
First-Principles Study on Mechanical and Optical Behavior of Plutonium Oxide under Typical Structural Phases and Vacancy Defects
 ,
,
Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
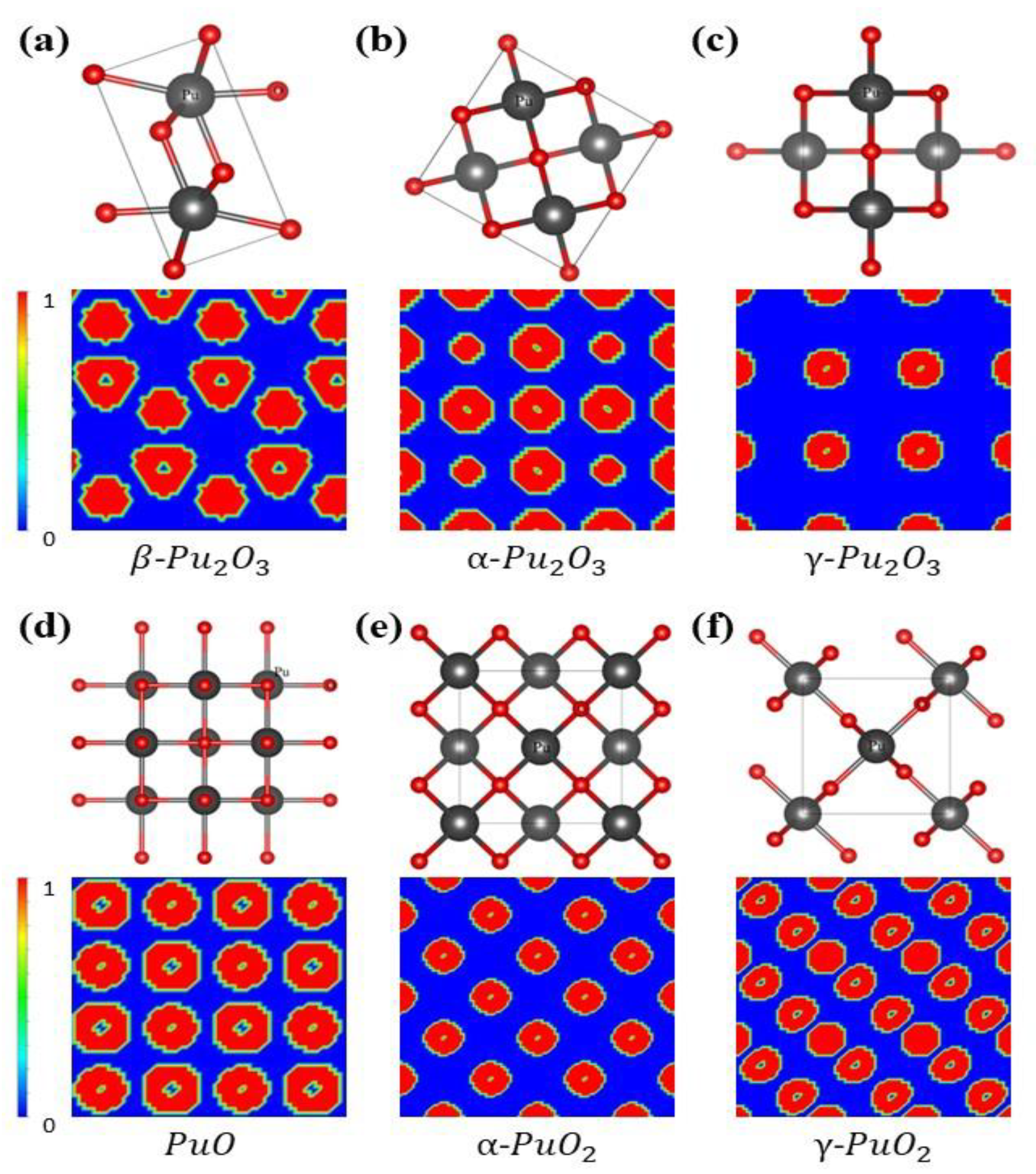
3.1. Crystal Structure Analysis
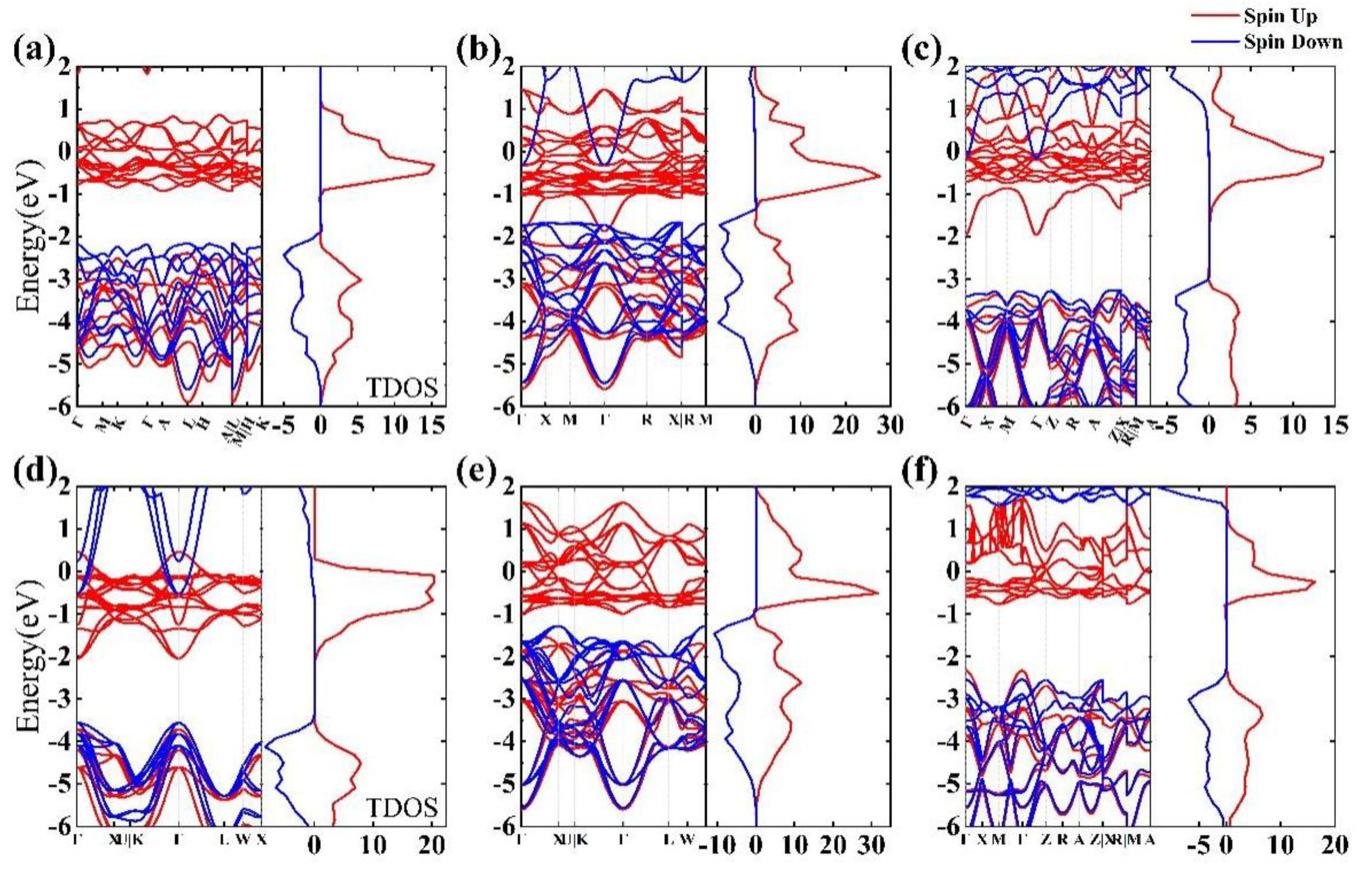
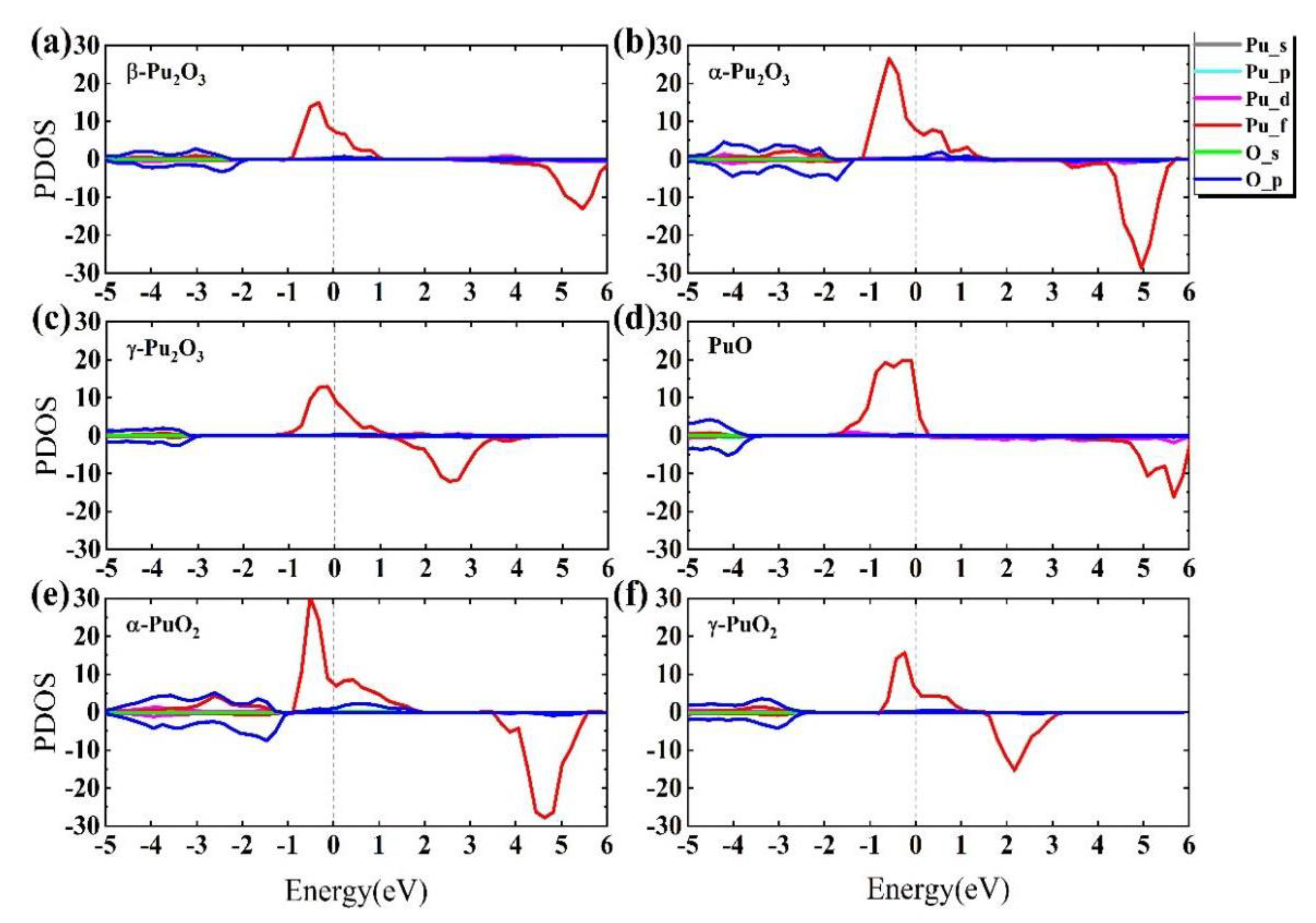
3.2. Band Structure and Density of Electronic States
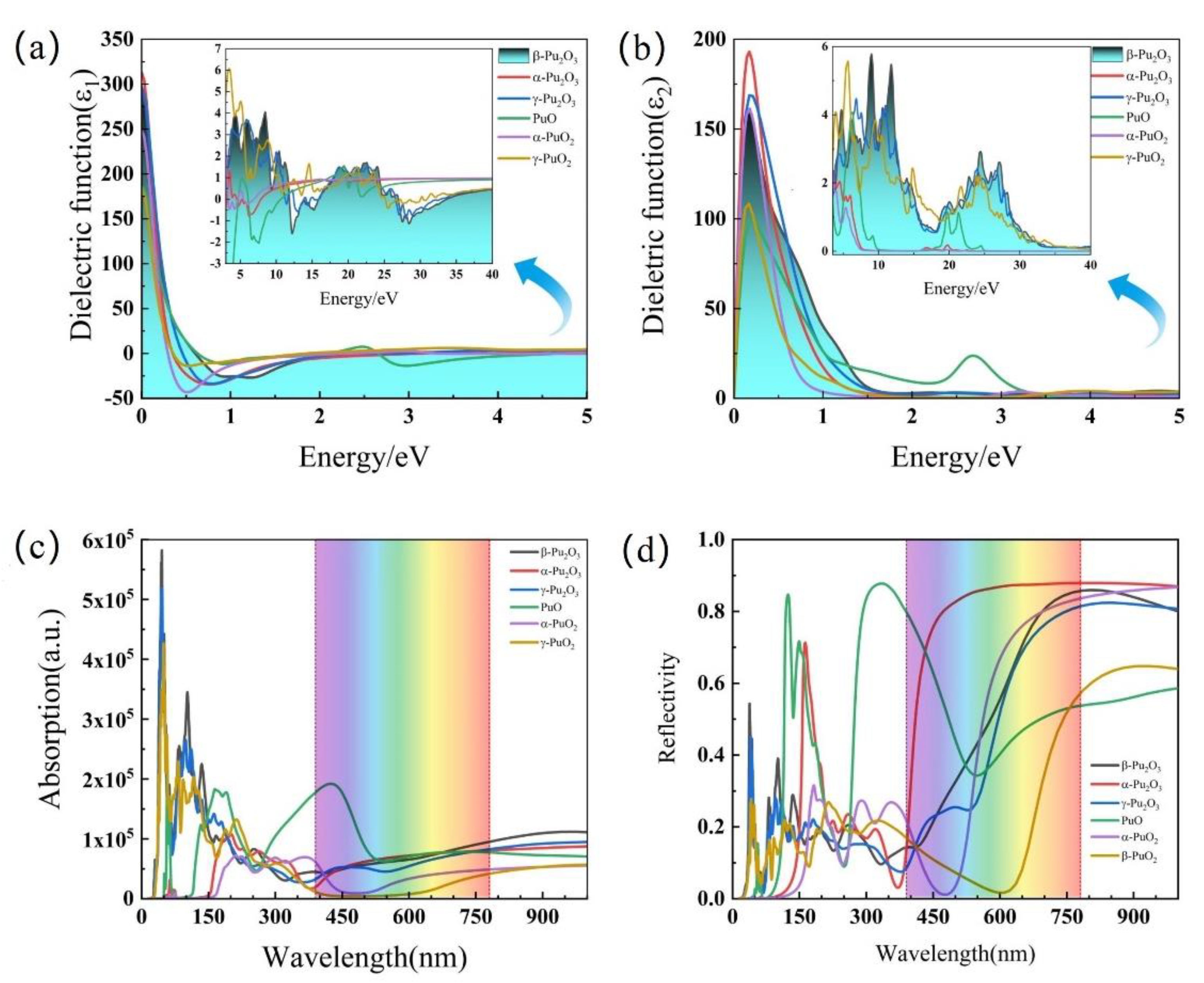
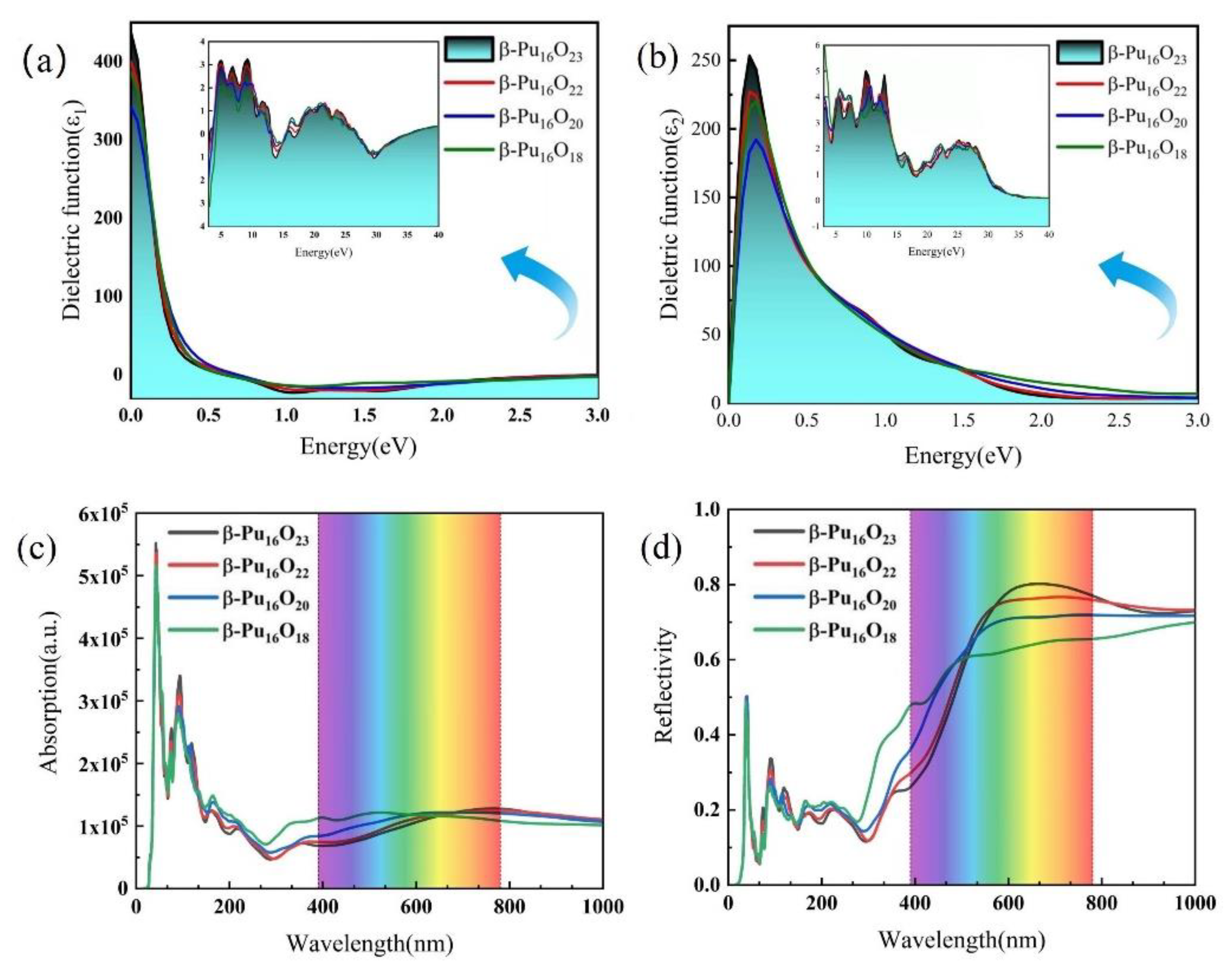
3.3. Mechanical and Optical Properties
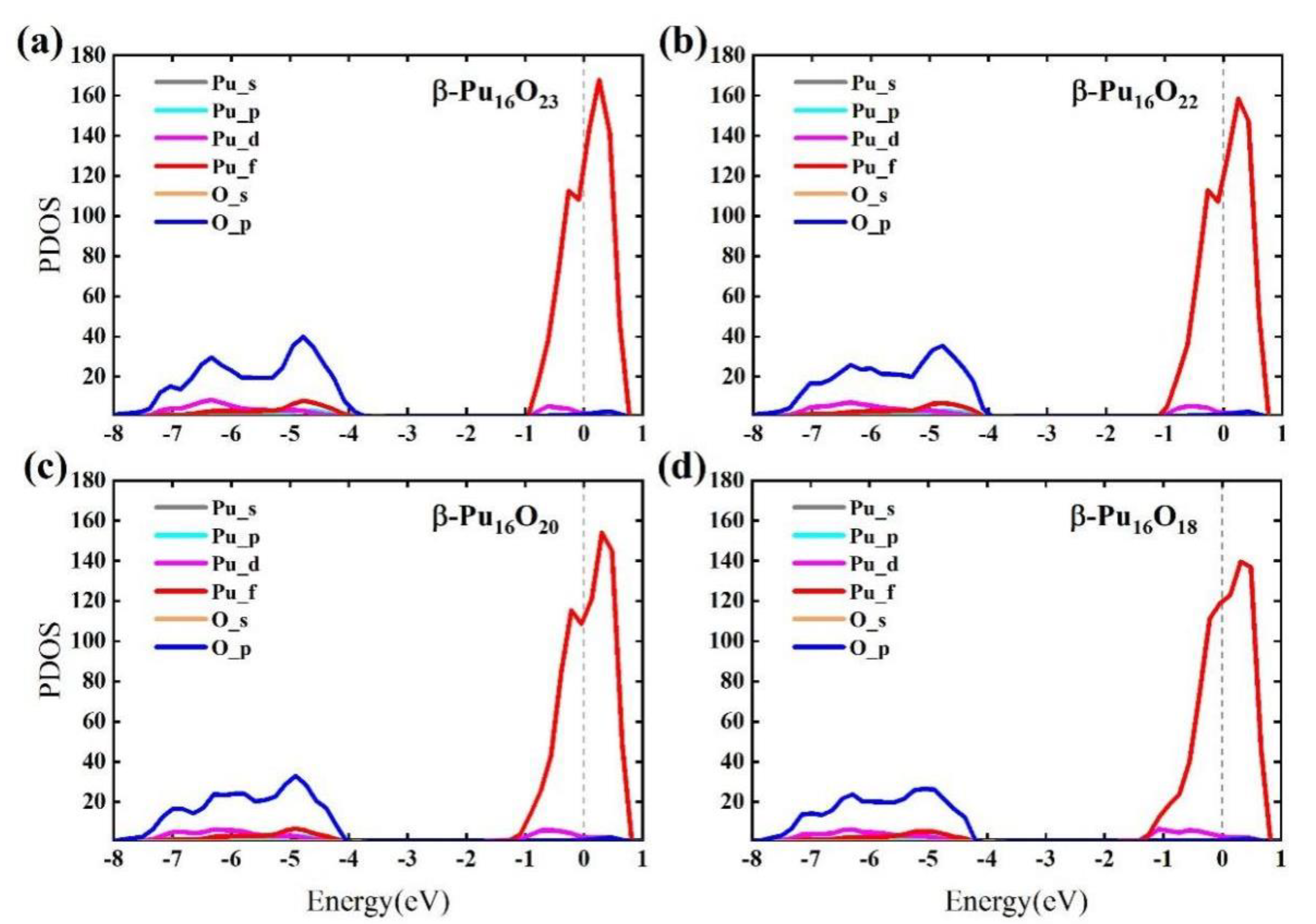
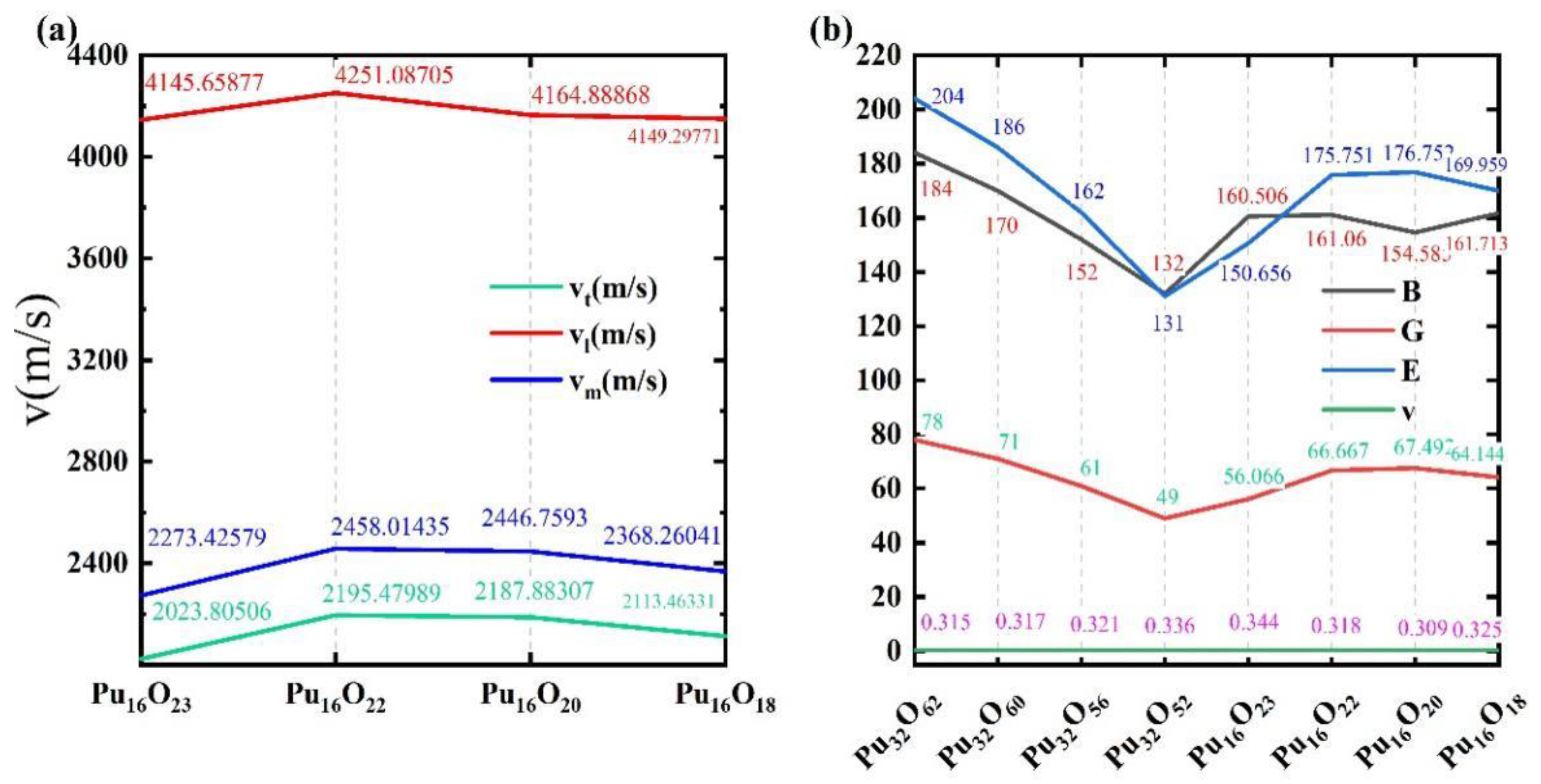
3.4. Mechanical and Optical Properties at Different Oxygen Vacancy Concentrations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Q.; Lu, H. Structure and Thermodynamic Stability of PuC2(g) Molecules. Chin. J. Chem. Phys. 2003, 16, 368–370. Available online: http://cjcp.ustc.edu.cn/hxwlxb/en/article/doi/10.1088/1674-0068/16/5/368-370 (accessed on 12 October 2022).
- Butterfield, M.T.; Durakiewicz, T.; Guziewicz, E.; Joyce, J.J.; Arko, A.J.; Graham, K.S.; Moore, D.P.; Morales, L.A. Photoemission of surface oxides and hydrides of delta plutonium. Surf. Sci. 2004, 571, 74–82. Available online: https://www.sciencedirect.com/science/article/pii/S0039602804010404 (accessed on 12 October 2022). [CrossRef]
- Chikalla, T.D.; McNeilly, C.E.; Skavdahl, R.E. The plutonium-oxygen system. J. Nucl. Mater. 1964, 12, 131–141. Available online: https://www.sciencedirect.com/science/article/pii/0022311564901321 (accessed on 12 October 2022). [CrossRef]
- Chen, J.; Meng, D.Q.; Du, J.G.; Jiang, G.; Gao, T.; Zhu, Z.H. Molecular structure and spectroscopic study of plutonium oxides. Acta Phys. Sin. (Chin. Ed.) 2010, 59, 1658. [Google Scholar] [CrossRef]
- Ghosh, P.S.; Somayajulu, P.S.; Arya, A.; Dey, G.K.; Dutta, B.K. Thermal expansion and thermal conductivity of (Th,Ce)O2 mixed oxides: A molecular dynamics and experimental study. J. Alloys Compd. 2015, 638, 172–181. Available online: https://www.sciencedirect.com/science/article/pii/S0925838815007653 (accessed on 12 October 2022). [CrossRef]
- Ghosh, P.S.; Somayajulu, P.S.; Krishnan, K.; Pathak, N.; Arya, A.; Dey, G.K. Thermal expansion and thermal conductivity of (Th,U)O2 mixed oxides: A molecular dynamics and experimental study. J. Alloys Compd. 2015, 650, 165–177. Available online: https://www.sciencedirect.com/science/article/pii/S0925838815306587 (accessed on 12 October 2022). [CrossRef]
- Ghosh, P.S.; Kuganathan, N.; Galvin, C.O.T.; Arya, A.; Dey, G.K.; Dutta, B.K.; Grimes, R.W. Melting behavior of (Th,U)O2 and (Th,Pu)O2 mixed oxides. J. Nucl. Mater. 2016, 479, 112–122. Available online: https://www.sciencedirect.com/science/article/pii/S0022311516302884 (accessed on 12 October 2022). [CrossRef] [Green Version]
- Ghosh, P.S.; Arya, A.; Dey, G.K.; Kuganathan, N.; Grimes, R.W. A computational study on the superionic behaviour of ThO2. Phys. Chem. Chem. Phys. 2016, 18, 31494–31504. [Google Scholar] [CrossRef]
- Somayajulu, P.S.; Ghosh, P.S.; Arya, A.; Devi, K.V.V.; Sathe, D.B.; Banerjee, J.; Dutta, B.K. Thermal expansion and thermal conductivity of (Th,Pu)O2 mixed oxides: A molecular dynamics and experimental study. J. Alloys Compd. 2016, 664, 291–303. Available online: https://www.sciencedirect.com/science/article/pii/S092583881532034X (accessed on 12 October 2022). [CrossRef]
- Somayajulu, P.S.; Ghosh, P.S.; Banerjee, J.; Babu, K.L.N.C.; Danny, K.M.; Mandal, B.P.; Mahata, T.; Sengupta, P.; Sali, S.K.; Arya, A. Experimental and molecular dynamics study of thermo-physical and transport properties of ThO2-5wt.%CeO2 mixed oxides. J. Nucl. Mater. 2015, 467, 644–659. Available online: https://www.sciencedirect.com/science/article/pii/S0022311515302993 (accessed on 12 October 2022). [CrossRef]
- Shi, J.; Li, G.; Wan, L.; Gao, T.; Luo, W. New insights into the interfacial interactions of O2 and H2O molecules with PuH2 (110) and (111) surfaces from first-principles calculations. Int. J. Hydrog. Energy. 2022. Available online: https://www.sciencedirect.com/science/article/pii/S036031992203823X (accessed on 12 October 2022).
- Meng, D.Q.; Jiang, G.; Liu, X.L.; Luo, D.L.; Zhang, W.X.; Zhu, Z.H. Structure and potential function of Pu3 system. Acta Phys. Sin. (Chin. Ed.) 2001, 50, 1268. [Google Scholar] [CrossRef]
- Alexandrov, V.; Shvareva, T.Y.; Hayun, S.; Asta, M.; Navrotsky, A. Actinide Dioxides in Water: Interactions at the Interface. J. Phys. Chem. Lett. 2011, 2, 3130–3134. [Google Scholar] [CrossRef]
- Eloirdi, R.; Gouder, T.; Wastin, F.; Huber, F.; Shick, A.B.; Kolorenč, J. Dilution effect on the U 5f states in Au matrix: A photoemission spectroscopy study. Phys. Rev. B 2011, 84, 235143. [Google Scholar] [CrossRef]
- Flores, H.G.G.; Pugmire, D.L. The growth and evolution of thin oxide films on δ-plutonium surfaces. IOP Conf. Ser. Mater. Sci. Engi. 2010, 9, 012038. [Google Scholar] [CrossRef] [Green Version]
- Dinh, L.N.; Haschke, J.M.; Saw, C.K.; Allen, P.G.; McLean, W. Pu2O3 and the plutonium hydriding process. J. Nucl. Mater. 2011, 408, 171–175. Available online: https://www.sciencedirect.com/science/article/pii/S0022311510007178 (accessed on 12 October 2022). [CrossRef] [Green Version]
- Ravat, B.; Jolly, L.; Oudot, B.; Fabas, A.; Guerault, H.; Popa, I.; Delaunay, F. New insight into δ-Pu alloy oxidation kinetics highlighted by using in-situ X-ray diffraction coupled with an original Rietveld refinement method. Corros. Sci. 2018, 138, 66–74. Available online: https://www.sciencedirect.com/science/article/pii/S0010938×17321911 (accessed on 12 October 2022). [CrossRef]
- Wang, L.L.; Wan, M.J.; Ma, J.J.; Jiang, G. Molecular dynamics simulation of U1-xPuxO2 thermal expansion. Acta Phys. Sin. (Chin. Ed.) 2014, 63, 083103. Available online: https://wulixb.iphy.ac.cn/cn/article/id/58990 (accessed on 12 October 2022). [CrossRef]
- Xie, A.D.; Meng, D.Q.; Luo, D.L.; Ma, M.Z.; Zhu, Z.H. Effect of external electric field on the excited states of plutonium dihydride. Acta Phys. Sin. (Chin. Ed.) 2006, 55, 2180–2184. [Google Scholar] [CrossRef]
- Atlas, L.M.; Schlehman, G.J.; Readey, D.W. Defects in PuO2-x: Density Measurements at High Temperature. J. Am. Ceram Soc. 1966, 49, 624. [Google Scholar] [CrossRef]
- Sari, C.; Benedict, U.; Blank, H. A study of the ternary system UO2-PuO2-Pu2O3. J. Nucl. Mater. 1970, 35, 267–277. Available online: https://www.sciencedirect.com/science/article/pii/0022311570902114 (accessed on 12 October 2022). [CrossRef]
- Zhang, L.; Sun, B.; Song, H.F. First principles study of hydrogen behavior in plutonium oxides. Chin. J. Comput. Phys. 2020, 5, 595–602. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?doi=10.19596/j.cnki.1001-246x.8118 (accessed on 12 October 2022).
- Anderson, G.G.; Palermo, J.J.; Schilling, J.D.; Roth, R.; Heuser, J.; Hultgren, S.J. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 2003, 301, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Skriver, H.L.; Andersen, O.K.; Johansson, B. Calculated Bulk Properties of the Actinide Metals. Phys. Rev. Lett. 1978, 41, 42–45. Available online: https://link.aps.org/doi/10.1103/PhysRevLett.41.42 (accessed on 12 October 2022). [CrossRef]
- Petit, L.; Svane, A.; Szotek, Z.; Temmerman, W.M. First-principles calculations of PuO(2+/−x). Science 2003, 301, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Prodan, I.D.; Scuseria, G.E.; Martin, R.L. Covalency in the actinide dioxides: Systematic study of the electronic properties using screened hybrid density functional theory. Phys. Rev. B 2007, 76, 033101. Available online: https://link.aps.org/doi/10.1103/PhysRevB.76.033101 (accessed on 12 October 2022). [CrossRef]
- Svane, A.; Petit, L.; Szotek, Z.; Temmerman, W.M. Self-interaction-corrected local spin density theory of 5f electron localization in actinides. Phys. Rev. B 2007, 76, 115116. Available online: https://link.aps.org/doi/10.1103/PhysRevB.76.115116 (accessed on 12 October 2022). [CrossRef] [Green Version]
- Prodan, I.D.; Scuseria, G.E.; Sordo, J.A.; Kudin, K.N.; Martin, R.L. Lattice defects and magnetic ordering in plutonium oxides: A hybrid density-functional-theory study of strongly correlated materials. J. Chem. Phys. 2005, 123, 014703. [Google Scholar] [CrossRef]
- Jomard, G.; Amadon, B.; Bottin, F.; Torrent, M. Structural, thermodynamic, and electronic properties of plutonium oxides from first principles. Phys. Rev. B 2008, 78, 075125. Available online: https://link.aps.org/doi/10.1103/PhysRevB.78.075125 (accessed on 12 October 2022). [CrossRef]
- Yang, Y.; Lu, Y.; Zhang, P. Optical properties of PuO2 and α-Pu2O3 by GGA+U+QA studies. J. Nucl. Mater. 2014, 452, 414–418. Available online: https://www.sciencedirect.com/science/article/pii/S002231151400347X (accessed on 12 October 2022). [CrossRef]
- Shi, H.; Zhang, P. First-principles study of α-Pu2O3. J. Nucl. Mater. 2012, 420, 159–163. Available online: https://www.sciencedirect.com/science/article/pii/S002231151100818X (accessed on 12 October 2022). [CrossRef] [Green Version]
- Lu, Y.; Yang, Y.; Zheng, F.; Zhang, P. Electronic and thermodynamic properties of α-Pu2O3. Phys. Lett. A 2014, 378, 3060–3065. Available online: https://www.sciencedirect.com/science/article/pii/S0375960114008391 (accessed on 12 October 2022). [CrossRef]
- Zhang, P.; Wang, B.-T.; Zhao, X.-G. Ground-state properties and high-pressure behavior of plutonium dioxide: Density functional theory calculations. Phys. Rev. B 2010, 82, 144110. Available online: https://link.aps.org/doi/10.1103/PhysRevB.82.144110 (accessed on 12 October 2022). [CrossRef]
- Ao, B.; Qiu, R.; Lu, H.; Chen, P. First-principles DFT+U calculations on the energetics of Ga in Pu, Pu2O3 and PuO2. Comput. Mate. Sci. 2016, 122, 263–271. Available online: https://www.sciencedirect.com/science/article/pii/S0927025616302737 (accessed on 12 October 2022). [CrossRef]
- Ao, B.; Tang, J.; Ye, X.; Tao, R.; Qiu, R. Phase Segregation, Transition, or New Phase Formation of Plutonium Dioxide: The Roles of Transition Metals. Inorg. Chem. 2019, 58, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Ao, B.; Lu, H.; Qiu, R.; Ye, X.; Shi, P.; Chen, P.; Wang, X. First-Principles Energetics of Some Nonmetallic Impurity Atoms in Plutonium Dioxide. J. Phys. Chem. C 2015, 119, 14879–14889. [Google Scholar] [CrossRef]
- Atta-Fynn, R.; Ray, A.K. Bulk and (112¯0) surface properties of β-Pu2O3: A theoretical study using DFT with exact exchange for correlated electrons. Chem. Phys. Lett. 2013, 583, 42–48. Available online: https://www.sciencedirect.com/science/article/pii/S0009261413009500 (accessed on 12 October 2022). [CrossRef]
- Lu, Y.; Yang, Y.; Zhang, P. Charge states of point defects in plutonium oxide: A first-principles study. J. Alloys. Compds. 2015, 649, 544–552. Available online: https://www.sciencedirect.com/science/article/pii/S0925838815306174 (accessed on 12 October 2022). [CrossRef]
- Agarwal, R.; Sen, B.K.; Venugopal, V. Phase diagram analysis of (U,Pu)O2−x sub-system. J. Nucl. Mater. 2009, 385, 112–116. [Google Scholar] [CrossRef]
- Ma, B.L.; Wu, Y.Y.; Guo, Y.H.; Yin, W.; Zhan, Q.; Yang, H.G.; Wang, B.T. Effects of Monovacancy and Divacancies on Hydrogen Solubility, Trapping and Diffusion Behaviors in fcc-Pd by First Principles. Materials 2020, 13, 4876. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. Available online: https://link.aps.org/doi/10.1103/PhysRevB.54.11169 (accessed on 12 October 2022). [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. Available online: https://link.aps.org/doi/10.1103/PhysRevLett.77.3865 (accessed on 12 October 2022). [CrossRef] [PubMed]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Zhang, S.X.; Liu, S.Y.; Yan, D.L.; Yu, Q.; Ren, H.T.; Yu, B.; Li, D.J. First principles study on structural stability and mechanical properties of Ta1-xHfxC and Ta1-xZrxC solid solutions. Acta Phys. Sin. (Chin. Ed.) 2021, 70, 117102. Available online: https://wulixb.iphy.ac.cn/cn/article/id/9e8d0b59-fb1e-45d0-abd6-a9fd0bef7357 (accessed on 12 October 2022). [CrossRef]
- Ma, J.-J.; Zhang, C.-B.; Qiu, R.; Zhang, P.; Ao, B.; Wang, B.-T. Pressure-induced structural and electronic phase transitions of uranium trioxide. Phys. Rev. B 2021, 104, 174103. Available online: https://link.aps.org/doi/10.1103/PhysRevB.104.174103 (accessed on 12 October 2022). [CrossRef]
- Ghosh, P.S.; Arya, A. First-principles study of phase stability, electronic and mechanical properties of plutonium sub-oxides. Phys. Chem. Chem. Phys. 2019, 21, 16818–16829. [Google Scholar] [CrossRef]
- Huang, S.S.; Ma, J.J.; Lai, K.; Zhang, C.B.; Yin, W.; Qiu, R.Z.; Zhang, P.; Wang, B.T. Point defects stability, hydrogen diffusion, electronic structure, and mechanical properties of defected equiatomic γ(U,Zr) from first-principles. Materials 2022, 15, 7452. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plutonium Oxide | Formation Energy (eV) | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) |
|---|---|---|---|---|---|---|---|
| −3.411 | 3.76 | 3.76 | 5.97 | 90 | 90 | 120 | |
| −3.357 | 5.38 | 5.38 | 5.38 | 90 | 90 | 90 | |
| −3.401 | 3.76 | 3.76 | 5.97 | 90 | 90 | 120 | |
| −2.808 | 4.97 | 4.97 | 4.97 | 90 | 90 | 90 | |
| −3.673 | 3.76 | 3.76 | 5.97 | 90 | 90 | 120 | |
| −3.507 | 3.76 | 3.76 | 5.97 | 90 | 90 | 120 |
| C11 | C12 | C13 | C14 | C33 | C44 | C66 | |
|---|---|---|---|---|---|---|---|
| 253.275 | 122.725 | 151.414 | 8.55 | 228.163 | −8.55 | 65.275 | |
| 305.341 | 4.798 | 4.798 | - | 305.341 | −17.313 | −17.313 | |
| 226.968 | 127.209 | 92.974 | - | 259.606 | 54.821 | 96.290 | |
| 425.817 | 112.785 | 112.785 | - | 425.817 | 45.283 | 45.283 | |
| 3925.219 | 2276.845 | 2276.845 | - | 3925.219 | 65.254 | 65.254 | |
| 4966.565 | 5302.567 | 112.485 | - | 572.217 | −433.486 | 445.562 |
| Plutonium Oxide | Bulk Modulus (GPa) | Young’s Modulus (GPa) | Shear Modulus (GPa) | Poisson’s Ratio |
|---|---|---|---|---|
| - | 176.2 | 155.78 | 57.58 | 0.35 |
| - | 104.98 | 128.82 | 49.72 | 0.3 |
| - | 148.87 | 171.67 | 65.63 | 0.31 |
| 217.13 | 205.43 | 76.52 | 0.34 | |
| -Pu | 209.49 | 281.5 | 110.3 | 0.28 |
| -Pu | 175.13 | 153.61 | 56.73 | 0.35 |
| Plutonium Oxide | Debye Temperature (K) | Lattice Thermal Conductivity |
|---|---|---|
| - | 285.864 | 0.8124 |
| - | 307.852 | 0.8501 |
| - | 303.668 | 0.8250 |
| - | 301.879 | 0.8535 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, J.-X.; Yang, F.; Wang, Q.-B.; He, Y.-Y.; Liu, Y.-N.; Hu, Z.-Y.; Wen, W.-W.; Wu, Y.-P.; Zheng, C.-Y.; Yu, A.; et al. First-Principles Study on Mechanical and Optical Behavior of Plutonium Oxide under Typical Structural Phases and Vacancy Defects. Materials 2022, 15, 7785. https://doi.org/10.3390/ma15217785
Cheng J-X, Yang F, Wang Q-B, He Y-Y, Liu Y-N, Hu Z-Y, Wen W-W, Wu Y-P, Zheng C-Y, Yu A, et al. First-Principles Study on Mechanical and Optical Behavior of Plutonium Oxide under Typical Structural Phases and Vacancy Defects. Materials. 2022; 15(21):7785. https://doi.org/10.3390/ma15217785
Chicago/Turabian StyleCheng, Jin-Xing, Fei Yang, Qing-Bo Wang, Yuan-Yuan He, Yi-Nuo Liu, Zi-Yu Hu, Wei-Wei Wen, You-Peng Wu, Cheng-Yin Zheng, Ai Yu, and et al. 2022. "First-Principles Study on Mechanical and Optical Behavior of Plutonium Oxide under Typical Structural Phases and Vacancy Defects" Materials 15, no. 21: 7785. https://doi.org/10.3390/ma15217785