
Review of the Developments and Difficulties in Inorganic Solid-State Electrolytes
Abstract
:1. Introduction
2. Solid-State Electrolytes
2.1. Garnet-Type Electrolytes
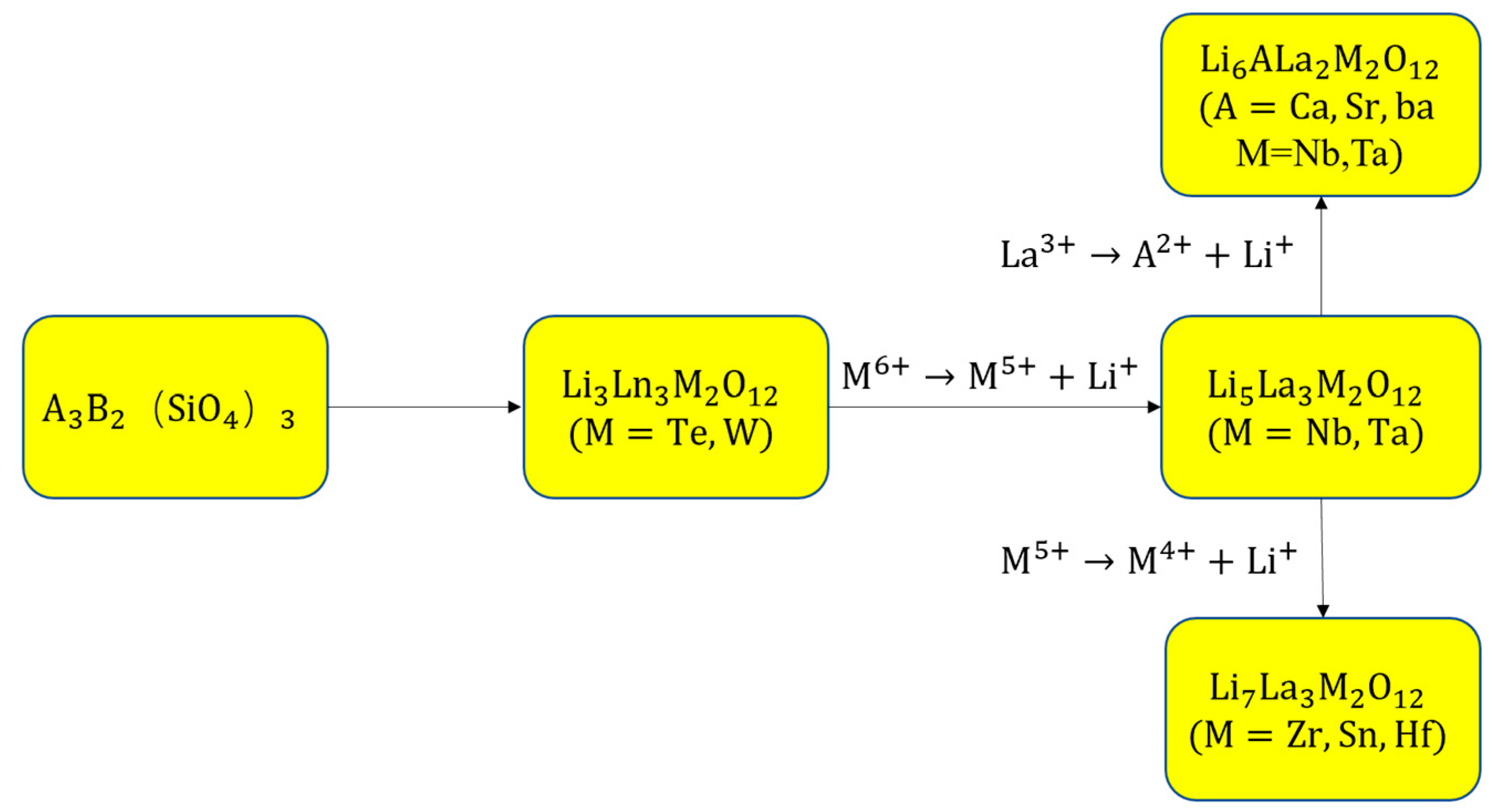
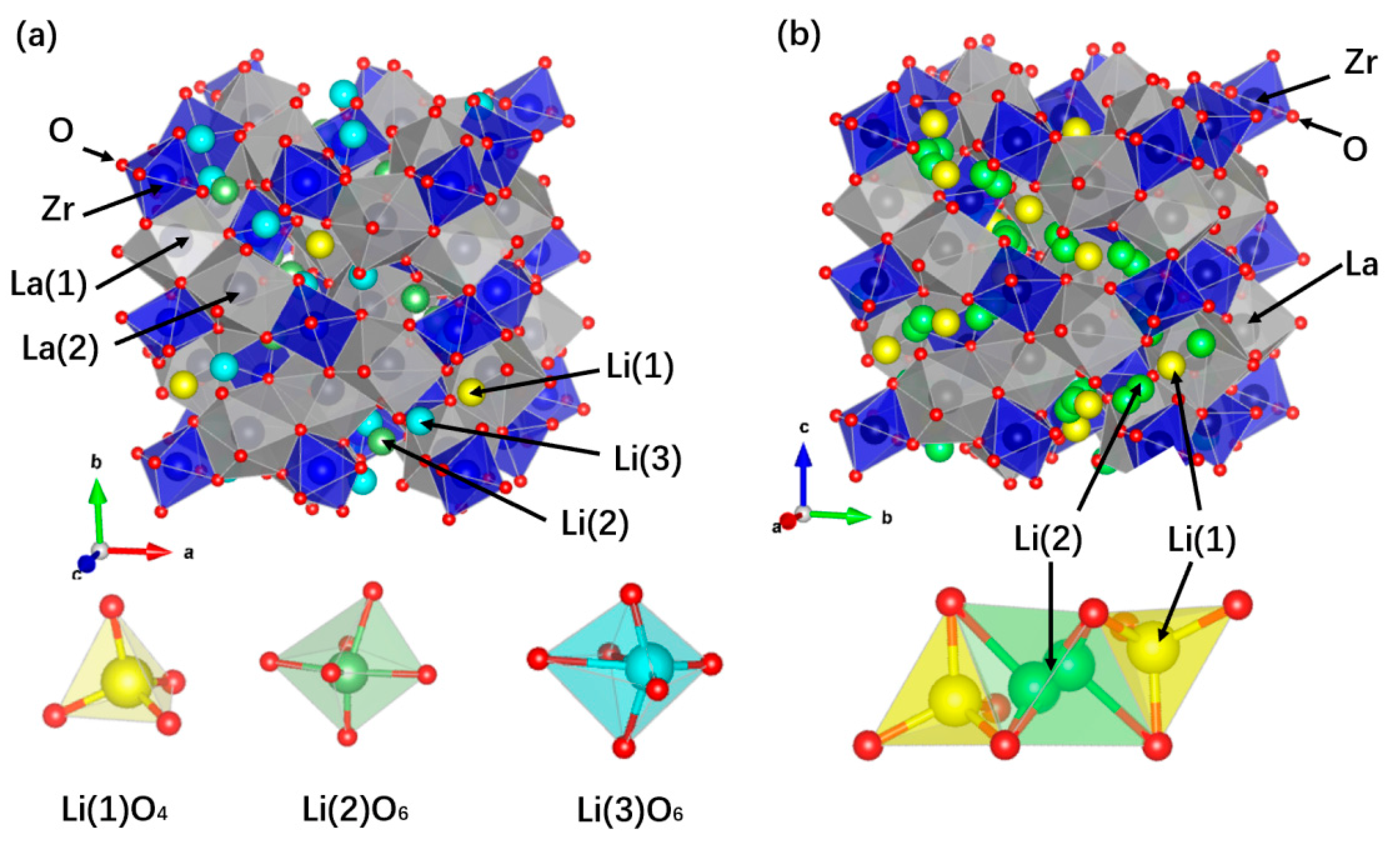
2.1.1. Historical Process, Structure, and Li-ion Diffusion Mechanisms
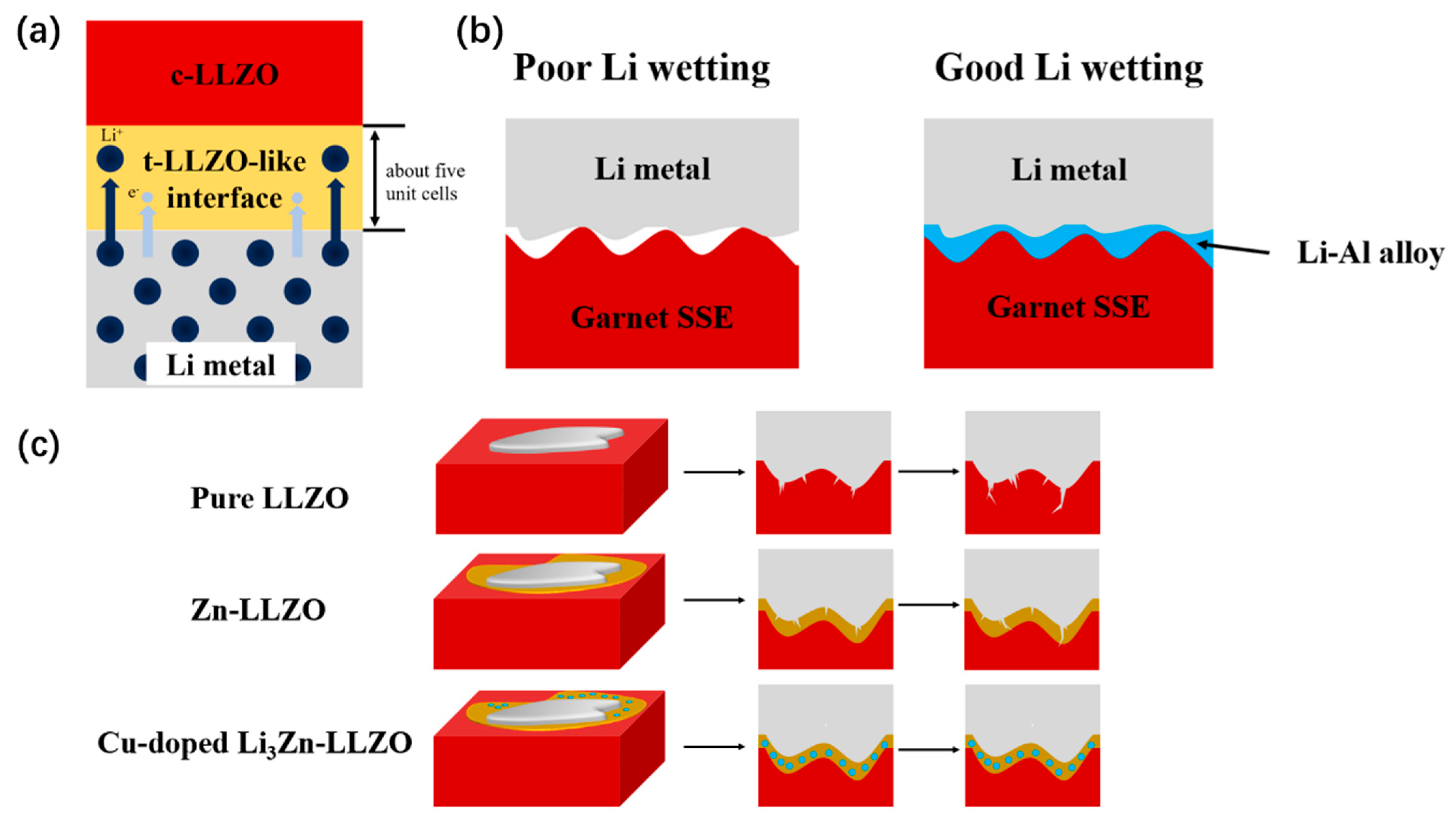
2.1.2. Stability toward Li Anode
2.1.3. Stability toward Cathodes
2.1.4. Air Stability
2.2. Perovskite-Type Electrolytes
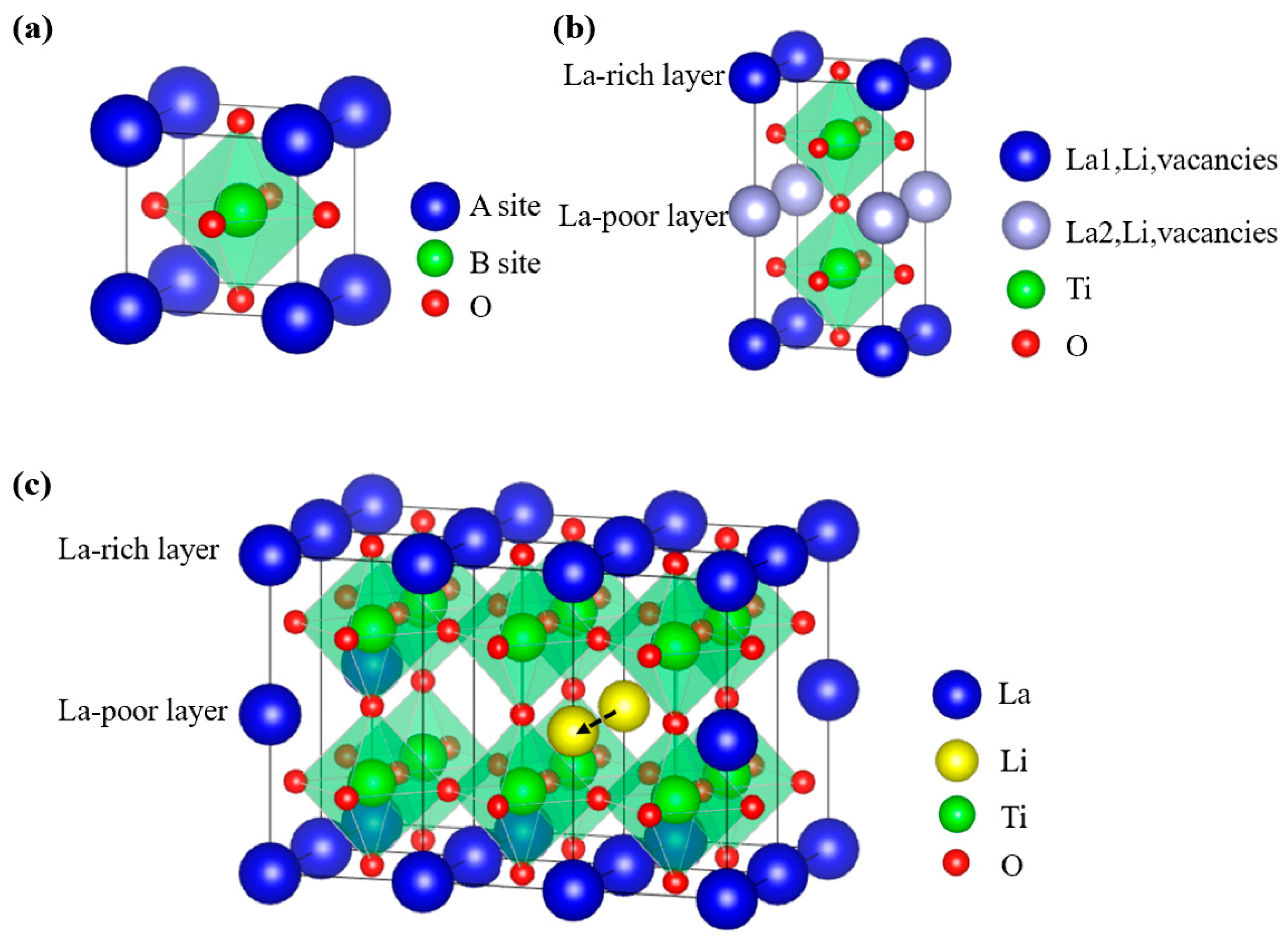
2.2.1. Historical Process, Structure, and Li-ion Diffusion Mechanisms
2.2.2. Stability toward Li Anode
2.2.3. Air Stability
2.3. NASICON-Type Electrolytes
2.3.1. Historical Process, Structure, and Li-Ion Diffusion Mechanisms
2.3.2. Stability toward Li Anode
2.3.3. Stability toward Cathodes
2.4. Thio-/LISICON System
2.4.1. LISICON Structure Conductors
2.4.2. Thio-LISICON Structure Conductors
2.4.3. Stability toward Li Anode
2.4.4. Stability toward Cathodes
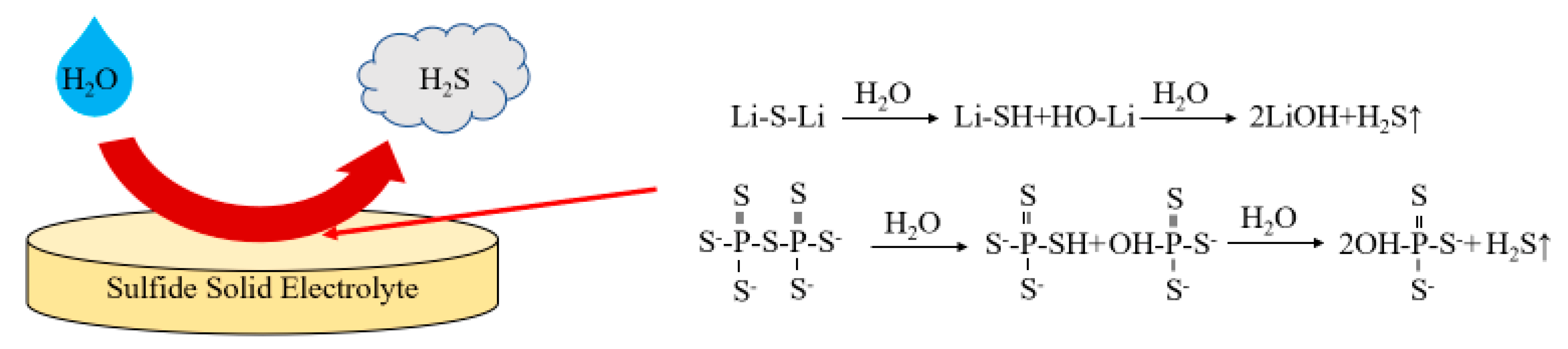
2.4.5. Air Stability
3. Summary
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Palacín, M.R.; De Guibert, A. Batteries: Why do batteries fail? Science 2016, 351, 1253292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, Y.; Wu, C.; Pang, W.K.; Mao, J.; Guo, Z. Constructing nitrided interfaces for stabilizing Li metal electrodes in liquid electrolytes. Chem. Sci. 2021, 12, 8945–8966. [Google Scholar] [CrossRef]
- Li, S.; Jiang, M.; Xie, Y.; Xu, H.; Jia, J.; Li, J. Developing High-Performance Lithium Metal Anode in Liquid Electrolytes: Challenges and Progress. Adv. Mater. 2018, 30, 1706375. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Kotobuki, M.; Song, S.; Lai, M.O.; Lu, L. Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power Sources 2018, 389, 198–213. [Google Scholar] [CrossRef]
- Meng, Y.S.; Srinivasan, V.; Xu, K. Designing better electrolytes. Science 2022, 378, eabq3750. [Google Scholar] [CrossRef]
- Yao, X.; Huang, B.; Yin, J.; Peng, G.; Huang, Z.; Gao, C.; Liu, D.; Xu, X. All-solid-state lithium batteries with inorganic solid electrolytes: Review of fundamental science. Chin. Phys. B 2015, 25, 018802. [Google Scholar] [CrossRef]
- Takada, K. Progress and prospective of solid-state lithium batteries. Acta Mater. 2013, 61, 759–770. [Google Scholar] [CrossRef]
- Dirican, M.; Yan, C.; Zhu, P.; Zhang, X. Composite solid electrolytes for all-solid-state lithium batteries. Mater. Sci. Eng. R Rep. 2019, 136, 27–46. [Google Scholar] [CrossRef]
- Subramanian, K.; Alexander, G.V.; Karthik, K.; Patra, S.; Indu, M.S.; Sreejith, O.V.; Viswanathan, R.; Narayanasamy, J.; Murugan, R. A brief review of recent advances in garnet structured solid electrolyte based lithium metal batteries. J. Energy Storage 2021, 33, 102157. [Google Scholar] [CrossRef]
- Yu, T.; Yang, X.; Yang, R.; Bai, X.; Xu, G.; Zhao, S.; Duan, Y.; Wu, Y.; Wang, J. Progress and perspectives on typical inorganic solid-state electrolytes. J. Alloys Compd. 2021, 885, 161013. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, H.; Ma, J.; Xu, G.; Dong, T.; Chen, J.; Cui, G. Overcoming the Challenges of 5 v Spinel LiNi0.5Mn1.5O4 Cathodes with Solid Polymer Electrolytes. ACS Energy Lett. 2019, 4, 2871–2886. [Google Scholar] [CrossRef]
- Mishra, A.K.; Chaliyawala, H.A.; Patel, R.; Paneliya, S.; Vanpariya, A.; Patel, P.; Ray, A.; Pati, R.; Mukhopadhyay, I. Review-Inorganic Solid State Electrolytes: Insights on Current and Future Scope. J. Electrochem. Soc. 2021, 168, 080536. [Google Scholar] [CrossRef]
- Tao, B.; Ren, C.; Li, H.; Liu, B.; Jia, X.; Dong, X.; Zhang, S.; Chang, H. Thio-/LISICON and LGPS-Type Solid Electrolytes for All-Solid-State Lithium-Ion Batteries. Adv. Funct. Mater. 2022, 32, 2203551. [Google Scholar] [CrossRef]
- Kasper, H. A new series of rare earth garnets. Inorg. Chem. 1968, 765, 1000–1002. [Google Scholar]
- Mazza, D. Remarks on a ternary phase in the La2O3Me2O5Li2O system (Me=Nb, Ta). Mater. Lett. 1988, 7, 205–207. [Google Scholar] [CrossRef]
- Thangadurai, V.; Kaack, H.; Weppner WJ, F. Novel Fast Lithium Ion Conduction in Garnet-Type Li5La3M2O12 (M: Nb, Ta). ChemInform 2003, 34, 437–440. [Google Scholar] [CrossRef]
- Thangadurai, V.; Weppner, W. Li6ALa2Nb2O12 (A = Ca, Sr, Ba): A new class of fast lithium ion conductors with garnet-like structure. J. Am. Ceram. Soc. 2005, 88, 411–418. [Google Scholar] [CrossRef]
- Murugan, R.; Thangadurai, V.; Weppner, W. Fast lithium ion conduction in garnet-type Li7La 3Zr2O12. Angew. Chemie—Int. Ed. 2007, 46, 7778–7781. [Google Scholar] [CrossRef]
- Wang, C.; Fu, K.; Kammampata, S.P.; McOwen, D.W.; Samson, A.J.; Zhang, L.; Hitz, G.T.; Nolan, A.M.; Wachsman, E.D.; Mo, Y.; et al. Garnet-Type Solid-State Electrolytes: Materials, Interfaces, and Batteries. Chem. Rev. 2020, 120, 4257–4300. [Google Scholar] [CrossRef]
- Xue, W.; Yang, Y.; Yang, Q.; Liu, Y.; Wang, L.; Chen, C.; Cheng, R. The effect of sintering process on lithium ionic conductivity of Li6.4Al0.2La3Zr2O12 garnet produced by solid-state synthesis. RSC Adv. 2018, 8, 13083–13088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Tao, X.; Huang, X.; Zou, C.; Yi, L.; Chen, X.; Zang, Z.; Luo, Z.; Wang, X. Efficient Mutual-Compensating Li-Loss Strategy toward Highly Conductive Garnet Ceramics for Li-Metal Solid-State Batteries. ACS Appl. Mater. Interfaces 2021, 13, 56054–56063. [Google Scholar] [CrossRef] [PubMed]
- Awaka, J.; Kijima, N.; Hayakawa, H.; Akimoto, J. Synthesis and structure analysis of tetragonal Li7La3Zr2O12 with the garnet-related type structure. J. Solid State Chem. 2009, 182, 2046–2052. [Google Scholar] [CrossRef]
- Meier, K.; Laino, T.; Curioni, A. Solid-state electrolytes: Revealing the mechanisms of Li-Ion conduction in tetragonal and cubic LLZO by first-principles calculations. J. Phys. Chem. C 2014, 118, 6668–6679. [Google Scholar] [CrossRef]
- Awaka, J.; Takashima, A.; Kataoka, K.; Kijima, N.; Idemoto, Y.; Akimoto, J. Crystal structure of fast lithium-ion-conducting cubic Li7La3Zr2O12. Chem. Lett. 2011, 40, 60–62. [Google Scholar] [CrossRef]
- Xu, M.; Park, M.S.; Lee, J.M.; Kim, T.Y.; Park, Y.S.; Ma, E. Mechanisms of Li + transport in garnet-type cubic Li3+xLa3M2O12 (M = Te, Nb, Zr). Phys. Rev. B 2012, 85, 052301. [Google Scholar] [CrossRef]
- Geiger, C.A.; Alekseev, E.; Lazic, B.; Fisch, M.; Armbruster, T.; Langner, R.; Fechtelkord, M.; Kim, N.; Pettke, T.; Weppner, W. Crystal chemistry and stability of ‘Li7La3Zr2O12’ garnet: A fast lithium-ion conductor. Inorg. Chem. 2011, 50, 1089–1097. [Google Scholar] [CrossRef]
- Pesci, F.M.; Bertei, A.; Brugge, R.H.; Emge, S.P.; Hekselman AK, O.; Marbella, L.E.; Grey, C.P.; Aguadero, A. Establishing Ultralow Activation Energies for Lithium Transport in Garnet Electrolytes. ACS Appl. Mater. Interfaces 2020, 12, 32806–32816. [Google Scholar] [CrossRef]
- Ahn, J.H.; Park, S.Y.; Lee, J.M.; Park, Y.; Lee, J.H. Local impedance spectroscopic and microstructural analyses of Al-in-diffused Li7La3Zr2O12. J. Power Sources 2014, 254, 287–292. [Google Scholar] [CrossRef]
- Shin, D.O.; Oh, K.; Kim, K.M.; Park, K.-Y.; Lee, B.; Lee, Y.-G.; Kang, K. Synergistic multi-doping effects on the Li7La3Zr2O12 solid electrolyte for fast lithium ion conduction. Sci. Rep. 2015, 5, 18053. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Kang, J.-H.; Song, K.; Kang, B. Simultaneously Improved Cubic Phase Stability and Li-Ion Conductivity in Garnet-Type Solid Electrolytes Enabled by Controlling the Al Occupation Sites. ACS Appl. Mater. Interfaces 2022, 14, 12331–12339. [Google Scholar] [CrossRef]
- Rettenwander, D.; Langer, J.; Schmidt, W.; Arrer, C.; Harris, K.J.; Terskikh, V.; Goward, G.R.; Wilkening, M.; Amthauer, G. Site occupation of Ga and Al in stabilized cubic Li7-3(x + y)GaxAlyLa3Zr2O12 garnets as deduced from 27Al and 71Ga MAS NMR at ultrahigh magnetic fields. Chem. Mater. 2015, 27, 3135–3142. [Google Scholar] [CrossRef]
- Chen, C.; Sun, Y.; He, L.; Kotobuki, M.; Hanc, E.; Chen, Y.; Zeng, K.; Lu, L. Microstructural and Electrochemical Properties of Al- And Ga-Doped Li7La3Zr2O12 Garnet Solid Electrolytes. ACS Appl. Energy Mater. 2020, 3, 4708–4719. [Google Scholar] [CrossRef]
- Huang, X.; Su, J.; Song, Z.; Xiu, T.; Jin, J.; Badding, M.E.; Wen, Z. Synthesis of Ga-doped Li7La3Zr2O12 solid electrolyte with high Li+ ion conductivity. Ceram. Int. 2021, 47, 2123–2130. [Google Scholar] [CrossRef]
- Su, J.; Huang, X.; Song, Z.; Xiu, T.; Badding, M.E.; Jin, J.; Wen, Z. Overcoming the abnormal grain growth in Ga-doped Li7La3Zr2O12 to enhance the electrochemical stability against Li metal. Ceram. Int. 2019, 45, 14991–14996. [Google Scholar] [CrossRef]
- Ni, L.; Wu, Z.; Zhang, C. Effect of Sintering Process on Ionic Conductivity of Li7-xLa3Zr2-xNbxO12 (x = 0, 0.2, 0.4, 0.6) Solid Electrolytes. Materials 2021, 14, 1671. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, C.; Lin, F.; Li, B.; Yang, F.; Zhu, H.; Duan, J.; Chen, Z. Submicron-Sized Nb-Doped Lithium Garnet for High Ionic Conductivity Solid Electrolyte and Performance of Quasi-Solid-State Lithium Battery. Materials 2020, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Enkhbayar, E.; Kim, J. Study of Codoping Effects of Ta5+ and Ga3+ on Garnet Li7La3Zr2O12. ACS Omega 2022, 7, 47265–47273. [Google Scholar] [CrossRef]
- Wu, J.-F.; Pang, W.K.; Peterson, V.K.; Wei, L.; Guo, X. Garnet-Type Fast Li-Ion Conductors with High Ionic Conductivities for All-Solid-State Batteries. ACS Appl. Mater. Interfaces 2017, 9, 12461–12468. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.; Redhammer, G.J.; Rettenwander, D.; Senyshyn, A.; Schmidt, W.; Wilkening, M.; Amthauer, G. Crystal Structure of Garnet-Related Li-Ion Conductor Li7-3xGaxLa3Zr2O12: Fast Li-Ion Conduction Caused by a Different Cubic Modification? Chem. Mater. 2016, 28, 1861–1871. [Google Scholar] [CrossRef]
- Fritsch, C.; Zinkevich, T.; Indris, S.; Etter, M.; Baran, V.; Bergfeldt, T.; Knapp, M.; Ehrenberg, H.; Hansen, A.-L. Garnet to hydrogarnet: Effect of post synthesis treatment on cation substituted LLZO solid electrolyte and its effect on Li ion conductivity. RSC Adv. 2021, 11, 30283–30294. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, K.; Samson, A.J.; Dai, J.; Gritton, J.E.; Hu, L.; Wachsman, E.D.; Thangadurai, V. Electrochemical Stability of Garnet-Type Li7La2.75Ca0.25Zr1.75Nb0.25O12 with and without Atomic Layer Deposited-Al2O3 under CO2 and Humidity. J. Electrochem. Soc. 2019, 166, 1844–1852. [Google Scholar] [CrossRef]
- Han, F.; Zhu, Y.; He, X.; Mo, Y.; Wang, C. Electrochemical Stability of Li10GeP2S12 and Li7La3Zr2O12 Solid Electrolytes. Adv. Energy Mater. 2016, 6, 1501590. [Google Scholar] [CrossRef]
- Yang, L.; Lu, Z.; Qin, Y.; Wu, C.; Fu, C.; Gao, Y.; Liu, J.; Jiang, L.; Du, Z.; Xie, Z.; et al. Interrelated interfacial issues between a Li7La3Zr2O12-based garnet electrolyte and Li anode in the solid-state lithium battery: A review. J. Mater. Chem. A 2021, 9, 5952–5979. [Google Scholar] [CrossRef]
- Pervez, S.A.; Kim, G.; Vinayan, B.P.; Cambaz, M.A.; Kuenzel, M.; Hekmatfar, M.; Fichtner, M.; Passerini, S. Overcoming the Interfacial Limitations Imposed by the Solid-Solid Interface in Solid-State Batteries Using Ionic Liquid-Based Interlayers. Small 2020, 16, 2000279. [Google Scholar] [CrossRef] [Green Version]
- Golozar, M.; Paolella, A.; Demers, H.; Savoie, S.; Girard, G.; Delaporte, N.; Gauvin, R.; Guerfi, A.; Lorrmann, H.; Zaghib, K. Direct observation of lithium metal dendrites with ceramic solid electrolyte. Sci. Rep. 2020, 10, 18410. [Google Scholar] [CrossRef]
- Ma, C.; Cheng, Y.; Yin, K.; Luo, J.; Sharafi, A.; Sakamoto, J.; Li, J.; More, K.L.; Dudney, N.J.; Chi, M. Interfacial Stability of Li Metal-Solid Electrolyte Elucidated via in Situ Electron Microscopy. Nano Lett. 2016, 16, 7030–7036. [Google Scholar] [CrossRef]
- Jung, S.-K.; Gwon, H.; Kim, H.; Yoon, G.; Shin, D.; Hong, J.; Jung, C.; Kim, J.-S. Unlocking the hidden chemical space in cubic-phase garnet solid electrolyte for efficient quasi-all-solid-state lithium batteries. Nat. Commun. 2022, 13, 7638. [Google Scholar] [CrossRef]
- Müller, M.; Schmieg, J.; Dierickx, S.; Joos, J.; Weber, A.; Gerthsen, D.; Ivers-Tiffée, E. Reducing Impedance at a Li-Metal Anode/Garnet-Type Electrolyte Interface Implementing Chemically Resolvable in Layers. ACS Appl. Mater. Interfaces 2022, 14, 14739–14752. [Google Scholar] [CrossRef]
- Jiang, W.; Dong, L.; Liu, S.; Ai, B.; Zhao, S.; Zhang, W.; Pan, K.; Zhang, L. Improvement of the Interface between the Lithium Anode and a Garnet-Type Solid Electrolyte of Lithium Batteries Using an Aluminum-Nitride Layer. Nanomaterials 2022, 12, 2023. [Google Scholar] [CrossRef]
- Fu, K.K.; Gong, Y.; Liu, B.; Zhu, Y.; Xu, S.; Yao, Y.; Luo, W.; Wang, C.; Lacey, S.D.; Dai, J.; et al. Toward garnet electrolyte-based Li metal batteries: An ultrathin, highly effective, artificial solid-state electrolyte/metallic Li interface. Sci. Adv. 2017, 3, 1601659. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, G.; Wang, K.; Dong, W.; Han, J.; Yu, Y.; Min, Z.; Yang, C.; Lu, Z. Electrochemically-Matched and Nonflammable Janus Solid Electrolyte for Lithium-Metal Batteries. ACS Appl. Mater. Interfaces 2021, 13, 39271–39281. [Google Scholar] [CrossRef]
- Tsai, C.; Thuy Tran, N.T.; Schierholz, R.; Liu, Z.; Windmüller, A.; Lin, C.; Xu, Q.; Lu, X.; Yu, S.; Tempel, H.; et al. Instability of Ga-substituted Li7La3Zr2O12 toward metallic Li. J. Mater. Chem. A 2022, 10, 10998–11009. [Google Scholar] [CrossRef]
- Li, J.; Luo, H.; Liu, K.; Zhang, J.; Zhai, H.; Su, X.; Wu, J.; Tang, X.; Tan, G. Excellent Stability of Ga-Doped Garnet Electrolyte against Li Metal Anode via Eliminating LiGaO2 Precipitates for Advanced All-Solid-State Batteries. ACS Appl. Mater. Interfaces 2023, 15, 7165–7174. [Google Scholar] [CrossRef]
- Shen, F.; Guo, W.; Zeng, D.; Sun, Z.; Gao, J.; Li, J.; Zhao, B.; He, B.; Han, X. A Simple and Highly Efficient Method toward High-Density Garnet-Type LLZTO Solid-State Electrolyte. ACS Appl. Mater. Interfaces 2020, 12, 30313–30319. [Google Scholar] [CrossRef]
- Botros, M.; Scherer, T.; Popescu, R.; Kilmametov, A.; Clemens, O.; Hahn, H. Microstrain and electrochemical performance of garnet solid electrolyte integrated in a hybrid battery cell. RSC Adv. 2019, 9, 31102–31114. [Google Scholar] [CrossRef] [Green Version]
- Grissa, R.; Seidl, L.; Dachraoui, W.; Sauter, U.; Battaglia, C. Li7La3Zr2O12 Protonation as a Means to Generate Porous/Dense/Porous-Structured Electrolytes for All-Solid-State Lithium-Metal Batteries. ACS Appl. Mater. Interfaces 2022, 14, 46001–46009. [Google Scholar] [CrossRef]
- Liu, M.; Xie, W.; Li, B.; Wang, Y.; Li, G.; Zhang, S.; Wen, Y.; Qiu, J.; Chen, J.; Zhao, P. Garnet Li7La3Zr2O12-Based Solid-State Lithium Batteries Achieved by In Situ Thermally Polymerized Gel Polymer Electrolyte. ACS Appl. Mater. Interfaces 2022, 14, 43116–43126. [Google Scholar] [CrossRef]
- He, X.; Yan, F.; Gao, M.; Shi, Y.; Ge, G.; Shen, B.; Zhai, J. Cu-Doped Alloy Layer Guiding Uniform Li Deposition on a Li-LLZO Interface under High Current Density. ACS Appl. Mater. Interfaces 2021, 13, 42212–42219. [Google Scholar] [CrossRef]
- Ihrig, M.; Finsterbusch, M.; Laptev, A.M.; Tu, C.-H.; Tran NT, T.; Lin, C.-A.; Kuo, L.-Y.; Ye, R.; Sohn, Y.J.; Kaghazchi, P.; et al. Study of LiCoO2/Li7La3Zr2O12:Ta Interface Degradation in All-Solid-State Lithium Batteries. ACS Appl. Mater. Interfaces 2022, 14, 11288–11299. [Google Scholar] [CrossRef]
- Zhang, N.; Long, X.; Wang, Z.; Yu, P.; Han, F.; Fu, J.; Ren, G.; Wu, Y.; Zheng, S.; Huang, W.; et al. Mechanism Study on the Interfacial Stability of a Lithium Garnet-Type Oxide Electrolyte against Cathode Materials. ACS Appl. Energy Mater. 2018, 1, 5968–5976. [Google Scholar] [CrossRef]
- Sastre, J.; Chen, X.; Aribia, A.; Tiwari, A.N.; Romanyuk, Y.E. Fast Charge Transfer across the Li7La3Zr2O12 Solid Electrolyte/LiCoO2 Cathode Interface Enabled by an Interphase-Engineered All-Thin-Film Architecture. ACS Appl. Mater. Interfaces 2020, 12, 36196–36207. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Liu, Y.; Li, W.; Liu, X.; Xie, J.; Jiang, J.; Jiang, Y.; Zhao, B.; Zhang, J. One-pot synthesis and multifunctional surface modification of lithium-rich manganese-based cathode for enhanced structural stability and low-temperature performance. J. Colloid Interface Sci. 2022, 615, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.; Jian, Z.; Zhou, J.; Zheng, Y.; Chen, W. Boosting the Electrochemical Performance of Li1.2Ni0.13Co0.13Mn0.54O2 by Rough Coating with the Superionic Conductor Li7La3Zr2O12. ACS Appl. Mater. Interfaces 2021, 13, 54916–54923. [Google Scholar] [CrossRef] [PubMed]
- Grissa, R.; Payandeh, S.; Heinz, M.; Battaglia, C. Impact of Protonation on the Electrochemical Performance of Li7La3Zr2O12 Garnets. ACS Appl. Mater. Interfaces 2021, 13, 14700–14709. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, C.H.; Jarry, A.; Chen, W.; Ye, Y.; Zhu, J.; Kostecki, R.; Persson, K.; Guo, J.; Salmeron, M.; et al. Interrelationships among Grain Size, Surface Composition, Air Stability, and Interfacial Resistance of Al-Substituted Li7La3Zr2O12 Solid Electrolytes. ACS Appl. Mater. Interfaces 2015, 7, 17649–17655. [Google Scholar] [CrossRef]
- Smetaczek, S.; Limbeck, A.; Zeller, V.; Ring, J.; Ganschow, S.; Rettenwander, D.; Fleig, J. Li+/H+ exchange of Li7La3Zr2O12 single and polycrystals investigated by quantitative LIBS depth profiling. Mater. Adv. 2022, 3, 8760–8770. [Google Scholar] [CrossRef]
- Abrha, L.H.; Hagos, T.T.; Nikodimos, Y.; Bezabh, H.K.; Berhe, G.B.; Hagos, T.M.; Huang, C.-J.; Tegegne, W.A.; Jiang, S.-K.; Weldeyohannes, H.H.; et al. Dual-Doped Cubic Garnet Solid Electrolytes with Superior Air Stability. ACS Appl. Mater. Interfaces 2020, 12, 25709–25717. [Google Scholar] [CrossRef]
- Cheng, L.; Crumlin, E.J.; Chen, W.; Qiao, R.; Hou, H.; Franz Lux, S.; Zorba, V.; Russo, R.; Kostecki, R.; Liu, Z.; et al. The origin of high electrolyte-electrode interfacial resistances in lithium cells containing garnet type solid electrolytes. Phys. Chem. Chem. Phys. 2014, 16, 18294–18300. [Google Scholar] [CrossRef]
- Belous, A.G. Lithium ion conductors based on the perovskite La2/3-xLi3xTiO3. J. Eur. Ceram. Soc. 2001, 21, 1797–1800. [Google Scholar] [CrossRef]
- Inaguma, Y.; Chen, L.; Itoh, M.; Nakamura, T. High lithium ion conductivity in the perovskite-type compounds. Solid State Commun. 1993, 86, 689–693. [Google Scholar] [CrossRef]
- Harada, Y.; Ishigaki, T.; Kawai, H.; Kuwano, J. Lithium ion conductivity of polycrystalline perovskite La0.67-xLi3xTiO3 with ordered and disordered arrangements of the A-site ions. Solid State Ionics 1998, 108, 407–413. [Google Scholar] [CrossRef]
- Takada, K. Nazca Lines by La ordering in La2/3−xLi3xTiO3 ion-conductive perovskite. Appl. Phys. Lett. 2014, 101, 073903. [Google Scholar]
- Inaguma, Y.; Chen, L.; Itoh, M.; Nakamura, T. Candidate compounds with perovskite structure for high lithium ionic conductivity. Solid State Ion. 1994, 70, 196–202. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Hu, Y.; Luo, L. Structural origin of low Li-ion conductivity in perovskite solid-state electrolyte. Nano Energy 2022, 92, 106758. [Google Scholar] [CrossRef]
- Adachi, G.Y.; Imanaka, N.; Tamura, S. Ionic conducting lanthanide oxides. Chem. Rev. 2002, 102, 2405–2429. [Google Scholar] [CrossRef]
- La, L.; Sn MO, M.; Chung, H.; Kim, J.; Kim, H. Dependence of the lithium ionic conductivity on the B-site ion substitution in (Li0.5La0.5)Ti1−xMxO (M=Sn, Zr, Mn, Ge). Solid State Ion. 1998, 107, 153–160. [Google Scholar]
- Ling, M.; Jiang, Y.; Huang, Y.; Zhou, Y.; Zhu, X. Enhancement of ionic conductivity in Li0.5La0.5TiO3 with Ag nanoparticles. J. Mater. Sci. 2020, 55, 3750–3759. [Google Scholar] [CrossRef]
- Yan, S.; Yim, C.H.; Pankov, V.; Bauer, M.; Baranova, E.; Weck, A.; Merati, A.; Abu-Lebdeh, Y. Perovskite solid-state electrolytes for Lithium metal batteries. Batteries 2021, 7, 75–78. [Google Scholar] [CrossRef]
- Kimura, K.; Wagatsuma, K.; Tojo, T.; Inada, R.; Sakurai, Y. Effect of composition on lithium-ion conductivity for perovskite-type lithium-strontium-tantalum-zirconium-oxide solid electrolytes. Ceram. Int. 2016, 42, 5546–5552. [Google Scholar] [CrossRef]
- Huang, B.; Xu, B.; Li, Y.; Zhou, W.; You, Y.; Zhong, S.; Wang, C.-A.; Goodenough, J.B. Li-Ion Conduction and Stability of Perovskite Li3/8Sr7/16Hf1/4Ta3/4O3. ACS Appl. Mater. Interfaces 2016, 8, 14552–14557. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Du, Q.; Zou, B.; Wen, Z.; Chen, C. Synthesis and characterization of perovskite-type (Li, Sr)(Zr, Nb) O3 quaternary solid electrolyte for all-solid-state batteries. J. Power Sources 2016, 306, 623–629. [Google Scholar] [CrossRef]
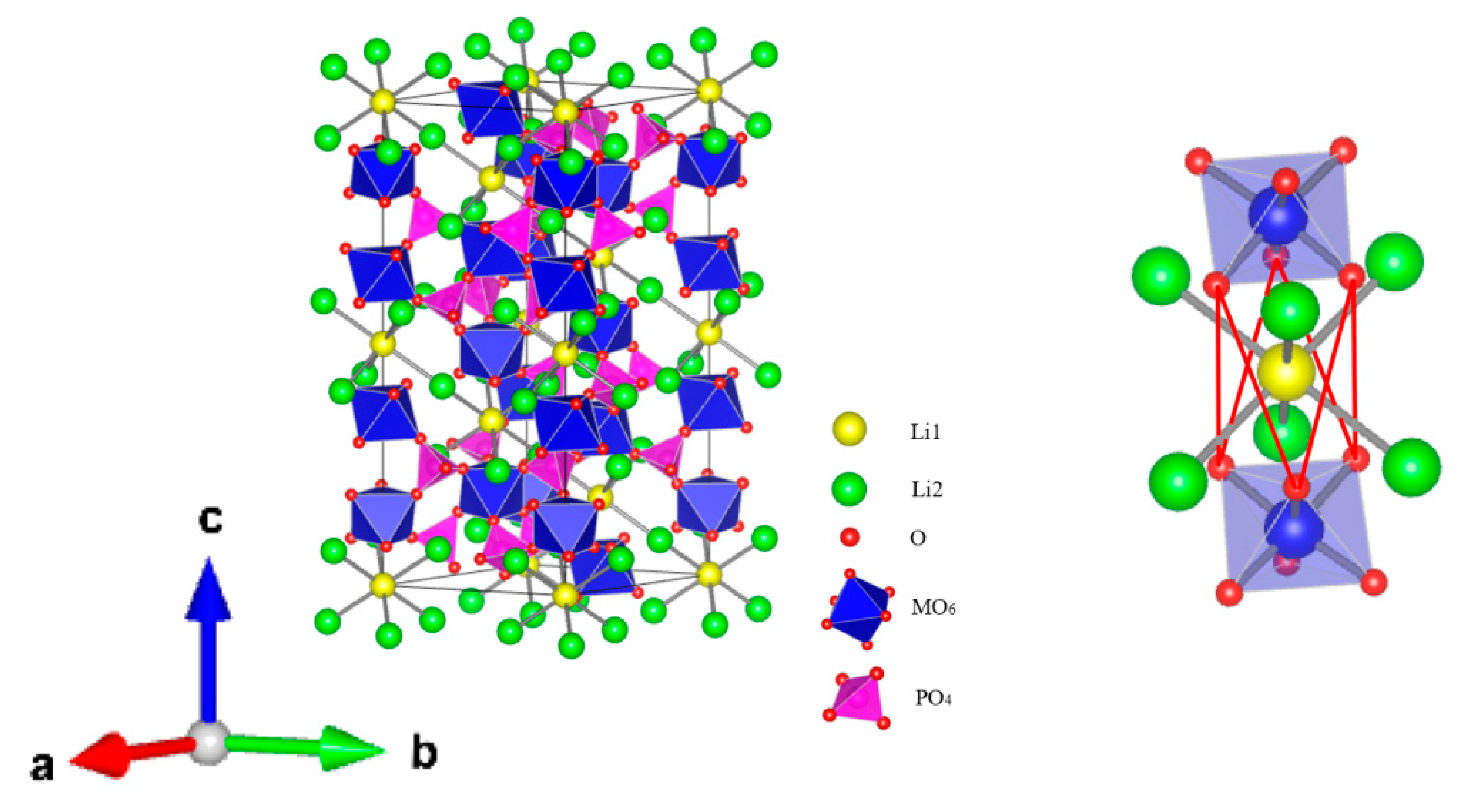
- Amores, M.; El-Shinawi, H.; McClelland, I.; Yeandel, S.R.; Baker, P.J.; Smith, R.I.; Playford, H.Y.; Goddard, P.; Corr, S.A.; Cussen, E.J. Li1.5La1.5MO6 (M=W6+, Te6+) as a new series of lithium-rich double perovskites for all-solid-state lithium-ion batteries. Nat. Commun. 2020, 11, 6392. [Google Scholar] [CrossRef]
- Xu, L.; Feng, T.; Huang, J.; Hu, Y.; Zhang, L.; Luo, L. Structural Heterogeneity Induced Li Dendrite Growth in Li0.33La0.56TiO3Solid-State Electrolytes. ACS Appl. Energy Mater. 2022, 5, 3741–3747. [Google Scholar] [CrossRef]
- Galvez-Aranda, D.E.; Seminario, J.M. Solid electrolyte interphase formation between the Li0.29La0.57TiO3 solid-state electrolyte and a Li-metal anode: An ab initio molecular dynamics study. RSC Adv. 2020, 10, 9000–9015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Zhang, R.; Sun, J.; Wu, M.; Zhao, T. Polyoxyethylene (PEO)|PEO-Perovskite|PEO Composite Electrolyte for All-Solid-State Lithium Metal Batteries. ACS Appl. Mater. Interfaces 2019, 11, 46930–46937. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, S.; Chen, X.; Yang, W.; Yao, X.; Hu, X.; Han, Q.; Wang, H. Tape-Casting Li0.34La0.56TiO3 Ceramic Electrolyte Films Permit High Energy Density of Lithium-Metal Batteries. Adv. Mater. 2020, 32, 1906221. [Google Scholar] [CrossRef]
- Yan, S.; Al-Salih, H.; Yim, C.-H.; Merati, A.; Baranova, E.A.; Weck, A.; Abu-Lebdeh, Y. Engineered interfaces between perovskite La2/3xLi3xTiO3 electrolyte and Li metal for solid-state batteries. Front. Chem. 2022, 10, 966274. [Google Scholar] [CrossRef]
- Jia, M.; Bi, Z.; Shi, C.; Zhao, N.; Guo, X. Polydopamine Coated Lithium Lanthanum Titanate in Bilayer Membrane Electrolytes for Solid Lithium Batteries. ACS Appl. Mater. Interfaces 2020, 12, 46231–46238. [Google Scholar] [CrossRef]
- Xu, H.; Chien, P.-H.; Shi, J.; Li, Y.; Wu, N.; Liu, Y.; Hu, Y.-Y.; Goodenough, J.B. High-performance all-solid-state batteries enabled by salt bonding to perovskite in poly(ethylene oxide). Proc. Natl. Acad. Sci. USA 2019, 116, 18815–18821. [Google Scholar] [CrossRef] [Green Version]
- Durán, T.; Climent-Pascual, E.; Pérez-Prior, M.T.; Levenfeld, B.; Varez, A.; Sobrados, I.; Sanz, J. Aqueous and non-aqueous Li+/H+ ion exchange in Li0.44La0.52TiO3 perovskite. Adv. Powder Technol. 2017, 28, 514–520. [Google Scholar] [CrossRef]
- Bohnke, O.; Nghi, Q.; Boulant, A.; Emery, J.; Tomas, Š.; Barré, M. H+/Li+ exchange property of Li3XLa2/3−XTiO3 in water and in humid atmosphere. Solid State Ion. 2011, 188, 144–147. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Chien, P.-H.; Wu, N.; Xin, S.; Xue, L.; Park, K.; Hu, Y.-Y.; Goodenough, J.B. A Perovskite Electrolyte That Is Stable in Moist Air for Lithium-Ion Batteries. Angew. Chem. Int. Ed. Engl. 2018, 57, 8587–8591. [Google Scholar] [CrossRef]
- Thangadurai, V.; Weppner, W. Recent progress in solid oxide and lithium ion conducting electrolytes research. Ionics 2006, 12, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Aono, H.; Sugimoto, E.; Sadaoka, Y.; Imanaka, N.; Adachi, G. The Electrical Properties of Ceramic Electrolytes for LiMxTi2−x(PO4)3+yLi2O, M = Ge, Sn, Hf, and Zr Systems. J. Electrochem. Soc. 1993, 140, 1827–1832. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Hong, H.-P.; Kafalas, J.A. Fast Na+-ion transport in skeleton structures. Mater. Res. Bull. 1976, 11, 203–220. [Google Scholar] [CrossRef]
- Subramanian, M.A.; Subramanian, R.; Clearfield, A. Lithium ion conductors in the system AB(IV)2(PO4)3 (B = Ti, Zr and Hf). Solid State Ion. 1986, 18, 562–569. [Google Scholar] [CrossRef]
- Francisco, B.E.; Stoldt, C.R.; M’Peko, J.-C. Lithium-Ion Trapping from Local Structural Distortions in Sodium Super Ionic Conductor (NASICON) Electrolytes. Chem. Mater. 2014, 26, 4741–4749. [Google Scholar] [CrossRef]
- Casciola, M.; Costantino, U.; Merlini, L.; Andersen IG, K.; Andersen, E.K. Preparation, structural characterization and conductivity of LiZr2(PO4)3. Solid State Ion. 1988, 26, 229–235. [Google Scholar] [CrossRef]
- Hou, M.; Liang, F.; Chen, K.; Dai, Y.; Xue, D. Challenges and perspectives of NASICON-type solid electrolytes for all-solid-state lithium batteries. Nanotechnology 2020, 31, 132003. [Google Scholar] [CrossRef]
- Lu, Y.; Hu, E.; Yousaf, M.; Ma, L.; Wang, J.; Wang, F.; Lund, P. NASICON-Type Lithium-Ion Conductor Materials with High Proton Conductivity Enabled by Lithium Vacancies. Energy Fuels 2022, 36, 15154–15164. [Google Scholar] [CrossRef]
- Aono, H.; Imanaka, N.; Adachi, G. High Li+ Conducting Ceramics. Acc. Chem. Res. 1994, 27, 265–270. [Google Scholar] [CrossRef]
- Bachman, J.C.; Muy, S.; Grimaud, A.; Chang, H.; Pour, N.; Lux, S.F.; Paschos, O.; Maglia, F.; Lupart, S.; Lamp, P.; et al. Inorganic Solid-State Electrolytes for Lithium Batteries: Mechanisms and Properties Governing Ion Conduction. Chem. Rev. 2016, 116, 140–162. [Google Scholar] [CrossRef]
- Aono, H.; Sugimoto, E.; Sadaoka, Y.; Imanaka, N.; Adachi, G.Y. Ionic conductivity and sinterability of lithium titanium phosphate system. Solid State Ion. 1990, 40, 38–42. [Google Scholar] [CrossRef]
- Lang, B.; Ziebarth, B.; Elsässer, C. Lithium Ion Conduction in LiTi2(PO4)3 and Related Compounds Based on the NASICON Structure: A First-Principles Study. Chem. Mater. 2015, 27, 5040–5048. [Google Scholar] [CrossRef]
- Fu, J. Superionic conductivity of glass-ceramics in the system Li2O-Al2O3-TiO2-P2O5. Solid State Ionics 1997, 96, 195–200. [Google Scholar] [CrossRef]
- Hamao, N.; Yamaguchi, Y.; Hamamoto, K. Densification of a NASICON-Type LATP Electrolyte Sheet by a Cold-Sintering Process. Materials 2021, 14, 4737. [Google Scholar] [CrossRef]
- Xu, X.; Wen, Z.; Yang, X.; Chen, L. Dense nanostructured solid electrolyte with high Li-ion conductivity by spark plasma sintering technique. Mater. Res. Bull. 2008, 43, 2334–2341. [Google Scholar] [CrossRef]
- Fu, J. Fast Li+ ion conducting glass-ceramics in the system Li2O-Al2O3-GeO2-P2O5. Solid State Ion. 1997, 104, 191–194. [Google Scholar] [CrossRef]
- Zallocco, V.M.; Freitas, J.M.; Bocchi, N.; Rodrigues AC, M. Electrochemical stability of a NASICON solid electrolyte from the lithium aluminum germanium phosphate (LAGP) series. Solid State Ion. 2022, 378, 115888. [Google Scholar] [CrossRef]
- Cruz, A.M.; Ferreira, E.B.; Rodrigues AC, M. Controlled crystallization and ionic conductivity of a nanostructured LiAlGePO4 glass-ceramic. J. Non-Cryst. Solids 2009, 355, 2295–2301. [Google Scholar] [CrossRef]
- Zhu, H.; Prasad, A.; Doja, S.; Bichler, L.; Liu, J. Spark Plasma Sintering of Lithium Aluminum Germanium Phosphate Solid Electrolyte and its Electrochemical Properties. Nanomaterials 2019, 9, 1086. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wen, Z.; Wu, X.; Yang, X.; Gu, Z. Lithium ion-conducting glass-ceramics of Li1.5Al0.5Ge1.5(PO4)3−xLi2O (x=0.0–0.20) with good electrical and electrochemical properties. J. Am. Ceram. Soc. 2007, 90, 2802–2806. [Google Scholar] [CrossRef]
- Nikodimos, Y.; Tsai, M.C.; Abrha, L.H.; Weldeyohannis, H.H.; Chiu, S.F.; Bezabh, H.K.; Shitaw, K.N.; Fenta, F.W.; Wu, S.H.; Su, W.N.; et al. Al-Sc dual-doped LiGe2(PO4)3- a NASICON-type solid electrolyte with improved ionic conductivity. J. Mater. Chem. A 2020, 8, 11302–11313. [Google Scholar] [CrossRef]
- Feng, J.K.; Lu, L.; Lai, M.O. Lithium storage capability of lithium ion conductor Li1.5Al0.5Ge1.5(PO4)3. J. Alloys Compd. 2010, 501, 255–258. [Google Scholar] [CrossRef]
- He, L.; Sun, Q.; Chen, C.; Oh JA, S.; Sun, J.; Li, M.; Tu, W.; Zhou, H.; Zeng, K.; Lu, L. Failure Mechanism and Interface Engineering for NASICON-Structured All-Solid-State Lithium Metal Batteries. ACS Appl. Mater. Interfaces 2019, 11, 20895–20904. [Google Scholar] [CrossRef]
- Kaboli, S.; Girard, G.; Zhu, W.; Gheorghe Nita, A.; Vijh, A.; George, C.; Trudeau, M.L.; Paolella, A. Thermal evolution of NASICON type solid-state electrolytes with lithium at high temperature via in situ scanning electron microscopy. Chem. Commun. 2021, 57, 11076–11079. [Google Scholar] [CrossRef]
- Chen, R.; Yao, C.; Yang, Q.; Pan, H.; Yu, X.; Zhang, K.; Li, H. Enhancing the Thermal Stability of NASICON Solid Electrolyte Pellets against Metallic Lithium by Defect Modification. ACS Appl. Mater. Interfaces 2021, 13, 18743–18749. [Google Scholar] [CrossRef]
- Mashekova, A.; Baltash, Y.; Yegamkulov, M.; Trussov, I.; Bakenov, Z.; Mukanova, A. Polycationic doping of the LATP ceramic electrolyte for Li-ion batteries. RSC Adv. 2022, 12, 29595–29601. [Google Scholar] [CrossRef]
- Stegmaier, S.; Reuter, K.; Scheurer, C. Exploiting Nanoscale Complexion in LATP Solid-State Electrolyte via Interfacial Mg2+ Doping. Nanomaterials 2022, 12, 2912. [Google Scholar] [CrossRef]
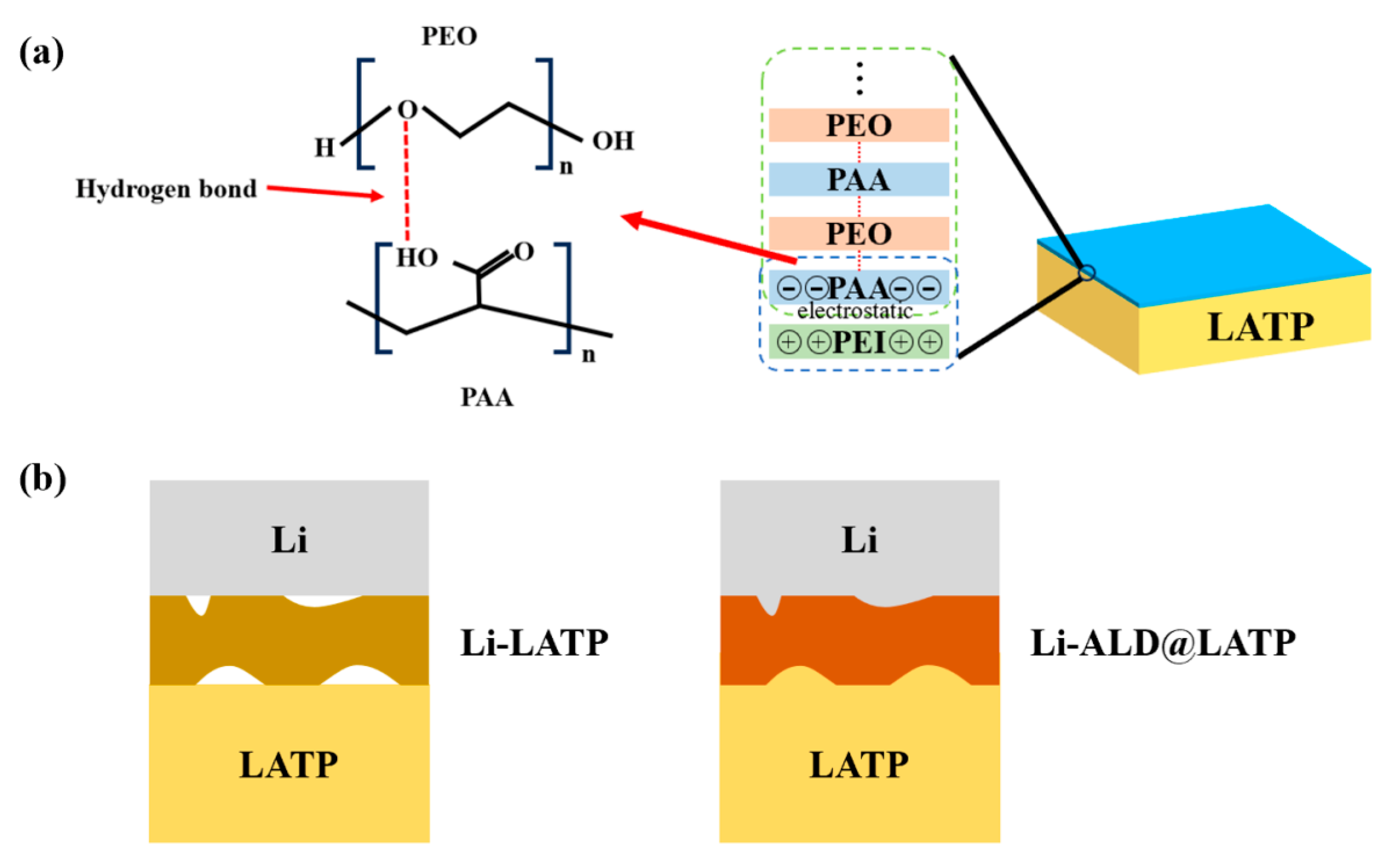
- Tolganbek, N.; Sarsembina, M.; Nurpeissova, A.; Kanamura, K.; Bakenov, Z.; Mentbayeva, A. Effect of a layer-by-layer assembled ultra-thin film on the solid electrolyte and Li interface. Nanoscale Adv. 2022, 4, 4606–4616. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Q.; Zhao, Y.; Wang, B.; Kaghazchi, P.; Adair, K.R.; Li, R.; Zhang, C.; Liu, J.; Kuo, L.-Y.; et al. Stabilizing the Interface of NASICON Solid Electrolyte against Li Metal with Atomic Layer Deposition. ACS Appl. Mater. Interfaces 2018, 10, 31240–31248. [Google Scholar] [CrossRef]
- Huang, C.; Li, Z.; Duan, S.; Xie, S.; Yuan, S.; Hou, S.; Cao, G.; Jin, H. Improving the stability of NASICON-type electrolyte with Li metal anode by interfacial modification. J. Power Sources 2022, 536, 231491. [Google Scholar] [CrossRef]
- Cortes FJ, Q.; Lewis, J.A.; Tippens, J.; Marchese, T.S.; McDowell, M.T. How Metallic Protection Layers Extend the Lifetime of NASICON-Based Solid-State Lithium Batteries. J. Electrochem. Soc. 2020, 167, 050502. [Google Scholar] [CrossRef]
- Gao, L.; Zhao, R.; Han, S.; Li, S.; Zou, R.; Zhao, Y. Antiperovskite Ionic Conductor Layer for Stabilizing the Interface of NASICON Solid Electrolyte Against Li Metal in All-Solid-State Batteries. Batter. Supercaps 2021, 4, 1491–1498. [Google Scholar] [CrossRef]
- Tian, H.-K.; Jalem, R.; Gao, B.; Yamamoto, Y.; Muto, S.; Sakakura, M.; Iriyama, Y.; Tateyama, Y. Electron and Ion Transfer across Interfaces of the NASICON-Type LATP Solid Electrolyte with Electrodes in All-Solid-State Batteries: A Density Functional Theory Study via an Explicit Interface Model. ACS Appl. Mater. Interfaces 2020, 12, 54752–54762. [Google Scholar] [CrossRef]
- Kim, H.-S.; Oh, Y.; Kang, K.H.; Kim, J.H.; Kim, J.; Yoon, C.S. Characterization of Sputter-Deposited LiCoO2 Thin Film Grown on NASICON-type Electrolyte for Application in All-Solid-State Rechargeable Lithium Battery. ACS Appl. Mater. Interfaces 2017, 9, 16063–16070. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Choi, J.; Anandan, V.; Kim, J.H. High-Temperature Chemical Stability of Li1.4Al0.4Ti1.6(PO4)3Solid Electrolyte with Various Cathode Materials for Solid-State Batteries. J. Phys. Chem. C 2020, 124, 14963–14971. [Google Scholar] [CrossRef]
- Wang, L.; Gong, D.; Niu, S.; Wang, L.; Shi, Q.; Wang, X.; Qiao, J.; Liu, G.; Zhan, C. Origin and regulation of interfacial instability for nickel-rich cathodes and NASICON-type Li1+xAlxTi2−x(PO4)3 solid electrolytes in solid-state lithium batteries. Appl. Surf. Sci. 2023, 619, 156741. [Google Scholar] [CrossRef]
- Hong HY, P. Crystal structure and ionic conductivity of Li14Zn(GeO4)4 and other new Li+ superionic conductors. Mater. Res. Bull. 1978, 13, 117–124. [Google Scholar] [CrossRef]
- Alpen U v Bell, M.F.; Wichelhaus, W.; Cheung, K.Y.; Dudley, G.J. Ionic conductivity of Li14Zn(GeO4)4 (Lisicon). Electrochim. Acta 1978, 23, 1395–1397. [Google Scholar] [CrossRef]
- Hu, Y.-W.; Raistrick, I.D.; Huggins, R.A. Ionic Conductivity of Lithium Orthosilicate-Lithium Phosphate Solid Solutions. J. Electrochem. Soc. 1977, 124, 1240–1248. [Google Scholar] [CrossRef]
- Kuwano, J.; West, A.R. New Li+ ion conductors in the system, Li4GeO4-Li3VO4. Mater. Res. Bull. 1980, 15, 1661–1667. [Google Scholar] [CrossRef]
- Deng, Y.; Eames, C.; Fleutot, B.; David, R.; Chotard, J.N.; Suard, E.; Masquelier, C.; Islam, M.S. Enhancing the Lithium Ion Conductivity in Lithium Superionic Conductor (LISICON) Solid Electrolytes through a Mixed Polyanion Effect. ACS Appl. Mater. Interfaces 2017, 9, 7050–7058. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Suzuki, K.; Yonemura, M.; Hirayama, M.; Kanno, R. Enhancing Fast Lithium Ion Conduction in Li4GeO4-Li3PO4 Solid Electrolytes. ACS Appl. Energy Mater. 2019, 2, 6608–6615. [Google Scholar] [CrossRef]
- Song, S.; Lu, J.; Zheng, F.; Duong, H.M.; Lu, L. A facile strategy to achieve high conduction and excellent chemical stability of lithium solid electrolytes. RSC Adv. 2015, 5, 6588–6594. [Google Scholar] [CrossRef]
- Fujimura, K.; Seko, A.; Koyama, Y.; Kuwabara, A.; Kishida, I.; Shitara, K.; Fisher CA, J.; Moriwake, H.; Tanaka, I. Accelerated materials design of lithium superionic conductors based on first-principles calculations and machine learning algorithms. Adv. Energy Mater. 2013, 3, 980–985. [Google Scholar] [CrossRef]
- Ong, S.P.; Mo, Y.; Richards, W.D.; Miara, L.; Lee, H.S.; Ceder, G. Phase stability, electrochemical stability and ionic conductivity of the Li10±1MP2X12 (M = Ge, Si, Sn, Al or P, and X = O, S or Se) family of superionic conductors. Energy Environ. Sci. 2013, 6, 148–156. [Google Scholar] [CrossRef]
- Kanno, R.; Hata, T.; Kawamoto, Y.; Irie, M. Synthesis of a new lithium ionic conductor, thio-LISICON-lithium germanium sulfide system. Solid State Ion. 2000, 130, 97–104. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, F.; Yang, J.; Wang, B.; Sun, J. New lithium ion conductor, thio-LISICON lithium zirconium sulfide system. Solid State Ion. 2008, 179, 1714–1716. [Google Scholar] [CrossRef]
- Murayama, M.; Sonoyama, N.; Yamada, A.; Kanno, R. Material design of new lithium ionic conductor, thio-LISICON, in the Li2S-P2S5 system. Solid State Ion. 2004, 170, 173–180. [Google Scholar] [CrossRef]
- Murayama, M.; Kanno, R.; Irie, M.; Ito, S.; Hata, T.; Sonoyama, N.; Kawamoto, Y. Synthesis of new lithium ionic conductor thio-LISICON—Lithium silicon sulfides system. J. Solid State Chem. 2002, 168, 140–148. [Google Scholar] [CrossRef]
- Murayama, M.; Kanno, R.; Kawamoto, Y.; Kamiyama, T. Structure of the thio-LISICON, Li4GeS4. Solid State Ion. 2002, 154, 789–794. [Google Scholar] [CrossRef]
- Homma, K.; Yonemura, M.; Kobayashi, T.; Nagao, M.; Hirayama, M.; Kanno, R. Crystal structure and phase transitions of the lithium ionic conductor Li3PS4. Solid State Ion. 2011, 182, 53–58. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, W.; Payzant, E.A.; Yu, X.; Wu, Z.; Dudney, N.J.; Kiggans, J.; Hong, K.; Rondinone, A.J.; Liang, C. Anomalous high ionic conductivity of nanoporous β-Li3PS4. J. Am. Chem. Soc. 2013, 135, 975–978. [Google Scholar] [CrossRef]
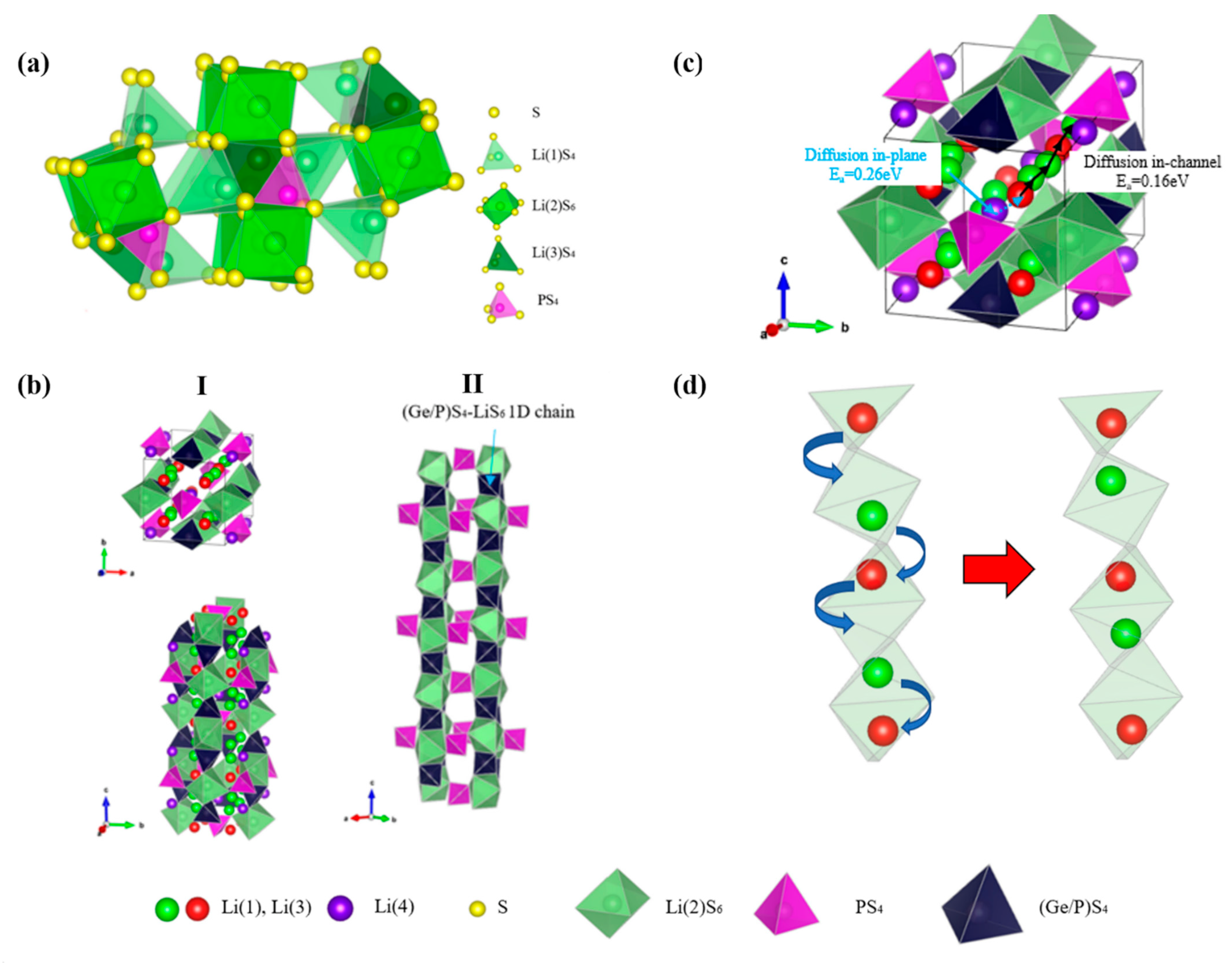
- Kamaya, N.; Homma, K.; Yamakawa, Y.; Hirayama, M.; Kanno, R.; Yonemura, M.; Kamiyama, T.; Kato, Y.; Hama, S.; Kawamoto, K.; et al. A lithium superionic conductor. Nat. Mater. 2011, 10, 682–686. [Google Scholar] [CrossRef]
- Kuhn, A.; Köhler, J.; Lotsch, B.V. Single-crystal X-ray structure analysis of the superionic conductor Li10GeP2S12. Phys. Chem. Chem. Phys. 2013, 15, 11620–11622. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Ong, S.P.; Ceder, G. First principles study of the Li10GeP2S12 lithium super ionic conductor material. Chem. Mater. 2012, 24, 15–17. [Google Scholar] [CrossRef]
- Liang, X.; Wang, L.; Jiang, Y.; Wang, J.; Luo, H.; Liu, C.; Feng, J. In-Channel and In-Plane Li Ion Diffusions in the Superionic Conductor Li10GeP2S12 Probed by Solid-State NMR. Chem. Mater. 2015, 27, 5503–5510. [Google Scholar] [CrossRef]
- Xu, M.; Ding, J.; Ma, E. One-dimensional stringlike cooperative migration of lithium ions in an ultrafast ionic conductor. Appl. Phys. Lett. 2012, 101, 2012–2015. [Google Scholar] [CrossRef]
- Liang, X.; Jiang, Y.; Cai, W.; Wu, S.; Wang, L.; Lei, Z.; Chen, J.; Lei, Y.; Yang, L.; Feng, J. New Li10GeP2S12 Structure Ordering and Li-Ion Dynamics Unveiled in Li4GeS4-Li3PS4 Superionic Conductors: A Solid-State Nuclear Magnetic Resonance Study. ACS Appl. Mater. Interfaces 2020, 12, 27029–27036. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Zhu, X.; Tang, Y.; Huang, F. Highly lithium-ion conductive thio-LISICON thin film processed by low-temperature solution method. J. Power Sources 2013, 224, 225–229. [Google Scholar] [CrossRef]
- Dawson, J.A.; Islam, S. Enhanced Li-Ion Conductivity in Nanosized Li10GeP2S12. ChemRxiv 2020. [Google Scholar] [CrossRef]
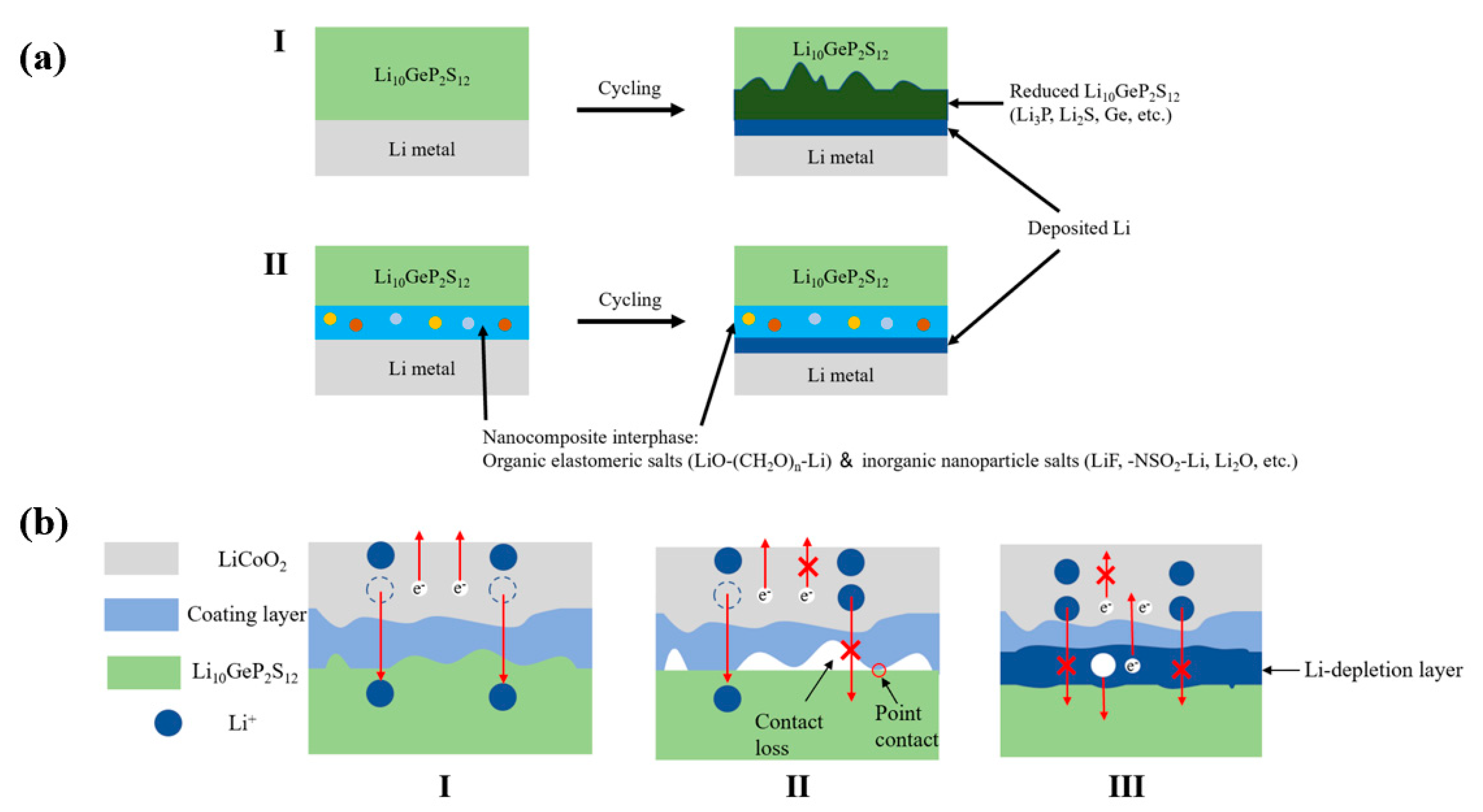
- Wenzel, S.; Randau, S.; Leichtweiß, T.; Weber, D.A.; Sann, J.; Zeier, W.G.; Janek, J. Direct Observation of the Interfacial Instability of the Fast Ionic Conductor Li10GeP2S12 at the Lithium Metal Anode. Chem. Mater. 2016, 28, 2400–2407. [Google Scholar] [CrossRef]
- Kanno, R.; Murayama, M.; Inada, T.; Kobayashi, T.; Sakamoto, K.; Sonoyama, N.; Yamada, A.; Kondo, S. A self-assembled breathing interface for all-solid-state ceramic lithium batteries. Electrochem. Solid-State Lett. 2004, 7, 455–458. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, M.; Cheng, Z.; Jiang, H.; Yang, J.; Wang, P.; He, P.; Zhou, H. Carbon-free and binder-free Li-Al alloy anode enabling an all-solid-state Li-S battery with high energy and stability. Sci. Adv. 2022, 8, eabn4372. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, D.; Li, Y.C.; Yu, Z.; Mallouk, T.E.; Wang, D. Salt-Based Organic-Inorganic Nanocomposites: Towards A Stable Lithium Metal/Li10GeP2S12 Solid Electrolyte Interface. Angew. Chem. Int. Ed. Engl. 2018, 57, 13608–13612. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, S.; Yang, J.; Wang, J.; Yao, L.; Yao, X.; Cui, P.; Xu, X. Interface Re-Engineering of Li10GeP2S12 Electrolyte and Lithium anode for All-Solid-State Lithium Batteries with Ultralong Cycle Life. ACS Appl. Mater. Interfaces 2018, 10, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Westover, A.S.; Yue, J.; Fan, X.; Wang, F.; Chi, M.; Leonard, D.N.; Dudney, N.J.; Wang, H.; Wang, C. High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes. Nat. Energy 2019, 4, 187–196. [Google Scholar] [CrossRef]
- Su, Y.; Ye, L.; Fitzhugh, W.; Wang, Y.; Gil-González, E.; Kim, I.; Li, X. A more stable lithium anode by mechanical constriction for solid state batteries. Energy Environ. Sci. 2020, 13, 908–916. [Google Scholar] [CrossRef]
- Li, Y.; Cao, D.; Arnold, W.; Ren, Y.; Liu, C.; Jasinski, J.B.; Druffel, T.; Cao, Y.; Zhu, H.; Wang, H. Regulated lithium ionic flux through well-aligned channels for lithium dendrite inhibition in solid-state batteries. Energy Storage Mater. 2020, 31, 344–351. [Google Scholar] [CrossRef]
- Zhang, W.; Richter, F.H.; Culver, S.P.; Leichtweiss, T.; Lozano, J.G.; Dietrich, C.; Bruce, P.G.; Zeier, W.G.; Janek, J. Degradation Mechanisms at the Li10GeP2S12/LiCoO2 Cathode Interface in an All-Solid-State Lithium-Ion Battery. ACS Appl. Mater. Interfaces 2018, 10, 22226–22236. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.-T.; Rueß, R.; Pan, R.; Walther, F.; Rohnke, M.; Hori, S.; Kanno, R.; Schröder, D.; Janek, J. A mechanistic investigation of the Li10GeP2S12|LiNi1-x-yCoxMnyO2 interface stability in all-solid-state lithium batteries. Nat. Commun. 2021, 12, 6669. [Google Scholar] [CrossRef]
- Yoon, K.; Kim, J.-J.; Seong, W.M.; Lee, M.H.; Kang, K. Investigation on the interface between Li10GeP2S12 electrolyte and carbon conductive agents in all-solid-state lithium battery. Sci. Rep. 2018, 8, 8066. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Li, C.; Zhao, Z.; Mu, D.; Wu, B. Air Stability and Interfacial Compatibility of Sulfide Solid Electrolytes for Solid-State Lithium Batteries: Advances and Perspectives. ChemElectroChem 2022, 9, e202101479. [Google Scholar] [CrossRef]
- Muramatsu, H.; Hayashi, A.; Ohtomo, T.; Hama, S.; Tatsumisago, M. Structural change of Li2S–P2S5 sulfide solid electrolytes in the atmosphere. Solid State Ion. 2011, 182, 116–119. [Google Scholar] [CrossRef]
- Lu, P.; Wu, D.; Chen, L.; Li, H.; Wu, F. Air Stability of Solid-State Sulfide Batteries and Electrolytes. Electrochem. Energy Rev. 2022, 5, 3–12. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Sun, Y.; Jia, H.; Peng, L.; Zhang, Y.; Xie, J. Li4-xSbxSn1-xS4 solid solutions for air-stable solid electrolytes. J. Energy Chem. 2020, 41, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Chen, N.; Li, X.; Li, X.; Adair, K.R.; Li, J.; Wang, C.; Yu, C.; Norouzi Banis, M.; Zhang, L.; et al. Li10Ge(P1−xSbx)2S12 Lithium-Ion Conductors with Enhanced Atmospheric Stability. Chem. Mater. 2020, 32, 2664–2672. [Google Scholar] [CrossRef]
- Tsukasaki, H.; Morimoto, H.; Mori, S. Ionic conductivity and thermal stability of Li2O–Li2S–P2S5 oxysulfide glass. Solid State Ion. 2020, 347, 1102–1105. [Google Scholar] [CrossRef]
- Xu, M.; Song, S.; Daikuhara, S.; Matsui, N.; Hori, S.; Suzuki, K.; Hirayama, M.; Shiotani, S.; Nakanishi, S.; Yonemura, M.; et al. Li10GeP2S12-Type Structured Solid Solution Phases in the Li9+δP3+δ′S12−kOk System: Controlling Crystallinity by Synthesis to Improve the Air Stability. Inorg. Chem. 2022, 61, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, Q.; Ma, X.; Liu, Q.; Zhou, X.; Lei, Z. Li10Sn0.95P2S11.9−xOx: A new sulfide solid electrolyte for all-solid-state batteries. J. Alloys Compd. 2022, 926, 166731. [Google Scholar] [CrossRef]
- Gao, J.; Sun, X.; Wang, C.; Zhang, Y.; Yang, L.; Song, D.; Wu, Y.; Yang, Z.; Ohsaka, T.; Matsumotoc, F.; et al. Sb- and O-Cosubstituted Li10SnP2S12 with High Electrochemical and Air Stability for All-Solid-State Lithium Batteries. ChemElectroChem 2022, 9, e202200156. [Google Scholar]
- Weng, W.; Zhou, D.; Liu, G.; Shen, L.; Li, M.; Chang, X.; Yao, X. Air exposure towards stable Li/Li10GeP2S12 interface for all-solid-state lithium batteries. Mater. Futur. 2022, 1, 021001. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte | Composition | Ionic Conductivity (S·cm−1) | Ref. |
|---|---|---|---|
| Garnet | Li5La3M2O12 (M = Nb, Ta) | 10−6 S·cm−1 (25 °C) | [17] |
| Li7La3Zr2O12 | 3 × 10−4 S·cm−1 (RT) | [19] | |
| Li7La3Zr2O12(tetragonal) | 1.63 × 10−6 S·cm−1 (RT) | [23] | |
| Li6.4La3Zr2Al0.2O12 | 3.8 × 10−4 S·cm−1 (RT) | [31] | |
| Li6.25La3Zr2Ga0.25O12 | 1.19 × 10−3 S·cm−1 (RT) | [33] | |
| Li6.4Ga0.2La3Zr2O12 | 1.25 × 10−3 S·cm−1 (25 °C) | [34] | |
| Li6.4Ga0.2La3Zr2O12 | 1.24 × 10−3 S·cm−1 (27 °C) | [35] | |
| Li6.2Al0.2La3Zr1.8Ta0.2O12 | 6.14 × 10−4 S·cm−1 (RT) | [30] | |
| Li6.6La3Zr1.6Nb0.4O12 | 3.86 × 10 −4 S·cm −1 (RT) | [36] | |
| Li6.4Ga0.133La3Zr1.8Ta0.2O12 | 6.141 × 10−4 S cm−1 (RT) | [38] | |
| Li6.20Ga0.30La2.95Rb0.05Zr2O12 | 1.62 × 10−3 S·cm−1 (RT) | [39] | |
| 4.56 × 10−3 S·cm−1 (60 °C) | [39] | ||
| Perovskite | Li0.34La0.51TiO0.294 | 2 × 10−5 S·cm−1 (RT) | [71] |
| Li0.5La0.5TiO3/5 wt% Ag | 4.2 × 10−5 S·cm−1 (RT) | [78] | |
| Li0.3Sr0.65Ta0.6Zr0.4O3 | 2.0 × 10−4 S·cm−1 (27 °C) | [80] | |
| Li3/8Sr7/16Hf1/4Ta3/4O3 | 3.8 × 10−4 S·cm−1 (RT) | [81] | |
| Li3/8Sr7/16Hf1/4Nb3/4O3 | 2.0 × 10−5 S·cm−1 (RT) | [82] | |
| Li0.34La0.56TiO3(film) | 2.0 × 10−5 S·cm−1 (RT) | [87] | |
| Li0.38Sr0.44Hf0.3O2.95F0.05 | 4.8 × 10−4 S·cm−1 (25 °C) | [93] | |
| NASICON | LiZr2(PO4)3 | 7 × 10−4 S·cm−1 (300 °C) | [99] |
| Li2.5Sr0.75Zr1.25(PO4)3 | 0.178S·cm−1 (550 °C) | [101] | |
| LiTi2(PO4)3 | 2 × 10−6 S·cm−1 (25 °C) | [104] | |
| Li1.3M0.3Ti1.7(PO4)3 M = Al or Sc | 7 × 10−4 S·cm−1 (25 °C) | [104] | |
| Li1+xAlxTi2-x(PO4)3 | 1.3 × 10−3 S·cm−1 (RT) | [106] | |
| Li1.3Al0.3Ti1.7(PO4)3 | 3 × 10−4 S·cm−1 (RT) | [107] | |
| Li1.4Al0.4Ti1.6(PO4)3 | 1.12 × 10−3 S·cm−1 (25 °C) | [108] | |
| Li1.5Al0.5Ge1.5(PO4)3 | 4 × 10−4 S·cm−1 (RT) | [109] | |
| Li1.5Al0.5Ge1.5(PO4)3 | 4.15 × 10−4 S·cm−1 (RT) | [110] | |
| Li1.5Al0.5Ge1.5(PO4)3 | 3.29 × 10−4 S cm−1 (RT) | [112] | |
| LAGP-0.05LiO2 | 7.25 × 10−4 S·cm−1 (RT) | [113] | |
| Li1.5Al0.33Sc0.17Ge1.5(PO4)3 | 5.826 × 10−3 S·cm−1 (bulk, RT) | [114] | |
| Li1.2Al0.2Zr0.1Ti1.7(PO4)3 | 4.07 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1Zr0.3Ti1.7(PO4)3 | 1.84 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.2Al0.2Hf0.1Ti1.7(PO4)3 | 2.68 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1Hf0.3Ti1.7(PO4)3 | 2.69 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.4Al0.2Mg0.1Ti1.7(PO4)3 | 1.13 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.5Al0.1Mg0.2Ti1.7(PO4)3 | 1.00 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.4Al0.2Ca0.1Ti1.7(PO4)3 | 2.80 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.5Al0.1Ca0.2Ti1.7(PO4)3 | 2.10 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.4Al0.2Sr0.1Ti1.7(PO4)3 | 1.10 × 10−4 S·cm−1 (40 °C) | [119] | |
| Li1.5Al0.1Sr0.2Ti1.7(PO4)3 | 8.10 × 10−5 S·cm−1 (40 °C) | [119] | |
| LISICON | Li14Zn(GeO4)4 | 0.125 S·cm−1 (300 °C) | [130] |
| Li14Zn(GeO4)4 | ~10−6 S·cm−1 (RT) | [131] | |
| Li3.6Ge0.6V0.4O4 | 4 × 10−5 S·cm−1 (18 °C) | [133] | |
| Li3.53(Ge0.75P0.25)0.7V0.3O4 | 5.1 × 10−5 S·cm−1 (25 °C) | [135] | |
| Li10.42Si1.5P1.5Cl0.08O11.92 | 1.03 × 10−5 S·cm−1 (27 °C) | [136] | |
| Li10.42Ge1.5P1.5Cl0.08O11.92 | 3.7 × 10−5 S·cm−1 (27 °C) | [136] | |
| Thio-LISICON | Li2.2Zn0.1Zr0.9S3 | 1.2 × 10−4 S·cm−1 (30 °C) | [140] |
| Li3.325P0.935S4 | 1.5 × 10−4 S·cm−1 (27 °C) | [141] | |
| Li3.4Si0.4P0.6S4 | 6.4 × 10−4 S·cm−1 (RT) | [142] | |
| β-Li3PS4(nanoporous) | 1.6 × 10−4 S·cm−1 (25 °C) | [145] | |
| Li10GeP2S12 | 1.2 × 10−2 S·cm−1 (RT) | [146] | |
| Li3.25Ge0.25P0.75S4 | 1.82 × 10−4 S·cm−1 (RT) | [152] | |
| Li3.8Sb0.2Sn0.8S4 | 3.5 × 10−4 S·cm−1 (RT) | [168] | |
| Li10Ge(P1–xSbx)2S12 | 12.1–15.7 × 10−3 S·cm−1 (RT) | [169] | |
| Li9+δP3+δ′S11.1O0.9 | 1.5 × 10−3 S·cm−1 (RT) | [171] | |
| Li10Sn0.95P2S11.4O0.5 | 3.96 × 10−3 S·cm−1 (RT) | [172] | |
| Li10SnP1.84Sb0.16S11.6O0.4 | 2.58 × 10−3 S·cm−1 (RT) | [173] |
| Electrolyte | Strategy | Cell Composition | Performance | Charge/Discharge Voltage | Ref. |
|---|---|---|---|---|---|
| LLZTO | Interfacial engineering | Li|AlN-LLZTO-AlN|Li (30 °C) | 3600 h at 0.01 mA·cm−2. | [50] | |
| Li|AlN-LLZTO|LFP (30 °C) | Initial discharge capacity of 131.1 mAh·g−1 at 0.1 C and 122.6 mAh·g−1 after 200 cycles. | ~3.55 V/~3.3 V | |||
| LLZO | Interfacial engineering | Li|Janus electrolyte|Li (RT) | About 300 h at 0.2 mA·cm−2. | [52] | |
| Li|Janus electrolyte|LFP (RT) | Discharge capacity of 140 mAh·g−1 at 0.1 C, 128 mAh·g−1 at 0.2 C and negligible decay after 100 cycles. | ~3.5 V/~3.3 V | |||
| Ga-LLZO-SiO2 | Engineering defect chemistry | Li|Ga-LLZO-SiO2(1 wt%)|Li (30 °C) | ∼500 h at 0.2 mA·cm−2 and then ∼1000 h at 0.3 mA·cm−2 | [54] | |
| Li|Ga-LLZO-SiO2(1 wt%)|LFP (30 °C) | Initial discharge capacity of 155 mAh·g−1 and ∼99% capacity retention after 20 cycles. | ~3.5 V/~3.3 V | |||
| Ga-LLZO | Grain refinement | Li|Ga-LLZO|Li (27 °C) | 600 h at 0.4 mA·cm−2. | [35] | |
| Li|Ga-LLZO|LFP (27 °C) | Capacity of 150 mAh·g−1 and negligible decay after 50 cycles | ~3.5 V/~3.3 V | |||
| LLZTO | Interfacial engineering | Li|GPE@LLZO|Li (30 °C) | 400 h at 0.2 mA·cm−2. | [58] | |
| Li|GPE@LLZO|LCO (30 °C) | Initial discharge capacity of 126.0 mAh·g−1 at 0.5 C, 104.1 mAh·g−1 after 100 cycles. | ~4 V/~3.8 V | |||
| LLZTO | Interfacial engineering | Li|Zn–Cu–LLZTO–Zn–Cu|Li (28 °C) | Over 450 h at 0.8 mA·cm−2 and a critical current density of 2.8 mA·cm−2. | [59] | |
| Li|Zn–Cu–LLZTO–Zn–Cu|LFP (28 °C) | Initial charge capacity of 146 mAh·g−1, 130.8 mAh·g−1 after 50 cycles. | ~3.6 V/~3.25 V | |||
| LLTO | Interfacial engineering | Li|PVDF:LLTO|Li (60 °C) | Fail after cycling at 0.1 mA cm−2 for 25 h. | [89] | |
| Li|PVDF:LLTO@PDA|Li (60 °C) | Over 800 h at 0.1 mA·cm−2. | ||||
| Li|PVDF:LLTO@PDA|NCM622 (60 °C) | Initial charge capacity of 158.2 mAh·g−1 and capacity retention of 83% after 100 cycles at 0.1 C. | 3.6~4.2 V/4.2~3.6 V | |||
| LSTZ | Interfacial engineering | Li|PEO/LSTZ|Li (45 °C) | Over 700 h at 0.1 mA·cm−2. | [90] | |
| Li|PEO/LSTZ|LFP (45 °C) | A stabilized capacity of 136 mAh·g−1 and capacity remains at 123 mAh·g−1 after 350 cycles. | ~3.6 V/~3.2 V | |||
| LATP | Interfacial engineering | Li|(PAA/PEO)30|LATP|(PAA/PEO)30|Li | 600 h at 0.1 mA·cm−2. | [121] | |
| Li|(PAA/PEO)30|LATP|(PAA/PEO)30|LFP | Initial discharge capacity of 115 mAh·g−1 at 0.1 C and 102 mAh·g−1 after 20 cycles. | ~3.5 V/~2.6 V | |||
| LATP | Interfacial engineering | Li|LATP@Al2O3|Li (RT) | 600 h with small voltage hysteresis. | [122] | |
| LATP | Interfacial engineering | Li|MoS2-LATP- MoS2|Li (60 °C) | More than 300 h at 0.05 mA·cm−2. | [123] | |
| LATP | Interfacial engineering | Li|Cr-LATP- Cr|Li (RT) | ~850 h at 0.2 mA·cm−2. | [124] | |
| LAGP | Interfacial engineering | Li|Li2OHBr|LAGP|Li2OHBr|Li (RT) | 300 h at 0.05 mA·cm−2. | [125] | |
| Li|Li2OHBr|LAGP|Li2OHBr|LFP (RT) | Initial specific capacity of 119.9 mAh·g−1, and remains the capacity of 110.1 mAh·g−1 after 20th cycle and the capacity of 96.3 mAh·g−1 after the 40th cycle at 0.1 C. | ~3.45 V/~3.35 V | |||
| LATP | Interfacial engineering | Li|5SnO2@LATP|LNLO|NCM (RT) | Initial discharge capacity of 171.49 mAh·g−1 and 89.47 % capacity retention after 100 cycles at 0.1C. | 3.6~4.3 V/4.3~3.3 V | [129] |
| LGPS | Interfacial engineering | Li0.8Al|LGPS|Li0.8Al | 2500 h at 0.5 mA·cm−2 | [156] | |
| LGPS | Interfacial engineering | Li|Nanocomposites-LGPS- Nanocomposites|Li | Over 1700 h with a Li deposition amount of 0.2 mAh·g−1. | [157] | |
| LGPS | Interfacial engineering | Li-LiH2PO4|LGPS|LiH2PO4-Li (25 °C) | Over 950 h at 0.1 mA·cm−2. | [158] | |
| Li-LiH2PO4|LGPS|LCO (25 °C) | 113.7 mAh·g−1 for the 500th cycle at 0.1 C with a retention of 86.7%. | ~3.9 V/~3.9 V | |||
| Li3.8Sb0.2Sn0.8S4 | LTO| Li3.8Sb0.2Sn0.8S4|LCO (RT) | Initial discharge capacity of up to 125 mAh·g−1, and gradually decreased to 105 mAh·g−1 (84% of the first discharge capacity) after 10 cycles. | [168] | ||
| Li10Ge(P0.925Sb0.075)2S12 | In|Li10Ge(P0.925Sb0.075)2S12|LNO@LCO (25 °C) | Initial discharge capacity of 128 mAh·g−1 at 0.1 C and 108 mAh·g−1 remained for over 50 cycles. | 3.3~3.6 V/3.6~3.3 V | [169] | |
| Li10Sn0.95P2S11.4O0.5 | Li-In|Li10Sn0.95P2S11.4O0.5|LNO@LCO (RT) | Specific discharge capacity of 133 mAh·g−1 in the first cycle. | 3.1~3.6 V/3.5~2.9 V | [172] | |
| Li10SnP1.84Sb0.16S11.6O0.4 | Li-In|Li10SnP1.84Sb0.16S11.6O0.4|LNO@LCO (25 °C) | Initial discharge capacity of 96 mAh·g−1 at 0.5 C, and maintains 85% capacity retention after 200 cycles. | 3.3~3.6 V/3.6~3.3 V | [173] | |
| LGPS | Interfacial engineering | Li|40s air-exposed Li10GeP2S12|Li (25 °C) | 1000 h with small polarization voltage of 26 mV at 0.1 mA·cm−2. | [174] | |
| Li|40s air-exposed Li10GeP2S12|LCO (25 °C) | 100 cycles with capacity retention of 80%, and discharge capacity of 113, 87, 66, 46 mAh·g−1 at 0.1, 0.2, 0.5 and 1 C, respectively. | ~3.9 V/~3.8 V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, T.; Yu, J.; Li, S.; Ma, H.; Liu, X. Review of the Developments and Difficulties in Inorganic Solid-State Electrolytes. Materials 2023, 16, 2510. https://doi.org/10.3390/ma16062510
Liu J, Wang T, Yu J, Li S, Ma H, Liu X. Review of the Developments and Difficulties in Inorganic Solid-State Electrolytes. Materials. 2023; 16(6):2510. https://doi.org/10.3390/ma16062510
Chicago/Turabian StyleLiu, Junlong, Tao Wang, Jinjian Yu, Shuyang Li, Hong Ma, and Xiaolong Liu. 2023. "Review of the Developments and Difficulties in Inorganic Solid-State Electrolytes" Materials 16, no. 6: 2510. https://doi.org/10.3390/ma16062510