Population Genetic Diversity and Structure of Ancient Tree Populations of Cryptomeria japonica var. sinensis Based on RAD-seq Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Sampling
2.2. DNA Extraction and RAD Sequencing
2.3. SNP Calling and Filtering
2.4. Genetic Diversity and Genetic Differentiation
2.5. Population Structure
2.6. Demographic History
3. Results
3.1. Genetic Diversity and Differentiation
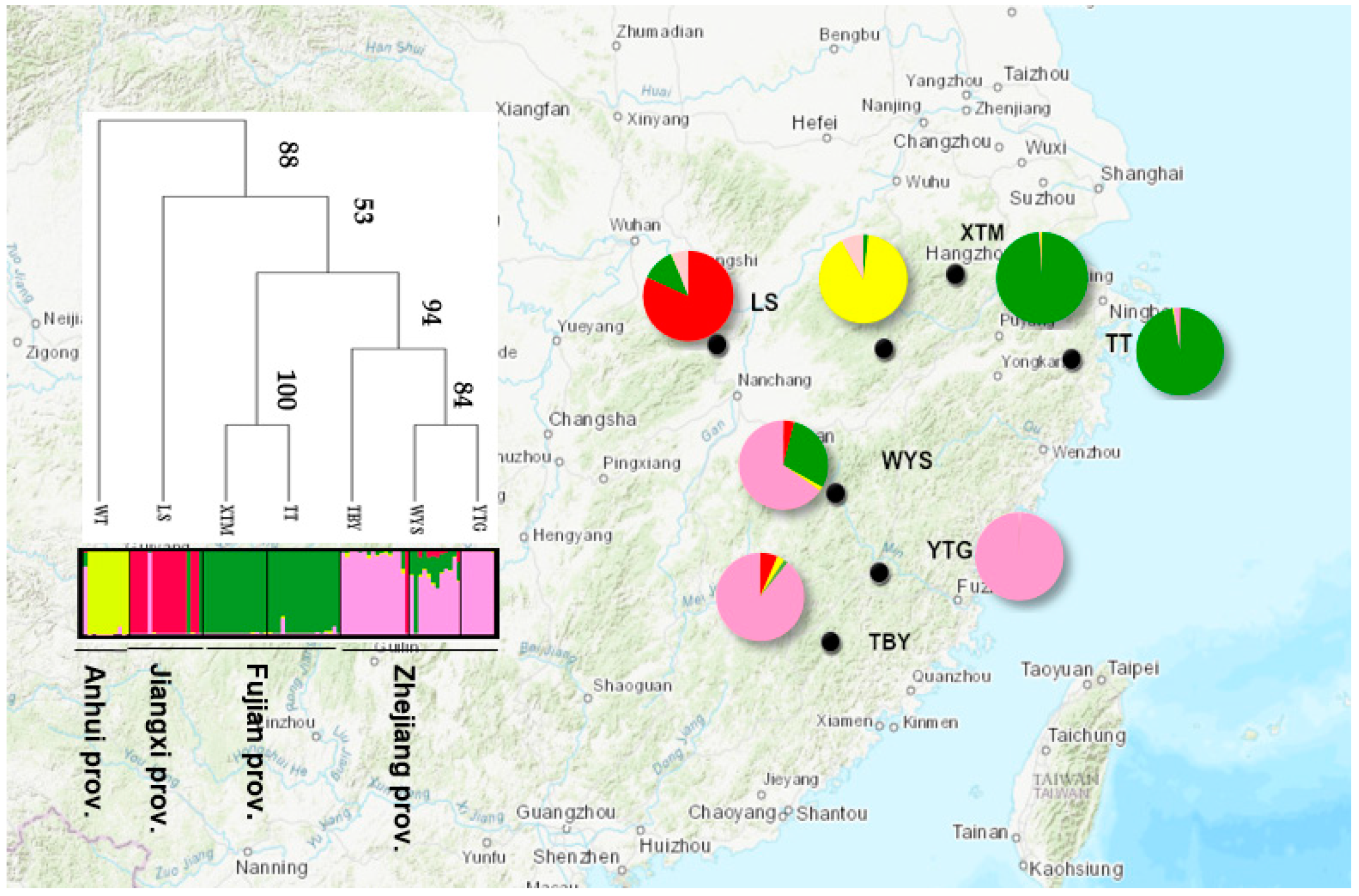
3.2. Genetic Structure
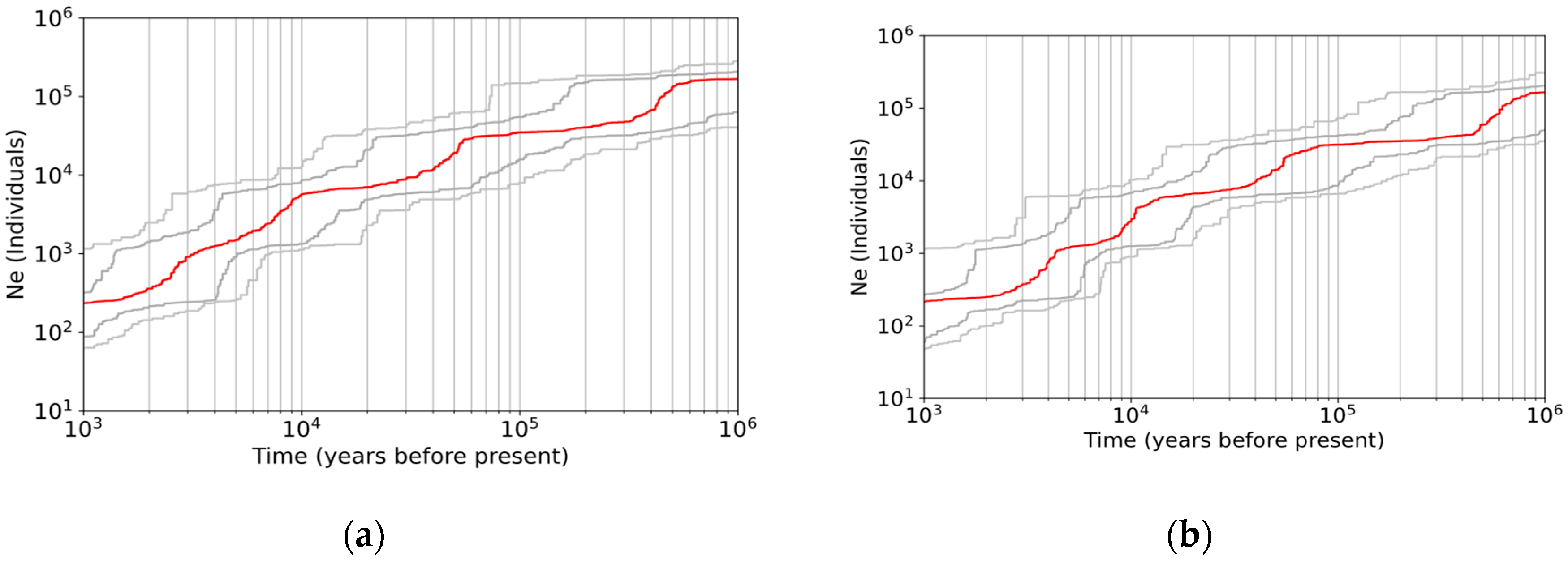
3.3. Demographic History
4. Discussion
4.1. An Old Plantation Derived from Japanese Origin
4.2. Low Genetic Diversity and High Genetic Differentiation in Chinese Cryptomeria
4.3. Specific Genetic Structure with an Absence of IBD
4.4. Continuous Decline of Population Size without Postglacial Recolonization
4.5. Conversation Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ding, W.-N.; Kunzmann, L.; Su, T.; Huang, J.; Zhou, Z.-K. A new fossil species of Cryptomeria (Cupressaceae) from the Rupelian of the Lühe Basin, Yunnan, East Asia: Implications for palaeobiogeography and palaeoecology. Rev. Palaeobot. Palynol. 2018, 248, 41–51. [Google Scholar] [CrossRef]
- Hayashi, Y. Taxonomical and Phytogeographical Study of Japanese Conifers; Norin-Shuppan: Tokyo, Japan, 1960. [Google Scholar]
- Tsumura, Y. Cryptomeria. In Wild Crop Relatives: Genomic and Breeding Resources: Forest Trees; Kole, C., Ed.; Springer: Berlin, Germany, 2011; pp. 49–63. [Google Scholar]
- Fu, L.G.; Yu, Y.F.; Mill, R.R. Taxodiaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China, 1999. [Google Scholar]
- Zheng, H.; Hu, D.; Wei, R.; Yan, S.; Wang, R. Chinese Fir Breeding in the High-Throughput Sequencing Era: Insights from SNPs. Forests 2019, 10, 681. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, S.Z.; Zhao, M.S.; Ni, B.Y.; Liu, L.; Chen, X.Y. Demographic genetic structure of Cryptomeria japonica var. sinensis in Tianmushan Nature Reserve, China. J. Integr. Plant Biol. 2008, 50, 1171–1177. [Google Scholar] [CrossRef]
- Luo, P.; Cao, Y.; Mo, J.; Wong, H.; Shi, J.; Xu, J. Analysis of genetic diversity and construction of DNA fingerprinting of clones in Cryptomeria fortune. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2017, 41, 191–196. [Google Scholar]
- Howe, G.T.; Yu, J.; Knaus, B.; Cronn, R.; Kolpak, S.; Dolan, P.; Lorenz, W.W.; Dean, J.F. A SNP resource for Douglas-fir: De novotranscriptome assembly and SNP detection and validation. BMC Genom. 2013, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-generation population genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Iwata, H.; Hayashi, T.; Tsumura, Y. Prospects for genomic selection in conifer breeding: A simulation study of Cryptomeria japonica. Tree Genet. Genom. 2011, 7, 747–758. [Google Scholar] [CrossRef]
- Zhou, W.; Ji, X.; Obata, S.; Pais, A.; Dong, Y.; Peet, R.; Xiang, Q.-Y.J. Resolving relationships and phylogeographic history of the Nyssa sylvatica complex using data from RAD-seq and species distribution modeling. Mol. Phylogen. Evol. 2018, 126, 1–16. [Google Scholar] [CrossRef]
- Brandrud, M.K.; Paun, O.; Lorenz, R.; Baar, J.; Hedrén, M. Restriction-site associated DNA sequencing supports a sister group relationship of Nigritella and Gymnadenia (Orchidaceae). Mol. Phylogenetics Evol. 2019, 136, 21–28. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Eaton, D.A.; Ree, R.H. Inferring phylogeny and introgression using RADseq data: An example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 2013, 62, 689–706. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogen. Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Huang, Y.; Yang, J. The origin and natural distribution of Cryptomeria. J. Sichuan For. Sci. Technol. 2007, 28, 92–94. [Google Scholar]
- Tsumura, Y.; Kimura, M.; Nakao, K.; Uchiyama, K.; Ujino-Ihara, T.; Wen, Y.; Tong, Z.; Han, W. Effects of the last glacial period on genetic diversity and genetic differentiation in Cryptomeria japonica in East Asia. Tree Genet. Genom. 2020, 16, 19. [Google Scholar] [CrossRef]
- Zhang, J. The Analysis on Genetic Diversity of Superior Cryptomehia Fortunei Resources and Screening of Hybrid Parent. Master’s Thesis, Zhejiang Agriculture and Forestry University, Zhejiang, China, 2014. [Google Scholar]
- Ueno, S.; Uchiyama, K.; Moriguchi, Y.; Ujino-Ihara, T.; Matsumoto, A.; Wei, F.-J.; Saito, M.; Higuchi, Y.; Futamura, N.; Kanamori, H. Scanning RNA-Seq and RAD-Seq approach to develop SNP markers closely linked to MALE STERILITY 1 (MS1) in Cryptomeria japonica D. Don. Breed. Sci. 2019, 69, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2014, 2, e431. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, S.P.; Jones, A.G. Constraints on the FST–heterozygosity outlier approach. J. Hered. 2017, 108, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006, 60, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Excoffier, L.; Smouse, P.; Quattro, J. Analysis of Molecular Variance Inferred From Metric Distances among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945. [Google Scholar]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. Neighbor-Net: An Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Wang, J. Estimating pairwise relatedness in a small sample of individuals. Heredity 2017, 119, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fu, Y.-X. Exploring population size changes using SNP frequency spectra. Nat. Genet. 2015, 47, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Khimoun, A.; Doums, C.; Molet, M.; Kaufmann, B.; Peronnet, R.; Eyer, P.; Mona, S. Urbanization without isolation: The absence of genetic structure among cities and forests in the tiny acorn ant Temnothorax nylanderi. Biol. Lett. 2020, 16, 20190741. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, N.; Uchiyama, K.; Miyagi, R.; Moritsuka, E.; Takahashi, A.; Tamura, K.; Tsumura, Y.; Teshima, K.M.; Tachida, H.; Kusumi, J. Inferring the demographic history of Japanese cedar, Cryptomeria japonica, using amplicon sequencing. Heredity 2019, 123, 371–383. [Google Scholar] [CrossRef]
- Suzuki, E.; Susukida, J. Age Structure and Regeration Process of Temperate Coniferous Stands in the Segire River Basin, Yakushima Island. Jpn. J. Ecol. 1989, 39, 45–51. [Google Scholar]
- Furlan, E.; Stoklosa, J.; Griffiths, J.; Gust, N.; Ellis, R.; Huggins, R.; Weeks, A. Small population size and extremely low levels of genetic diversity in island populations of the platypus, Ornithorhynchus anatinus. Ecol. Evol. 2012, 2, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Ledig, F.T. Genetic Variation in Pinus. In Ecology and Biogeography of Pinus; Richardson, D.M., Ed.; Cambridge University Press: Cambridge, UK, 1998; pp. 251–280. [Google Scholar]
- Potter, K.M.; Campbell, A.R.; Josserand, S.A.; Nelson, C.D.; Jetton, R.M. Population isolation results in unexpectedly high differentiation in Carolina hemlock (Tsuga caroliniana), an imperiled southern Appalachian endemic conifer. Tree Genet. Genom. 2017, 13, 105. [Google Scholar] [CrossRef]
- Dantas, L.G.; Esposito, T.; de Sousa, A.C.B.; Félix, L.; Amorim, L.L.; Benko-Iseppon, A.M.; Batalha-Filho, H.; Pedrosa-Harand, A. Low genetic diversity and high differentiation among relict populations of the neotropical gymnosperm Podocarpus sellowii (Klotz.) in the Atlantic Forest. Genetica 2015, 143, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vásquez, D.L.; Balslev, H.; Hansen, M.M.; Sklenář, P.; Romoleroux, K. Low genetic variation and high differentiation across sky island populations of Lupinus alopecuroides (Fabaceae) in the northern Andes. Alp. Bot. 2016, 126, 135–142. [Google Scholar] [CrossRef]
- Tsumura, Y.; Kado, T.; Takahashi, T.; Tani, N.; Ujino-Ihara, T.; Iwata, H. Genome scan to detect genetic structure and adaptive genes of natural populations of Cryptomeria japonica. Genetics 2007, 176, 2393–2403. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Ueno, S.; Kamijo, T.; Setoguchi, H.; Murakami, N.; Kato, M.; Tsumura, Y. Genetic differentiation and genetic diversity of Castanopsis (Fagaceae), the dominant tree species in Japanese broadleaved evergreen forests, revealed by analysis of EST-associated microsatellites. PLoS ONE 2014, 9, e87429. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Yamashita, K.; Saiki, S.-T.; Matsumoto, A.; Ujino-Ihara, T. Climate sensitivity of Cryptomeria japonica in two contrasting environments: Perspectives from QTL mapping. PLoS ONE 2020, 15, e0228278. [Google Scholar] [CrossRef]
- Cao, X.; Flament, N.; Müller, D.; Li, S. The dynamic topography of eastern China since the latest Jurassic Period. Tectonics 2018, 37, 1274–1291. [Google Scholar] [CrossRef]
- Warmuth, V.M.; Campana, M.G.; Eriksson, A.; Bower, M.; Barker, G.; Manica, A. Ancient trade routes shaped the genetic structure of horses in eastern Eurasia. Mol. Ecol. 2013, 22, 5340–5351. [Google Scholar] [CrossRef]
- Yang, S.; Xue, S.; Kang, W.; Qian, Z.; Yi, Z. Genetic diversity and population structure of Miscanthus lutarioriparius, an endemic plant of China. PLoS ONE 2019, 14, e0211471. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Z.; Li, H. Genetic diversity, population genetic structure and protection strategies for Houpoëa officinalis (Magnoliaceae), an endangered Chinese medical plant. J. Plant Biol. 2018, 61, 159–168. [Google Scholar] [CrossRef]
- Li, Z.-Z.; Gichira, A.W.; Wang, Q.-F.; Chen, J.-M. Genetic diversity and population structure of the endangered basal angiosperm Brasenia schreberi (Cabombaceae) in China. PeerJ 2018, 6, e5296. [Google Scholar] [CrossRef]
- Harrison, S.; Yu, G.; Takahara, H.; Prentice, I. Diversity of temperate plants in east Asia. Nature 2001, 413, 129–130. [Google Scholar] [CrossRef]
- Qiu, Y.; Sun, Y.; Zhang, X.; Lee, J.; Fu, C.-X.; Comes, H.P. Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to quaternary climate change and landbridge configurations. New Phytol. 2009, 183, 480–495. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, T.; Wang, Z.; Lu, Z.; Li, Y.; Fu, C.; Chen, X.; Zhao, M.; Olson, M.S.; Liu, J. Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nat. Commun. 2018, 9, 5449. [Google Scholar] [CrossRef]
- Li, E.-X.; Yi, S.; Qiu, Y.-X.; Guo, J.-T.; Comes, H.P.; Fu, C.-X. Phylogeography of two East Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol. Phylogen. Evol. 2008, 49, 702–714. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, T.; Zhang, L.; Kang, M.; Zhang, Z.; Zheng, Z.; Sun, P.; Shrestha, N.; Liu, J.; Yang, Y. Genomic analyses of a “living fossil”: The endangered dove-tree. Mol. Ecol. Resour. 2020, 20, 756–769. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Qiu, Y.-X.; Liu, Y.-H.; Qi, X.-S.; Kim, S.-H.; Han, J.; Takeuchi, Y.; Worth, J.R.P.; Yamasaki, M.; Sakurai, S.; et al. Climate oscillation during the Quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree Kalopanax septemlobus (Araliaceae) in East Asia. Mol. Ecol. 2012, 21, 3823–3838. [Google Scholar] [CrossRef]
- Tsukada, M. Altitudinal and latitudinal migration of Cryptomeria japonica for the past 20,000 years in Japan. Quatern. Res. 1986, 26, 135–152. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Dobeš, C.; Fu, C.-X.; Koch, M.A. Phylogeography of a living fossil: Pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol. Phylogenetics Evol. 2008, 48, 1094–1105. [Google Scholar] [CrossRef]
- Shen, Y.; Cheng, Y.; Li, K.; Li, H. Integrating Phylogeographic Analysis and Geospatial Methods to Infer Historical Dispersal Routes and Glacial Refugia of Liriodendron chinense. Forests 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-W.; Li, Y.; Zhang, Q.; Fang, Y.-M. Ancient east-west divergence, recent admixture, and multiple marginal refugia shape genetic structure of a widespread oak species (Quercus acutissima) in China. Tree Genet. Genom. 2018, 14, 88. [Google Scholar] [CrossRef]
- Zhang, P.; Shao, G.; Zhao, G.; Le Master, D.C.; Parker, G.R.; Dunning, J.B.; Li, Q. China’s forest policy for the 21st century. Science 2000, 288, 2135–2136. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Jiang, B.; Yuan, W.; Shen, A.; Yang, S.; Yao, S.; Liu, J. On the Management of Large-Diameter Trees in China’s Forests. Forests 2020, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Mo, Q.; Wang, H.; Zhang, Z.; Huang, G.; Ye, Q.; Zou, Q.; Kong, F.; Liu, Y.; Wang, G.G. Moso bamboo (Phyllostachys edulis (Carriere) J. Houzeau) invasion affects soil phosphorus dynamics in adjacent coniferous forests in subtropical China. Ann. For. Sci. 2018, 75, 24. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | Longitude (E) | Latitude (N) | Altitude (m) | n | |
|---|---|---|---|---|---|
| Province | location | ||||
| Jiangxi | Lushan (LS) | 115.5801 | 29.3259 | 895 m | 22 |
| Anhui | Huangshan (WT) | 118.1600 | 29.2743 | 436 m | 17 |
| Fujian | Tianbaoyan (TBY) | 117.3044 | 25.5756 | 1132 m | 17 |
| Nanping (YTG) | 118.0848 | 26.4418 | 846 m | 10 | |
| Wuyishan (WYS) | 117.4136 | 27.4535 | 892 m | 18 | |
| Zhejiang | Tiantaishan (TT) | 121.0541 | 29.1458 | 897 m | 17 |
| Tianmushan (XTM) | 119.2608 | 30.2017 | 1089 m | 21 | |
| Total | 122 | ||||
| Pop | N | Na | Ne | Ho | He | F | Ar | pAr | |
|---|---|---|---|---|---|---|---|---|---|
| LS | Mean | 16 | 1.939 | 1.532 | 0.307 | 0.316 | 0.023 * | 1.77 | 0.03 |
| SE | 0.008 | 0.011 | 0.006 | 0.005 | 0.009 | ||||
| WT | Mean | 11 | 1.550 | 1.309 | 0.187 | 0.174 | 0.023 * | 1.42 | 0.00 |
| SE | 0.016 | 0.013 | 0.009 | 0.007 | 0.015 | ||||
| TBY | Mean | 15 | 1.900 | 1.458 | 0.286 | 0.277 | −0.023 | 1.70 | 0.01 |
| SE | 0.010 | 0.011 | 0.006 | 0.005 | 0.009 | ||||
| YTG | Mean | 8 | 1.614 | 1.351 | 0.239 | 0.209 | −0.123 * | 1.53 | 0.00 |
| SE | 0.016 | 0.012 | 0.008 | 0.006 | 0.011 | ||||
| WYS | Mean | 11 | 1.871 | 1.466 | 0.306 | 0.279 | −0.087 * | 1.71 | 0.00 |
| SE | 0.011 | 0.011 | 0.007 | 0.006 | 0.009 | ||||
| TT | Mean | 17 | 1.838 | 1.429 | 0.266 | 0.257 | −0.033 | 1.64 | 0.01 |
| SE | 0.012 | 0.011 | 0.007 | 0.006 | 0.08 | ||||
| XTM | Mean | 15 | 1.807 | 1.427 | 0.279 | 0.257 | −0.082 * | 1.64 | 0.01 |
| SE | 0.013 | 0.011 | 0.007 | 0.006 | 0.007 | ||||
| Total | Mean | 13.3 | 1.789 | 1.424 | 0.267 | 0.253 | −0.042 | 1.63 | 0.0086 |
| SE | 0.005 | 0.004 | 0.003 | 0.002 | 0.004 |
| Source | df | SS | MS | Est. Var. | % |
|---|---|---|---|---|---|
| Among Pops | 6 | 4033.190 | 672.198 | 18.885 | 12% |
| Among Indiv | 89 | 14,142.633 | 158.906 | 18.552 | 12% |
| Within Indiv | 96 | 11,693.000 | 121.802 | 121.802 | 76% |
| Total | 191 | 29,868.823 | 159.239 | 100% | |
| FST | 0.119 | ||||
| Nm | 1.858 |
| LS | WT | TBY | YTG | WYS | TT | XTM | |
|---|---|---|---|---|---|---|---|
| LS | - | 1.014 | 2.460 | 1.251 | 2.630 | 1.533 | 1.963 |
| WT | 0.198 *** | - | 1.296 | 0.696 | 1.293 | 0.995 | 1.047 |
| TBY | 0.092 *** | 0.162 *** | - | 1.927 | 7.846 | 2.902 | 3.259 |
| YTG | 0.167 *** | 0.264 *** | 0.115 *** | - | 2.287 | 1.282 | 1.490 |
| WYS | 0.087 *** | 0.162 *** | 0.031 ** | 0.099 *** | - | 2.780 | 4.953 |
| TT | 0.140 *** | 0.201 *** | 0.079 *** | 0.163 *** | 0.083 *** | - | 4.788 |
| XTM | 0.113 *** | 0.193 *** | 0.071 *** | 0.144 *** | 0.048 ** | 0.050 *** | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, M.; Wen, Y.; Uchiyama, K.; Onuma, Y.; Tsumura, Y. Population Genetic Diversity and Structure of Ancient Tree Populations of Cryptomeria japonica var. sinensis Based on RAD-seq Data. Forests 2020, 11, 1192. https://doi.org/10.3390/f11111192
Cai M, Wen Y, Uchiyama K, Onuma Y, Tsumura Y. Population Genetic Diversity and Structure of Ancient Tree Populations of Cryptomeria japonica var. sinensis Based on RAD-seq Data. Forests. 2020; 11(11):1192. https://doi.org/10.3390/f11111192
Chicago/Turabian StyleCai, Mengying, Yafeng Wen, Kentaro Uchiyama, Yunosuke Onuma, and Yoshihiko Tsumura. 2020. "Population Genetic Diversity and Structure of Ancient Tree Populations of Cryptomeria japonica var. sinensis Based on RAD-seq Data" Forests 11, no. 11: 1192. https://doi.org/10.3390/f11111192