic Heritability
4.6. Factors Affecting the Accuracy of GS of S. platyclados
4.7. Potential Utility of GWAS and GS for Breeding Tropical Tree Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bawa, K. Conservation of genetic resources in the Dipterocarpaceae. In A Review of Dipterocarps: Taxonomy, Ecology and Silviculture; Appanah, S., Turnbull, J.M., Eds.; CIFOR: Bogor, Indonesia, 1998; pp. 45–56. [Google Scholar]
- Corlett, R.; Primack, R. Dipterocarps: Trees that dominate the Asian rain forest. Arnoldia 2005, 63, 2–7. [Google Scholar]
- Muslich, M.; Sumarni, G. Durability of 200 Indonesian wood species against marine borrers. J. For. Prod. Res. 2005, 23, 163–176. [Google Scholar] [CrossRef]
- Ghazoul, J. Dipterocarp Biology, Ecology, and Conservation; Oxford University Press: Oxford, UK, 2016; pp. 211–231. [Google Scholar]
- Appanah, S.; Turnbull, J.M. A Review of Dipterocarps: Taxonomy, Ecology, and Silviculture; CIFOR: Bogor, Indonesia, 1998; pp. 89–99. [Google Scholar]
- Inada, T.; Hardiwitono, S.; Purnomo, S.; Putra, I.; Kitajima, K.; Kanzaki, M. Dynamics of forest regeneration following logging management in a Bornean lowland Dipterocarp forest. J. Trop. For. Sci. 2017, 29, 185–197. [Google Scholar]
- Grattapaglia, D.; Silva-Junior, O.B.; Resende, R.T.; Cappa, E.P.; Müller, B.S.; Tan, B.; Isik, F.; Ratcliffe, B.; El-Kassaby, Y. Quantitative genetics and genomics converge to accelerate forest tree breeding. Front. Plant Sci. 2018, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, A.; Meilan, R.; Kirst, M.; Morgante, M.; Boerjan, W.; Sabatti, M.; Mugnozza, G.S. Accelerating the domestication of forest trees in a changing world. Trends Plant Sci. 2012, 17, 64–72. [Google Scholar] [CrossRef]
- Khan, M.A.; Korban, S.S. Association mapping in forest trees and fruit crops. J. Exp. Bot. 2012, 63, 4045–4060. [Google Scholar] [CrossRef] [Green Version]
- Heslot, N.; Jannink, J.-L.; Sorrells, M.E. Perspectives for genomic selection applications and research in plants. Crop Sci. 2015, 55, 1–12. [Google Scholar] [CrossRef]
- Iwata, H.; Hayashi, T.; Tsumura, Y. Prospects for genomic selection in conifer breeding: A simulation study of Cryptomeria japonica. Tree Genet. Genomes 2011, 7, 747–758. [Google Scholar] [CrossRef]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet 2016, 7, 221. [Google Scholar] [CrossRef] [Green Version]
- Kainer, D.; Lanfear, R.; Foley, W.J.; Külheim, C. Genomic approaches to selection in outcrossing perennials: Focus on essential oil crops. Theor. Appl. Genet. 2015, 128, 2351–2365. [Google Scholar] [CrossRef]
- Grinberg, N.F.; Orhobor, O.I.; King, R.D. An evaluation of Machine-learning for predicting phenotype: Studies in yeast, rice and wheat. Mach. Learn. 2019, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Grattapaglia, D.; Resende, M.D. Genomic selection in forest tree breeding. Tree Genet. Genomes 2011, 7, 241–255. [Google Scholar] [CrossRef]
- Howard, R.; Carriquiry, A.L.; Beavis, W.D. Parametric and nonparametric statistical methods for genomic selection of traits with additive and epistatic genetic architectures. G3 Genes Genomes Genet. 2014, 4, 1027–1046. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- De Los Campos, G.; Naya, H.; Gianola, D.; Crossa, J.; Legarra, A.; Manfredi, E.; Weigel, K.; Cotes, J.M. Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 2009, 182, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.H.E.; Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Baison, J.; Pan, J.; Karlsson, B.; Andersson, B.; Westin, J.; García-Gil, M.R.; Wu, H.X. Accuracy of genomic selection for growth and wood quality traits in two control-pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genom. 2018, 19, 946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.S.; Neves, L.G.; de Almeida Filho, J.E.; Resende, M.F.; Muñoz, P.R.; dos Santos, P.E.; Paludzyszyn Filho, E.; Kirst, M.; Grattapaglia, D. Genomic prediction in contrast to a genome-wide association study in explaining heritable variation of complex growth traits in breeding populations of Eucalyptus. BMC Genom. 2017, 18, 524. [Google Scholar] [CrossRef] [Green Version]
- Resende, M.F.R., Jr.; Muñoz, P.; Resende, M.D.V.; Garrick, D.J.; Fernando, R.L.; Davis, J.M.; Jokela, E.J.; Marti, T.A.; Peter, G.F.; Kirst, M. Accuracy of genomic selection methods in a standard data set of Loblolly pine (Pinus taeda L.). Genetics 2012, 190, 1503. [Google Scholar] [CrossRef] [Green Version]
- Chipman, H.A.; George, E.I.; McCulloch, R.E. BART: Bayesian additive regression trees. Ann. Appl. Stat. 2010, 4, 266–298. [Google Scholar] [CrossRef]
- Li, B.; Zhang, N.; Wang, Y.-G.; George, A.W.; Reverter, A.; Li, Y. Genomic prediction of breeding values using a subset of SNPs identified by three machine learning methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, P. Genome-wide prediction using Bayesian additive regression trees. Genet. Sel. Evol. 2016, 48, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef]
- Cappa, E.P.; El-Kassaby, Y.A.; Muñoz, F.; Garcia, M.N.; Villalba, P.V.; Klápště, J.; Poltri, S.N.M. Genomic-based multiple-trait evaluation in Eucalyptus grandis using dominant DArT markers. Plant Sci. 2018, 271, 27–33. [Google Scholar] [CrossRef]
- Tan, B.; Grattapaglia, D.; Martins, G.S.; Ferreira, K.Z.; Sundberg, B.; Ingvarsson, P.K. Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F 1 hybrids. BMC Plant Biol. 2017, 17, 110. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.; Doerksen, T.; Clément, S.; MacKay, J.; Bousquet, J. Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity 2014, 113, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Bartholomé, J.; Van Heerwaarden, J.; Isik, F.; Boury, C.; Vidal, M.; Plomion, C.; Bouffier, L. Performance of genomic prediction within and across generations in maritime pine. BMC Genom. 2016, 17, 604. [Google Scholar] [CrossRef]
- Uchiyama, K.; Iwata, H.; Moriguchi, Y.; Ujino-Ihara, T.; Ueno, S.; Taguchi, Y.; Tsubomura, M.; Mishima, K.; Iki, T.; Watanabe, A. Demonstration of genome-wide association studies for identifying markers for wood property and male strobili traits in Cryptomeria japonica. PLoS ONE 2013, 8, e79866. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, Y.; Fukatsu, E.; Mishima, K.; Hirao, T.; Teshima, K.M.; Tamura, M.; Tsubomura, M.; Iki, T.; Kurita, M.; Takahasi, M. Potential of genome-wide studies in unrelated plus trees of a coniferous species, Cryptomeria japonica (Japanese cedar). Front. Plant Sci. 2018, 9, 1322. [Google Scholar] [CrossRef]
- Murray, M.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrioko, S.; Gailing, O.; Finkeldey, R. Molecular phylogeny of Dipterocarpaceae in Indonesia based on chloroplast DNA. Plant Syst. Evol. 2006, 261, 99–115. [Google Scholar] [CrossRef]
- Somego, M. Cytogenetical study of Dipterocarpaceae. Malays. For. 1978, 41, 358–366. [Google Scholar]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2014, 2, e431. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:12073907. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK 1.9. Available online: https://www.cog-genomics.org/plink2/ (accessed on 17 January 2020).
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. pcaMethods—A bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef]
- Blyton, M.D.J.; Flanagan, N.S. A Comprehensive Guide to: GenAlEx 6.5; Australia (AU), Australian National University: Canberra, Australia, 2006. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Zegeye, H.; Rasheed, A.; Makdis, F.; Badebo, A.; Ogbonnaya, F.C. Genome-wide association mapping for seedling and adult plant resistance to stripe rust in synthetic hexaploid wheat. PLoS ONE 2014, 9, e105593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laido, G.; Marone, D.; Russo, M.A.; Colecchia, S.A.; Mastrangelo, A.M.; De Vita, P.; Papa, R. Linkage disequilibrium and genome-wide association mapping in tetraploid wheat (Triticum turgidum L.). PLoS ONE 2014, 9, e95211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Helmersson, A.; Westin, J.; Karlsson, B.; Wu, H.X. Efficiency of using spatial analysis for Norway spruce progeny tests in Sweden. Ann. For. Sci. 2018, 75, 2. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Ueno, S.; Ujino-Ihara, T.; Fujiwara, T.; Yamashita, K.; Kanetani, S.; Endo, R.; Matsumoto, A.; Uchiyama, K.; Matsui, Y. Mapping quantitative trait loci for growth and wood property traits in Cryptomeria japonica across multiple environments. Tree Genet. Genomes 2019, 15, 43. [Google Scholar] [CrossRef]
- Munoz, F. breedR: Statistical Methods for Forest Genetic Resources Analysts. 2015. Available online: https://prodinra.inra.fr/record/329057 (accessed on 22 February 2019).
- Verma, S.S.; De Andrade, M.; Tromp, G.; Kuivaniemi, H.; Pugh, E.; Namjou-Khales, B.; Mukherjee, S.; Jarvik, G.P.; Kottyan, L.C.; Burt, A. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014, 5, 370. [Google Scholar] [CrossRef]
- Isik, F.; Holland, J.; Maltecca, C. Genetic Data Analysis for Plant and Animal Breeding; Springer: Raleigh, NC, USA, 2017; pp. 287–309. [Google Scholar]
- Browning, B.L. Beagle 4.1. 2017. Available online: https://faculty.washington.edu/browning/beagle/b4_1.html (accessed on 24 February 2019).
- Wang, Y.; Lin, G.; Li, C.; Stothard, P. Genotype imputation methods and their effects on genomic predictions in cattle. Springer Sci. Rev. 2016, 4, 79–98. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pike, N. Using false discovery rates for multiple comparisons in ecology and evolution. Methods Ecol. Evol. 2011, 2, 278–282. [Google Scholar] [CrossRef]
- Endelman, J.; Endelman, M.J. Package ‘rrBLUP’. 2018. Available online: http://www2.uaem.mx/r-mirror/web/packages/rrBLUP/rrBLUP.pdf (accessed on 5 January 2019).
- Turner, S. qqman: QQ and Manhattan Plots for GWAS Data, R package version 0.1. 4; 2017. Available online: https://cran.r-project.org/web/packages/qqman/ (accessed on 5 January 2019).
- Minamikawa, M.F.; Takada, N.; Terakami, S.; Saito, T.; Onogi, A.; Kajiya-Kanegae, H.; Hayashi, T.; Yamamoto, T.; Iwata, H. Genome-wide association study and genomic prediction using parental and breeding populations of Japanese pear (Pyrus pyrifolia Nakai). Sci. Rep. 2018, 8, 11994. [Google Scholar] [CrossRef] [Green Version]
- De los Campos, G.; Sorensen, D.; Gianola, D. Genomic heritability: What is it? PLoS Genet. 2015, 11, e1005048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.H.; Martin, L.S.; Eskin, E. Population structure in genetic studies: Confounding factors and mixed models. PLoS Genet. 2018, 14, e1007309. [Google Scholar] [CrossRef] [PubMed]
- Allwright, M.R.; Payne, A.; Emiliani, G.; Milner, S.; Viger, M.; Rouse, F.; Kaurentjes, J.J.; Bérard, A.; Wildhagen, H.; Faivre-Rampant, P. Biomass traits and candidate genes for bioenergy revealed through association genetics in coppiced European Populus nigra (L.). Biotechnol. Biofuels 2016, 9, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanaga, H.; Teshima, K.M.; Khatab, I.A.; Inomata, N.; Finkeldey, R.; Siregar, I.Z.; Siregar, U.J.; Szmidt, A.E. Population structure and demographic history of a tropical lowland rainforest tree species Shorea parvifolia (Dipterocarpaceae) from Southeastern Asia. Ecol. Evol. 2012, 2, 1663–1675. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Nanami, S.; Kenzo, T.; Yoneda, R.; Diway, B.; Chong, L.; Azani, M.A.; Majid, N.M.; Lum, S.K.; Wong, K.-M. Demographic history of Shorea curtisii (Dipterocarpaceae) inferred from chloroplast DNA sequence variations. Biotropica 2012, 44, 577–585. [Google Scholar] [CrossRef]
- Ng, C.H.; Lee, S.L.; Tnah, L.H.; Ng, K.K.S.; Lee, C.T.; Diway, B.; Khoo, E. Geographic origin and individual assignment of Shorea platyclados (Dipterocarpaceae) for forensic identification. PLoS ONE 2017, 12, e0176158. [Google Scholar] [CrossRef]
- Ng, C.H.; Lee, S.L.; Tnah, L.H.; Ng, K.K.; Lee, C.T.; Diway, B.; Khoo, E. Genetic diversity and demographic history of an upper hill Dipterocarp (Shorea platyclados): Implications for conservation. J. Hered. 2019, 110, 844–856. [Google Scholar] [CrossRef]
- Ohtani, M.; Kondo, T.; Tani, N.; Ueno, S.; Lee, L.S.; Ng, K.K.; Muhammad, N.; Finkeldey, R.; Na’iem, M.; Indrioko, S. Nuclear and chloroplast DNA phylogeography reveals Pleistocene divergence and subsequent secondary contact of two genetic lineages of the tropical rainforest tree species Shorea leprosula (Dipterocarpaceae) in Southeast Asia. Mol. Ecol. 2013, 22, 2264–2279. [Google Scholar] [CrossRef]
- Brzyski, D.; Peterson, C.B.; Sobczyk, P.; Candès, E.J.; Bogdan, M.; Sabatti, C. Controlling the rate of GWAS false discoveries. Genetics 2017, 205, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Kuo, K.H. Multiple testing in the context of gene discovery in Sickle Cell disease using Genome-Wide Association Studies. Genom. Insights 2017, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, W.S. How does multiple testing correction work? Nat. Biotechnol. 2009, 271, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F., Jr.; Vazquez, A.I.; de los Campos, G.; Dervinis, C.; Sykes, R.; Davis, M.; Davenport, R.; Barbazuk, W.B. Genome-wide association study reveals putative regulators of bioenergy traits in Populus deltoides. New Phytol. 2017, 213, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.T.; Resende, M.D.V.; Silva, F.F.; Azevedo, C.F.; Takahashi, E.K.; Silva-Junior, O.B. Regional heritability mapping and genome-wide association identify loci for complex growth, wood and disease resistance traits in Eucalyptus. New Phytol. 2017, 213, 1287–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinot, F.; Szafranski, M.; Ambroise, C.; Samson, F. Learning the optimal scale for GWAS through hierarchical SNP aggregation. BMC Bioinform. 2018, 19, 459. [Google Scholar] [CrossRef] [PubMed]
- Kaler, A.S.; Purcell, L.C. Estimation of a significance threshold for genome-wide association studies. BMC Genom. 2019, 20, 618. [Google Scholar] [CrossRef] [Green Version]
- Ingvarsson, P.K. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 2005, 169, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.R.; Gill, G.P.; Kuntz, R.J.; Langley, C.H.; Neale, D.B. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Nat. Acad. Sci. USA 2004, 101, 15255–15260. [Google Scholar] [CrossRef] [Green Version]
- Ballesta, P.; Maldonado, C.; Pérez-Rodríguez, P.; Mora, F. SNP and haplotype-based genomic selection of quantitative traits in Eucalyptus globulus. Plants 2019, 8, 331. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.M.; DiFazio, S.; Gunter, L.; Jawdy, S.; Boerjan, W.; Tuskan, G. Genetic and physical mapping of Melampsora rust resistance genes in Populus and characterization of linkage disequilibrium and flanking genomic sequence. New Phytol. 2004, 164, 95–105. [Google Scholar] [CrossRef]
- Moritsuka, E.; Hisataka, Y.; Tamura, M.; Uchiyama, K.; Watanabe, A.; Tsumura, Y.; Tachida, H. Extended linkage disequilibrium in noncoding regions in a conifer, Cryptomeria japonica. Genetics 2012, 190, 1145–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, B.S.; de Almeida Filho, J.E.; Lima, B.M.; Garcia, C.C.; Missiaggia, A.; Aguiar, A.; Takahashi, E.; Kirst, M.; Gezan, S.; Silva-Junior, O.B. Independent and Joint-GWAS for growth traits in Eucalyptus by assembling genome-wide data for 3373 individuals across four breeding populations. New Phytol. 2019, 221, 818–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.; Doerksen, T.; Boyle, B.; Clément, S.; Deslauriers, M.; Beauseigle, S.; Blais, S.; Poulin, P.-L.; Lenz, P.; Caron, S. Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics 2011, 188, 197–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, P.R.; Beaulieu, J.; Mansfield, S.D.; Clément, S.; Desponts, M.; Bousquet, J. Factors affecting the accuracy of genomic selection for growth and wood quality traits in an advanced-breeding population of black spruce (Picea mariana). BMC Genom. 2017, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Gapare, W.; Liu, S.; Conaty, W.; Zhu, Q.-H.; Gillespie, V.; Llewellyn, D.; Stiller, W.; Wilson, I. Historical datasets support genomic selection models for the prediction of cotton fiber quality phenotypes across multiple environments. G3 Genes Genomes Genet. 2018, 8, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Spiegelhalter, D.J.; Best, N.G.; Carlin, B.P.; Van Der Linde, A. Bayesian measures of model complexity and fit. J. R. Stat. Soc. B 2002, 64, 583–639. [Google Scholar] [CrossRef] [Green Version]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Azodi, C.B.; Bolger, E.; McCarren, A.; Roantree, M.; de los Campos, G.; Shiu, S.-H. Benchmarking parametric and Machine Learning models for genomic prediction of complex traits. G3 Genes Genomes Genet. 2019, 9, 3691–3702. [Google Scholar] [CrossRef] [Green Version]
- Ogutu, J.O.; Piepho, H.-P.; Schulz-Streeck, T. A comparison of random forests, boosting and support vector machines for genomic selection. In Proceedings of the 14th European Workshop on QTL Mapping and Marker Assisted Selection (QTL-MAS), BMC Proc, Poznan, Poland, 17–18 May 2010. [Google Scholar]
- Montes, C.S.; Weber, J.C. Genetic variation in wood density and correlations with tree growth in Prosopis africana from Burkina Faso and Niger. Ann. For. Sci. 2009, 66, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Baltunis, B.S.; Gapare, W.; Wu, H. Genetic parameters and genotype by environment interaction in radiata pine for growth and wood quality traits in Australia. Silvae Genet. 2010, 59, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Gapare, W.J.; Ivković, M.; Baltunis, B.S.; Matheson, C.A.; Wu, H.X. Genetic stability of wood density and diameter in Pinus radiata D. Don plantation estate across Australia. Tree Genet. Genomes 2010, 6, 113–125. [Google Scholar] [CrossRef]
- Chen, Z.-Q.; Gil, M.R.G.; Karlsson, B.; Lundqvist, S.-O.; Olsson, L.; Wu, H.X. Inheritance of growth and solid wood quality traits in a large Norway spruce population tested at two locations in southern Sweden. Tree Genet. Genomes 2014, 10, 1291–1303. [Google Scholar] [CrossRef]
- Grattapaglia, D. Status and perspectives of genomic selection in forest tree breeding. In Genomic Selection for Crop Improvement; Varshney, R.K., Rookiwal, M., Sorrels, M.E., Eds.; Springer Nature: Cham, Switzerland, 2017; pp. 199–249. [Google Scholar]
- Arojju, S.K.; Cao, M.; Jahufer, M.Z.; Barrett, B.A.; Faville, M.J. Genomic predictive ability for foliar nutritive traits in perennial ryegrass. G3 Genes Genomes Genet. 2019, 9, 727958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Seefried, F.R.; Reinhardt, F.; Rensing, S.; Thaller, G.; Reents, R. Impacts of both reference population size and inclusion of a residual polygenic effect on the accuracy of genomic prediction. Genet. Sel. Evol. 2011, 43, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yu, Y.; Yuan, J.; Zhang, X.; Huang, H.; Li, F.; Xiang, J. Effects of marker density and population structure on the genomic prediction accuracy for growth trait in Pacific white shrimp Litopenaeus vannamei. BMC Genet. 2017, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Isik, F.; McKeand, S.E. Fourth cycle breeding and testing strategy for Pinus taeda in the NC State University Cooperative Tree Improvement Program. Tree Genet. Genomes 2019, 15, 70. [Google Scholar] [CrossRef]
- Dungey, H.; Brawner, J.; Burger, F.; Carson, M.; Henson, M.; Jefferson, P.; Matheson, A.C. A new breeding strategy for Pinus radiata in New Zealand and New South Wales. Silvae Genet. 2009, 58, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Ali, S. Manual for establishment of seed production areas in Dipterocarp forests in Peninsular Malaysia. In Malaysia–International Tropical Timber Organisation Joint Project: PD 185/91 Rev 2(F)-Phase II; Forestry Department Peninsular Malaysia: Kuala Lumpur, Malaysia, 2006. [Google Scholar]
- Wilcox, P.L.; Echt, C.E.; Burdon, R.D. Gene-assisted selection applications of association genetics for forest tree breeding. In Association Mapping in Plants; Oraguzie, N.C., Rikkerink, E.H.A., Gardiner, S.E., Nihal De Silva, H., Eds.; Springer: New York, NY, USA, 2007; pp. 211–247. [Google Scholar]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redona, E.; Atlin, G.; Jannink, J.; McCouch, S. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Begum, H.; Akdemir, D.; Collard, B.; Redoña, E.; Jannink, J.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395. [Google Scholar] [CrossRef] [Green Version]
- Isik, F.; Kumar, S.; Martinez-Garcia, P.J.; Iwata, H.; Yamamoto, T. Acceleration of forest and fruit tree domestication by genomic selection. In Advance in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 74, pp. 93–124. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | All SNPs | Number of SNPs on GWAS-Based Threshold | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −log10(P) > 0.5 | −log10(P) > 0.75 | −log10(P) > 1 | −log10(P) > 1.25 | −log10(P) > 1.5 | |||||||||||||||||
| Fold1 | Fold2 | Fold3 | Fold4 | Fold1 | Fold2 | Fold3 | Fold4 | Fold1 | Fold2 | Fold3 | Fold4 | Fold1 | Fold2 | Fold3 | Fold4 | Fold1 | Fold2 | Fold3 | Fold4 | ||
| Stem diameter_2014 | 5900 | 1869 | 1838 | 1906 | 1741 | 1034 | 1011 | 1112 | 975 | 576 | 556 | 566 | 520 | 314 | 331 | 314 | 282 | 163 | 182 | 158 | 135 |
| Total height_2014 | 5900 | 1681 | 1783 | 1711 | 1714 | 926 | 961 | 953 | 906 | 516 | 527 | 515 | 480 | 287 | 301 | 284 | 258 | 155 | 170 | 172 | 140 |
| Stem diameter_2017 | 5900 | 1770 | 1848 | 1807 | 1805 | 995 | 1011 | 980 | 1037 | 534 | 550 | 555 | 539 | 300 | 327 | 295 | 294 | 171 | 177 | 167 | 181 |
| Total height_2017 | 5900 | 1813 | 1810 | 1838 | 1803 | 1023 | 1040 | 1011 | 1010 | 541 | 584 | 583 | 570 | 312 | 312 | 319 | 306 | 181 | 170 | 177 | 169 |
| Clear bole height | 5900 | 1837 | 1760 | 1629 | 1787 | 1043 | 968 | 863 | 995 | 600 | 540 | 487 | 533 | 322 | 290 | 276 | 270 | 172 | 167 | 145 | 157 |
| Branch angle | 5900 | 1709 | 1839 | 1789 | 1665 | 899 | 1008 | 959 | 852 | 488 | 570 | 527 | 444 | 250 | 319 | 265 | 231 | 128 | 192 | 130 | 118 |
| Branch diameter ratio | 5900 | 1847 | 1871 | 1893 | 1841 | 1024 | 1071 | 1022 | 1032 | 594 | 599 | 574 | 590 | 331 | 314 | 327 | 321 | 188 | 187 | 187 | 191 |
| Wood density | 5900 | 1800 | 1807 | 1809 | 1781 | 975 | 987 | 1029 | 992 | 547 | 565 | 606 | 555 | 285 | 320 | 337 | 314 | 171 | 196 | 195 | 171 |
| Wood stiffness | 5900 | 1845 | 1806 | 1785 | 1814 | 1027 | 1007 | 994 | 1003 | 552 | 567 | 554 | 529 | 300 | 331 | 323 | 310 | 151 | 194 | 162 | 186 |
| Threshold | Model | Stem Diameter_2014 | Total Height_2014 | ||
|---|---|---|---|---|---|
| PredAbi | DIC | PredAbi | DIC | ||
| All SNPs | BL | 0.158 | 1182.032 | 0.075 | 459.510 |
| BRR | 0.145 | 1181.166 | 0.043 | 458.551 | |
| Bayes A | 0.150 | 1181.264 | 0.068 | 458.586 | |
| Bayes B | 0.155 | 1182.357 | 0.058 | 458.835 | |
| Bayes C | 0.153 | 1180.895 | 0.050 | 459.336 | |
| −log10(P) > 0.5 | BL | 0.080 | 1083.879 | −0.005 | 357.158 |
| BRR | 0.075 | 1078.096 | −0.003 | 352.719 | |
| Bayes A | 0.078 | 1080.453 | −0.005 | 355.732 | |
| Bayes B | 0.075 | 1083.867 | −0.005 | 358.664 | |
| Bayes C | 0.078 | 1079.938 | −0.005 | 355.619 | |
| −log10(P) > 0.75 | BL | 0.088 | 1068.282 | −0.005 | 342.287 |
| BRR | 0.085 | 1061.611 | −0.010 | 337.228 | |
| Bayes A | 0.083 | 1064.498 | −0.008 | 340.134 | |
| Bayes B | 0.088 | 1068.903 | −0.010 | 342.533 | |
| Bayes C | 0.088 | 1065.340 | −0.010 | 340.017 | |
| −log10(P) > 1 | BL | 0.080 | 1061.138 | −0.025 | 332.349 |
| BRR | 0.080 | 1055.108 | −0.025 | 328.483 | |
| Bayes A | 0.083 | 1057.265 | −0.025 | 329.535 | |
| Bayes B | 0.088 | 1062.515 | −0.030 | 333.926 | |
| Bayes C | 0.085 | 1060.258 | −0.028 | 332.131 | |
| −log10(P) > 1.25 | BL | 0.065 | 1065.320 | −0.003 | 326.738 |
| BRR | 0.068 | 1061.097 | −0.008 | 322.226 | |
| Bayes A | 0.068 | 1062.260 | −0.008 | 324.692 | |
| Bayes B | 0.075 | 1067.433 | −0.010 | 329.869 | |
| Bayes C | 0.075 | 1065.551 | −0.013 | 327.581 | |
| −log10(P) > 1.5 | BL | 0.083 | 1069.619 | −0.065 | 335.293 |
| BRR | 0.075 | 1066.736 | −0.065 | 331.438 | |
| Bayes A | 0.080 | 1067.921 | −0.068 | 332.917 | |
| Bayes B | 0.088 | 1072.385 | −0.068 | 338.721 | |
| Bayes C | 0.088 | 1070.623. | −0.068 | 337.967 | |
| Threshold | Model | Stem Diameter_2017 | Total Height_2017 | Clear Bole Height | Branch Angle | Branch Diameter Ratio | Wood Density | Wood Stiffness | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PredAbi | DIC | PredAbi | DIC | PredAbi | DIC | PredAbi | DIC | PredAbi | DIC | PredAbi | DIC | PredAbi | DIC | ||
| All SNPs | BL | 0.260 | 1526.615 | 0.165 | 574.750 | 0.005 | 1184.544 | −0.080 | 381.601 | 0.230 | 848.161 | 0.233 | 995.024 | 0.170 | 2083.834 |
| BRR | 0.255 | 1520.588 | 0.153 | 563.733 | −0.023 | 1185.003 | −0.078 | 385.408 | 0.220 | 839.370 | 0.223 | 992.683 | 0.165 | 2080.137 | |
| Bayes A | 0.258 | 1523.512 | 0.148 | 556.107 | −0.013 | 1184.597 | −0.088 | 382.937 | 0.225 | 842.294 | 0.230 | 995.637 | 0.165 | 2081.160 | |
| Bayes B | 0.255 | 1522.414 | 0.148 | 564.285 | −0.013 | 1184.720 | −0.080 | 383.137 | 0.220 | 843.542 | 0.228 | 993.401 | 0.173 | 2081.106 | |
| Bayes C | 0.255 | 1522.825 | 0.153 | 565.823 | −0.005 | 1185.273 | −0.078 | 383.844 | 0.230 | 840.545 | 0.220 | 993.448 | 0.165 | 2081.010 | |
| −log10(P) > 0.5 | BL | 0.233 | 1386.984 | 0.105 | 411.442 | −0.070 | 1086.862 | −0.063 | 308.514 | 0.188 | 678.869 | 0.180 | 872.559 | 0.105 | 1978.435 |
| BRR | 0.235 | 1380.652 | 0.103 | 404.501 | −0.070 | 1082.063 | −0.063 | 303.339 | 0.185 | 671.133 | 0.180 | 864.492 | 0.105 | 1971.373 | |
| Bayes A | 0.238 | 1382.296 | 0.103 | 406.776 | −0.073 | 1084.354 | −0.063 | 305.621 | 0.185 | 673.787 | 0.178 | 866.017 | 0.108 | 1973.105 | |
| Bayes B | 0.235 | 1387.565 | 0.105 | 411.455 | −0.073 | 1088.692 | −0.063 | 309.565 | 0.188 | 679.985 | 0.178 | 870.848 | 0.105 | 1975.335 | |
| Bayes C | 0.240 | 1383.941 | 0.100 | 407.718 | −0.075 | 1085.632 | −0.065 | 306.744 | 0.185 | 673.841 | 0.178 | 867.477 | 0.108 | 1973.510 | |
| −log10(P) > 0.75 | BL | 0.248 | 1360.589 | 0.068 | 385.929 | −0.068 | 1068.755 | −0.060 | 291.276 | 0.188 | 656.869 | 0.178 | 851.592 | 0.115 | 1955.416 |
| BRR | 0.246 | 1358.405 | 0.069 | 382.413 | −0.068 | 1066.210 | −0.058 | 289.408 | 0.186 | 652.858 | 0.178 | 848.949 | 0.114 | 1954.200 | |
| Bayes A | 0.245 | 1357.710 | 0.068 | 380.692 | −0.073 | 1065.465 | −0.060 | 290.056 | 0.185 | 651.084 | 0.175 | 848.884 | 0.115 | 1953.466 | |
| Bayes B | 0.243 | 1361.771 | 0.070 | 387.482 | −0.068 | 1070.474 | −0.060 | 293.956 | 0.183 | 656.417 | 0.175 | 852.915 | 0.120 | 1955.575 | |
| Bayes C | 0.248 | 1358.770 | 0.068 | 382.314 | −0.073 | 1067.379 | −0.060 | 290.647 | 0.183 | 653.112 | 0.175 | 848.861 | 0.110 | 1954.110 | |
| −log10(P) > 1 | BL | 0.238 | 1358.965 | 0.055 | 386.086 | −0.080 | 1056.541 | −0.078 | 276.519 | 0.205 | 654.709 | 0.163 | 839.495 | 0.120 | 1940.670 |
| BRR | 0.248 | 1353.824 | 0.058 | 378.666 | −0.083 | 1050.718 | −0.073 | 272.066 | 0.203 | 647.174 | 0.163 | 833.481 | 0.118 | 1937.816 | |
| Bayes A | 0.248 | 1355.459 | 0.055 | 382.559 | −0.083 | 1053.686 | −0.080 | 274.796 | 0.205 | 650.642 | 0.165 | 835.555 | 0.118 | 1937.764 | |
| Bayes B | 0.240 | 1359.282 | 0.053 | 388.164 | −0.080 | 1058.229 | −0.090 | 279.362 | 0.200 | 655.712 | 0.158 | 841.589 | 0.123 | 1940.677 | |
| Bayes C | 0.250 | 1356.864 | 0.055 | 384.505 | −0.080 | 1055.776 | −0.083 | 277.321 | 0.205 | 652.291 | 0.165 | 837.947 | 0.120 | 1938.827 | |
| −log10(P) > 1.25 | BL | 0.205 | 1362.100 | 0.073 | 387.631 | −0.083 | 1051.808 | −0.058 | 273.413 | 0.210 | 656.321 | 0.145 | 847.827 | 0.110 | 1940.075 |
| BRR | 0.208 | 1357.921 | 0.080 | 381.487 | −0.078 | 1047.357 | −0.053 | 268.808 | 0.210 | 650.858 | 0.150 | 842.105 | 0.108 | 1936.906 | |
| Bayes A | 0.208 | 1359.631 | 0.075 | 384.428 | −0.080 | 1049.106 | −0.058 | 271.239 | 0.208 | 652.110 | 0.143 | 844.628 | 0.110 | 1937.296 | |
| Bayes B | 0.203 | 1363.105 | 0.075 | 390.441 | −0.090 | 1055.538 | −0.070 | 276.558 | 0.210 | 656.525 | 0.140 | 851.635 | 0.115 | 1942.074 | |
| Bayes C | 0.200 | 1362.139 | 0.075 | 386.306 | −0.083 | 1053.154 | −0.065 | 274.552 | 0.210 | 654.503 | 0.140 | 847.205 | 0.118 | 1940.165 | |
| −log10(P) > 1.5 | BL | 0.208 | 1385.003 | 0.048 | 398.017 | −0.088 | 1064.260 | −0.090 | 304.395 | 0.185 | 670.608 | 0.150 | 859.403 | 0.095 | 1944.753 |
| BRR | 0.213 | 1378.850 | 0.045 | 392.367 | −0.088 | 1060.175 | −0.095 | 278.907 | 0.185 | 663.340 | 0.153 | 856.001 | 0.095 | 1941.782 | |
| Bayes A | 0.208 | 1381.038 | 0.045 | 394.956 | −0.090 | 1061.657 | −0.098 | 281.267 | 0.185 | 666.509 | 0.140 | 856.859 | 0.095 | 1941.671 | |
| Bayes B | 0.210 | 1388.738 | 0.045 | 401.640 | −0.095 | 1068.351 | −0.105 | 287.094 | 0.183 | 672.285 | 0.140 | 860.766 | 0.100 | 1946.401 | |
| Bayes C | 0.210 | 1385.915 | 0.048 | 399.274 | −0.100 | 1067.008 | −0.103 | 285.716 | 0.185 | 668.911 | 0.143 | 860.191 | 0.100 | 1946.039 | |
| Threshold | Model | Stem Diameter_2014 | Total Height_2014 | Stem Diameter_2017 | Total Height_2017 | Clear Bole Height | Branch Angle | Branch Diameter Ratio | Wood Density | Wood Stiffness |
|---|---|---|---|---|---|---|---|---|---|---|
| All SNPs | RF | 0.161 | 0.031 | 0.238 | 0.164 | −0.021 | −0.113 | 0.195 | 0.212 | 0.138 |
| XgBoost | 0.132 | −0.016 | 0.198 | 0.169 | −0.069 | −0.116 | 0.182 | 0.147 | 0.155 | |
| BART | 0.169 | 0.084 | 0.270 | 0.138 | 0.030 | −0.126 | 0.212 | 0.226 | 0.155 | |
| GWAS−based threshold | RF | 0.166 | 0.017 | 0.244 | 0.173 | −0.022 | −0.090 | 0.204 | 0.205 | 0.130 |
| XgBoost | 0.105 | 0.001 | 0.172 | 0.125 | 0.035 | −0.131 | 0.156 | 0.112 | 0.075 | |
| BART | 0.151 | 0.079 | 0.203 | 0.108 | −0.016 | −0.065 | 0.187 | 0.158 | 0.122 |
| Traits | Genomic Heritability | |
|---|---|---|
| All SNPs | Selected Marker | |
| Stem diameter_2014 | 0.247 | 0.475 |
| Total height_2014 | 0.224 | 0.457 |
| Stem diameter_2017 | 0.313 | 0.573 |
| Total height_2017 | 0.292 | 0.615 |
| Clear bole height | 0.211 | 0.466 |
| Branch angle | 0.190 | 0.342 |
| Branch diameter ratio | 0.337 | 0.526 |
| Wood density | 0.263 | 0.516 |
| Wood stiffness | 0.252 | 0.473 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawitri; Tani, N.; Na’iem, M.; Widiyatno; Indrioko, S.; Uchiyama, K.; Suwa, R.; Ng, K.K.S.; Lee, S.L.; Tsumura, Y. Potential of Genome-Wide Association Studies and Genomic Selection to Improve Productivity and Quality of Commercial Timber Species in Tropical Rainforest, a Case Study of Shorea platyclados. Forests 2020, 11, 239. https://doi.org/10.3390/f11020239
Sawitri, Tani N, Na’iem M, Widiyatno, Indrioko S, Uchiyama K, Suwa R, Ng KKS, Lee SL, Tsumura Y. Potential of Genome-Wide Association Studies and Genomic Selection to Improve Productivity and Quality of Commercial Timber Species in Tropical Rainforest, a Case Study of Shorea platyclados. Forests. 2020; 11(2):239. https://doi.org/10.3390/f11020239
Chicago/Turabian StyleSawitri, Naoki Tani, Mohammad Na’iem, Widiyatno, Sapto Indrioko, Kentaro Uchiyama, Rempei Suwa, Kevin Kit Siong Ng, Soon Leong Lee, and Yoshihiko Tsumura. 2020. "Potential of Genome-Wide Association Studies and Genomic Selection to Improve Productivity and Quality of Commercial Timber Species in Tropical Rainforest, a Case Study of Shorea platyclados" Forests 11, no. 2: 239. https://doi.org/10.3390/f11020239