
Accelerated First-Principles Calculations Based on Machine Learning for Interfacial Modification Element Screening of SiCp/Al Composites

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. First-Principles Calculations
2.2. Machine-Learning-Accelerated First-Principle Computations
3. Results
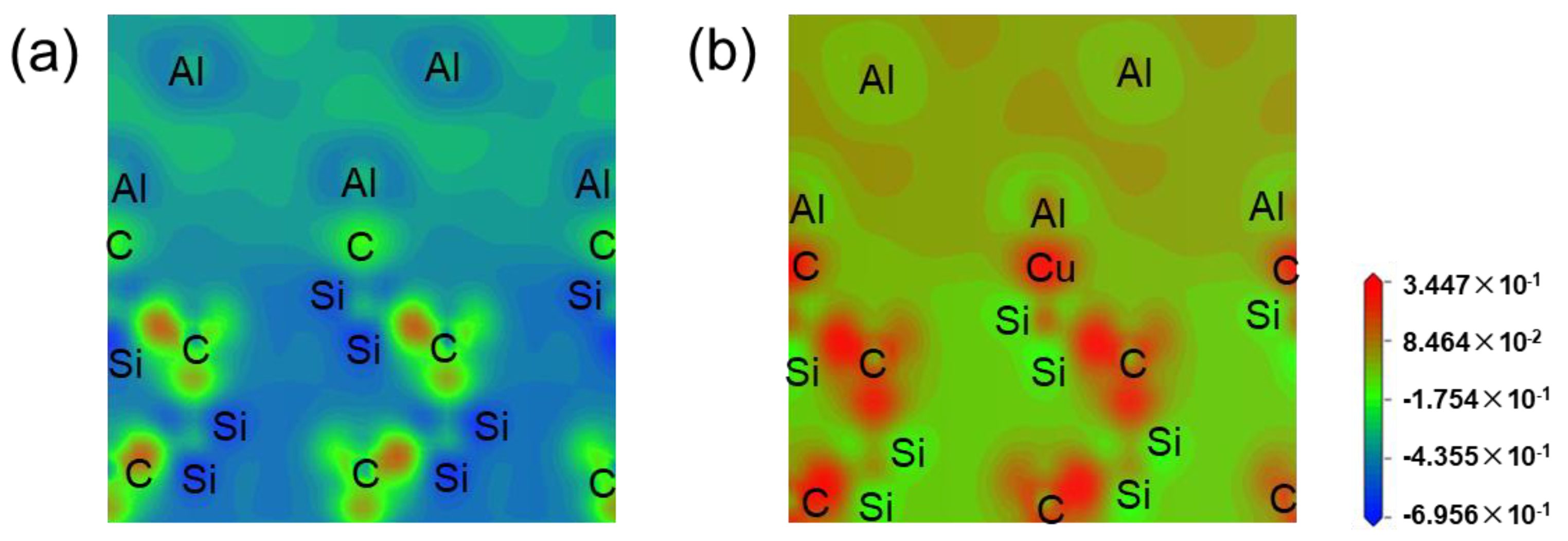
3.1. First-Principles Calculation Results
3.2. Database Establishment and Selection of Feature Values
3.3. Machine Learning Model Construction and Selection
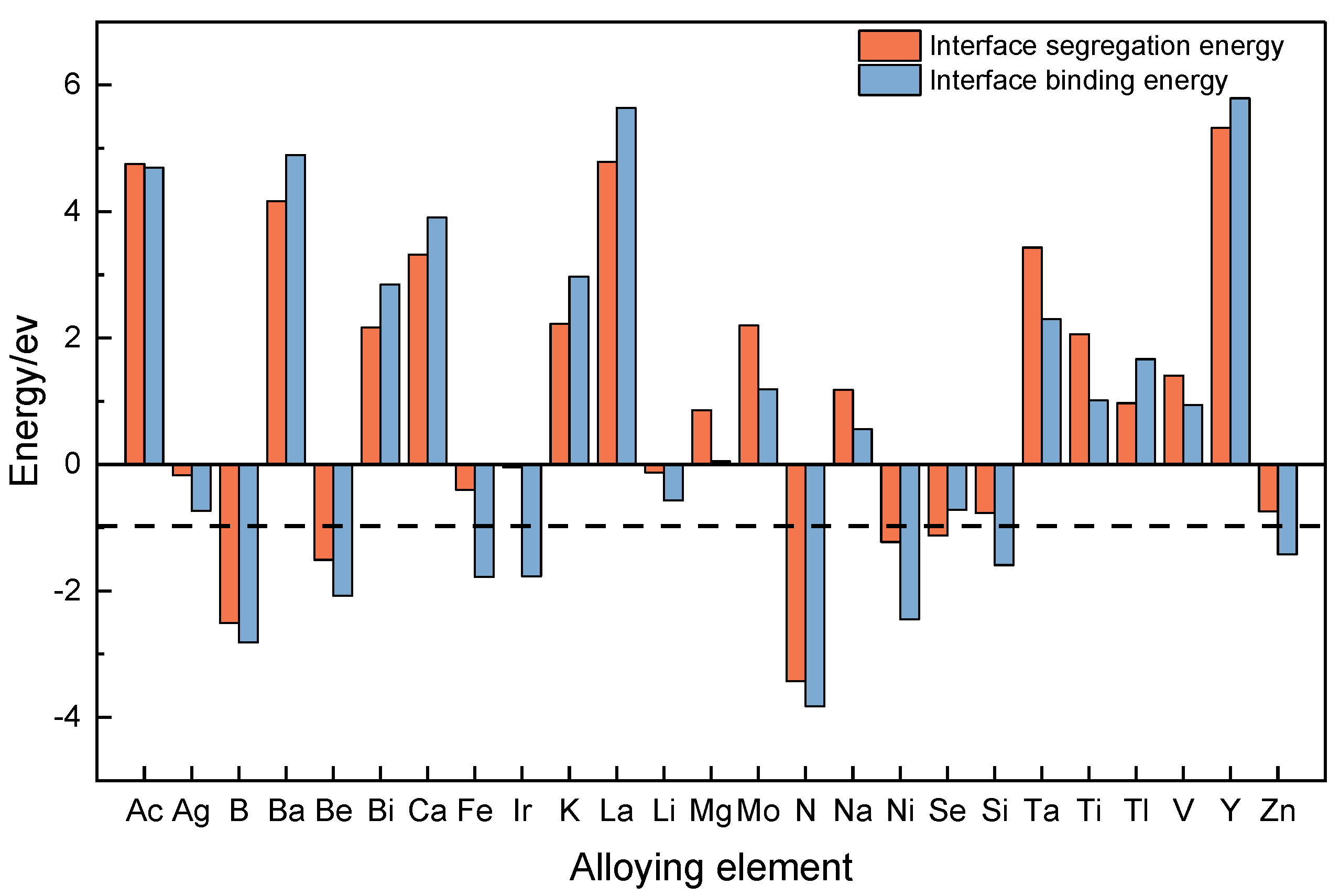
3.4. Screening of Alloying Elements for Interface Modulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Tahani, M.; Postek, E.; Sadowski, T. Investigating the Influence of Diffusion on the Cohesive Zone Model of the SiC/Al Composite Interface. Mloecules 2023, 28, 6757. [Google Scholar] [CrossRef]
- Ferraris, M.; Gili, F.; Lizarralde, X.; Igartua, A.; Mendoza, G.; Blugan, G.; Gorjan, L.; Casalegno, V. SiC particle reinforced Al matrix composites brazed on aluminum body for lightweight wear resistant brakes. Ceram. Int. 2022, 48, 10941–10951. [Google Scholar] [CrossRef]
- Gorbatyuk, S.M.; Pashkov, A.N.; Morozova, I.G.; Chicheneva, O.N. Technologies for applying Ni-Au coatings to heat sinks of SiC-Al metal matrix composite material. Mater. Today Proc. 2021, 38, 1889–1893. [Google Scholar] [CrossRef]
- Tiwari, A.; Agarwal, M.; Dixit, M.; Agrawal, P. Impact of SiC Addition on the Metallurgical and Microstructural Properties of Gravity Die Cast Composite for AA6061 Matrix. Trans. Indian Inst. Met. 2023, 76, 2635–2642. [Google Scholar] [CrossRef]
- Ahmadi, M.; Ansari, R.; Hassanzadeh-Aghdam, M.K. Micromechanical finite element analysis of Young’s modulus, yield strength and thermal expansion coefficient of nano-sized ceramic particle/metal matrix nanocomposites. J. Braz. Soc. Mech. Sci. Eng. 2023, 45, 478. [Google Scholar] [CrossRef]
- Shin, S.; Cho, S.; Lee, D.; Kim, Y.; Lee, S.B.; Lee, S.K.; Jo, I. Microstructural Evolution and Strengthening Mechanism of SiC/Al Composites Fabricated by a Liquid-Pressing Process and Heat Treatment. Materials 2019, 12, 3374. [Google Scholar] [CrossRef]
- Mi, G.; Xiang, Y.; Wang, C.; Xiong, L.; Ouyang, Q. Microstructure and mechanical properties of SiCp/Al composite fabricated by concurrent wire-powder feeding laser deposition. J. Mater. Res. Technol. 2023, 22, 66–79. [Google Scholar] [CrossRef]
- Hamasuna, K.; Iwamoto, C.; Satonaka, S.; Nishida, M.; Tomoshige, R.; Fujita, M. Development of resistance welding for silicon carbide. Mater. Trans. 2007, 48, 1060–1063. [Google Scholar] [CrossRef]
- Tahani, M.; Postek, E.; Motevalizadeh, L.; Sadowski, T. Effect of Vacancy Defect Content on the Interdiffusion of Cubic and Hexagonal SiC/Al Interfaces: A Molecular Dynamics Study. Molecules 2023, 28, 744. [Google Scholar] [CrossRef]
- Pech-Canul, M.I.; Katz, R.N.; Makhlouf, M.M.; Pickard, S. The role of silicon in wetting and pressureless infiltration of SiCp preforms by aluminum alloys. J. Mater. Sci. 2000, 35, 2167–2173. [Google Scholar] [CrossRef]
- Kajikawa, Y.; Nukami, T.; Flemings, M.C. Pressureless infiltration of aluminum metal-matrix composites. Metall. Mater. Trans. A 1995, 26, 2155–2159. [Google Scholar] [CrossRef]
- Lepen, E.; Le Pen, E.; Baptiste, D.; Hug, G. Multi-scale fatigue behaviour modelling of Al-Al2O3 short fibre composites. Int. J. Fatigue 2002, 24, 205–214. [Google Scholar] [CrossRef]
- Fouret, C.; Degallaix, S. Experimental and numerical study of the low-cycle fatigue behaviour of a cast metal matrix composite Al-SiCp. Int. J. Fatigue 2002, 24, 223–232. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, V. Effects of SiC, Al2O3, and ZrO2 particles on the LBMed characteristics of Al/SiC, Al/Al2O3, and Al/ZrO2 MMCs prepared by stir casting process. Part. Sci. Technol. 2019, 37, 766–776. [Google Scholar] [CrossRef]
- Jojith, R.; Radhika, N. Reciprocal dry sliding wear of SiCp/Al-7Si-0.3 Mg functionally graded composites: Influence of T6 treatment and process parameters. Ceram. Int. 2021, 47, 30459–30470. [Google Scholar] [CrossRef]
- Chong, S.-Y.; Atkinson, H.-V.; Jones, H. Effect of ceramic particle-size, melt superheat, impurities and alloy conditions on threshold pressure for infiltration of SiC powder compacts by aluminum-based melts. Mater. Sci. Eng. A-Struct. Mater. Prop. Microstruct. Process. 1993, 173, 233–237. [Google Scholar] [CrossRef]
- Ocelík, V.; Matthews, D.; De Hosson, J. Sliding wear resistance of metal matrix composite layers prepared by high power laser. Surf. Coat. Technol. 2005, 197, 303–315. [Google Scholar] [CrossRef]
- Prusov, E.S.; Kechin, V.A.; Deev, V.B.; Shurkin, P.K. Thermodynamics of the Effect of Alloying of Phase Formation during Crystallization of Aluminum Matrix Composites with Exogenous Reinforcement. Russ. J. Non-Ferr. Met. 2022, 63, 631–640. [Google Scholar] [CrossRef]
- Munir, K.S.; Li, Y.; Liang, D.; Qian, M.; Xu, W.; Wen, C. Effect of dispersion method on the deterioration, interfacial interactions and re-agglomeration of carbon nanotubes in titanium metal matrix composites. Mater. Des. 2015, 88, 138–148. [Google Scholar] [CrossRef]
- Fukumoto, A. First-principles calculations of p-type impurities in cubic SiC. Phys. Rev. B 1996, 53, 4458–4461. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, N.; Jun, Y. First-principles X-ray photoelectron spectroscopy binding energy shift calculation for boron and aluminum defects in 3C-silicon carbide. Jpn. J. Appl. Phys. 2019, 58, 31001. [Google Scholar] [CrossRef]
- Miyazato, I.; Yuzuru, T.; Keisuke, T. Accelerating the discovery of hidden two-dimensional magnets using machine learning and first principle calculations. J. Phys. Condens. Matter 2018, 30, 1L–6L. [Google Scholar] [CrossRef]
- Artrith, N.; Urban, A.; Ceder, G. Constructing first-principles phase diagrams of amorphous LixSi using machine-learning-assisted sampling with an evolutionary algorithm. J. Chem. Phys. 2018, 148, 241711. [Google Scholar] [CrossRef]
- Cytter, Y.; Rabani, E.; Neuhauser, D.; Preising, M.; Redmer, R.; Baer, R. Transition to metallization in warm dense helium-hydrogen mixtures using stochastic density functional theory within the Kubo-Greenwood formalism. Phys. Rev. B 2019, 100, 195101. [Google Scholar] [CrossRef]
- Rahmani-Ivriq, N.; Kordbacheh, A.A. Rectifying and spin filtering behavior of aluminum doped silicon carbide nanoribbons: The first principles study. J. Phys. D-Appl. Phys. 2021, 54, 165304. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Peng, P.; Gao, H.; Wang, J.; Sun, B. Preferred orientation at the Al/graphene interface: First-principles calculations and experimental observation. J. Alloys Compd. 2022, 900, 163304. [Google Scholar] [CrossRef]
- Zou, A.-H.; Zhou, X.-L.; Kang, Z.-B.; Rao, Y.-H.; Wu, K.-Y. Alloy Elements on SiC/Al Interface: A First-principle and Experimental Study. J. Inorg. Mater. 2019, 34, 1167–1174. [Google Scholar]
- Li, Y.F.; Xiao, B.; Wang, G.L.; Sun, L.; Zheng, Q.L.; Liu, Z.W.; Gao, Y.M. Revealing the novel fracture mechanism of the interfaces of TiB2/Fe composite from a first principles investigation. Acta Mater. 2018, 156, 228–244. [Google Scholar] [CrossRef]
- Xie, Y.P.; Zhao, S.J. First principles study of Al and Ni segregation to the α-Fe/Cu (100) coherent interface and their effects on the interfacial cohesion. Comput. Mater. Sci. 2012, 63, 329–335. [Google Scholar] [CrossRef]
- Liu, G.-L.; Guo, Y.-F.; Li, R.-D. Electronic theory of interface characteristics of ZA27/CNT. Acta Phys. Sin. 2007, 56, 4075–4078. [Google Scholar]
- Hasan, S.; Kordijazi, A.; Rohatgi, P.K.; Nosonovsky, M. Triboinformatic modeling of dry friction and wear of aluminum base alloys using machine learning algorithms. Tribol. Int. 2021, 161, 107065. [Google Scholar] [CrossRef]
- Chaluvaraju, B.V.; Afzal, A.; Vinnik, D.A.; Kaladgi, A.R.; Alamri, S.; Tirth, V. Mechanical and Corrosion Studies of Friction Stir Welded Nano Al2O3 Reinforced Al-Mg Matrix Composites: RSM-ANN Modelling Approach. Symmetry 2021, 13, 537. [Google Scholar]
- Majumdar, A.; Jindal, A.; Arora, S.; Bajya, M. Hybrid Neuro-Genetic Machine Learning Models for the Engineering of Ring-spun Cotton Yarns. J. Nat. Fibers 2022, 19, 15164–15175. [Google Scholar] [CrossRef]
- Sheikh-Ahmad, J.; Twomey, J. ANN constitutive model for high strain-rate deformation of Al 7075-T6. J. Mater. Process. Technol. 2007, 186, 339–345. [Google Scholar] [CrossRef]
- Abbas, A.T.; Sharma, N.; Anwar, S.; Luqman, M.; Tomaz, I.; Hegab, H. Multi-Response Optimization in High-Speed Machining of Ti-6Al-4V Using TOPSIS-Fuzzy Integrated Approach. Materials 2020, 13, 1104. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.G.; Kumar, R.L.; Ramalingam, V.V.; Viswanath, J.K.; Harikeshava, R.; Padmanaban, R. Corrosion and Tribological Characteristics of Friction Stir Processed Aluminium Alloy AA5052. Trans. Marit. Sci. Toms 2022, 11, 2. [Google Scholar]
- Ahmad, A.; Ostrowski, K.A.; Maślak, M.; Farooq, F.; Mehmood, I.; Nafees, A. Comparative Study of Supervised Machine Learning Algorithms for Predicting the Compressive Strength of Concrete at High Temperature. Materials 2021, 14, 4222. [Google Scholar] [CrossRef] [PubMed]
- Moayedi, H.; Bui, D.T.; Kalantar, B.; Foong, L.K. Machine-Learning-Based Classification Approaches toward Recognizing Slope Stability Failure. Appl. Sci. 2019, 9, 4638. [Google Scholar] [CrossRef]
- Dinaharan, I.; Palanivel, R.; Murugan, N.; Laubscher, R.F. Predicting the wear rate of AA6082 aluminum surface composites produced by friction stir processing via artificial neural network. Multidiscip. Model. Mater. Struct. 2020, 16, 409–423. [Google Scholar] [CrossRef]
- Koo, S.; Shin, D.; Kim, C. Application of Principal Component Analysis Approach to Predict Shear Strength of Reinforced Concrete Beams with Stirrups. Materials 2021, 14, 3471. [Google Scholar] [CrossRef]
- Ali, N.M.; Mustafah, Y.M.; Rashid, N.M. Performance analysis of robust road sign identification. Conf. Ser. Mater. Sci. Eng. 2013, 53, 012017. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alsurf | Esurf/(J·m−2) | SiCsurf | Esurf/(J·m−2) |
|---|---|---|---|
| (111) | 0.83107594 | (111) | 3.83029199 |
| (110) | 0.97319305 | (011) | 3.22173635 |
| (100) | 0.95097756 | (001) | 2.96169004 |
| (211) | 1.06830391 | (211) | 4.21076767 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Qu, N.; Zhang, X.; Chen, J.; Cui, P.; Huang, J.; Liu, Y.; Zhu, J. Accelerated First-Principles Calculations Based on Machine Learning for Interfacial Modification Element Screening of SiCp/Al Composites. Materials 2024, 17, 1322. https://doi.org/10.3390/ma17061322
Du X, Qu N, Zhang X, Chen J, Cui P, Huang J, Liu Y, Zhu J. Accelerated First-Principles Calculations Based on Machine Learning for Interfacial Modification Element Screening of SiCp/Al Composites. Materials. 2024; 17(6):1322. https://doi.org/10.3390/ma17061322
Chicago/Turabian StyleDu, Xiaoshuang, Nan Qu, Xuexi Zhang, Jiaying Chen, Puchang Cui, Jingtao Huang, Yong Liu, and Jingchuan Zhu. 2024. "Accelerated First-Principles Calculations Based on Machine Learning for Interfacial Modification Element Screening of SiCp/Al Composites" Materials 17, no. 6: 1322. https://doi.org/10.3390/ma17061322