
Stability of Strained Stanene Compared to That of Graphene

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
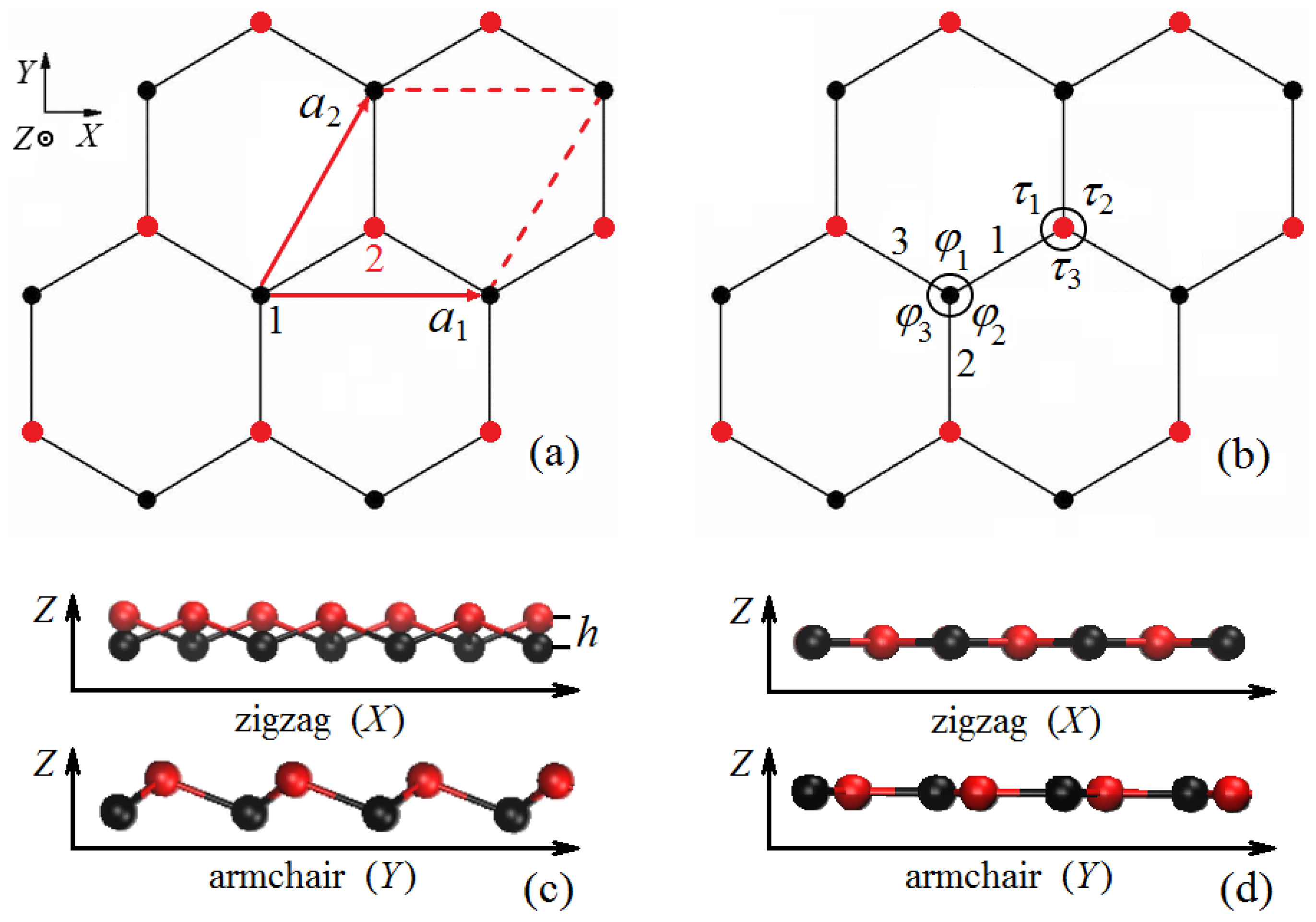
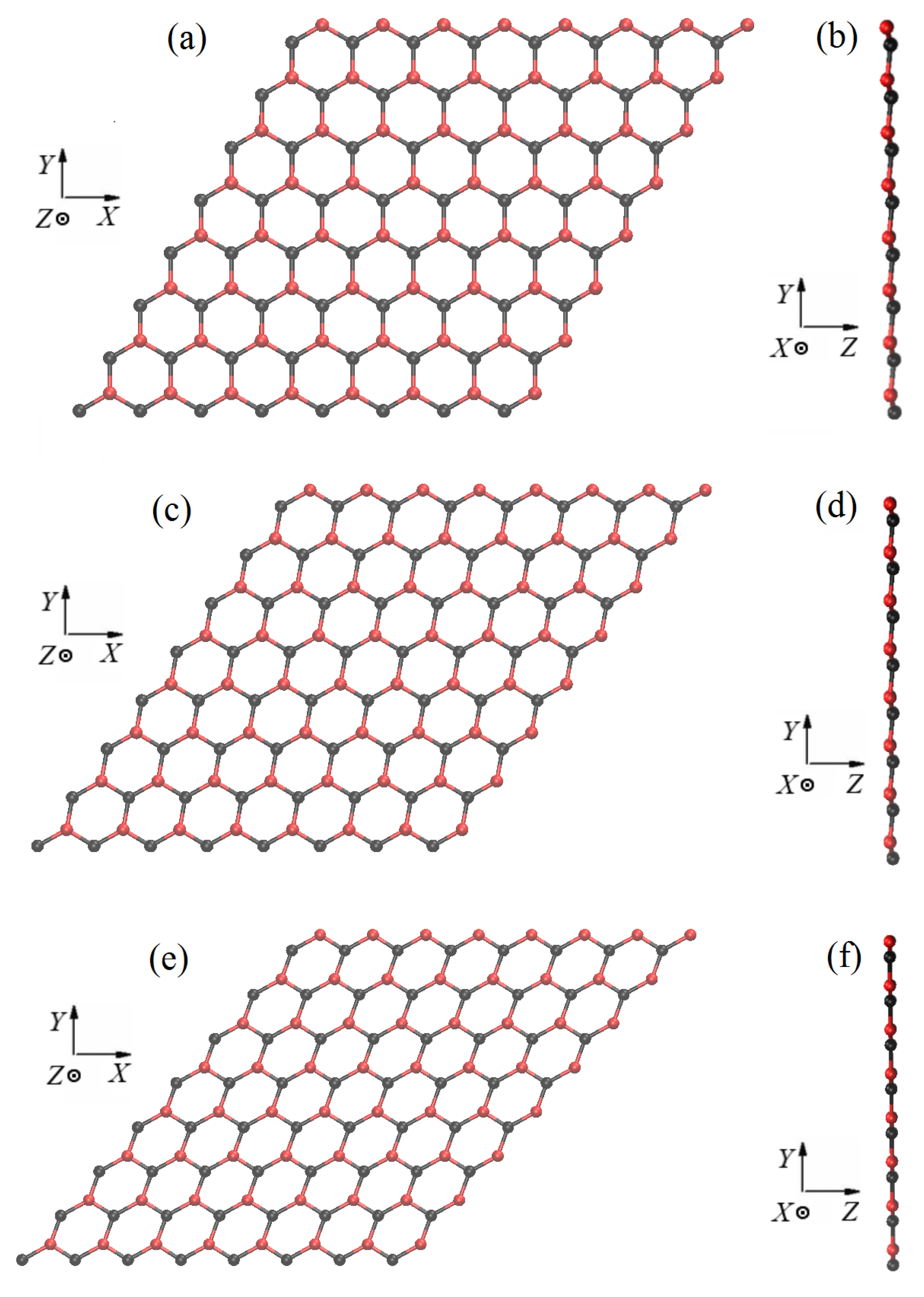
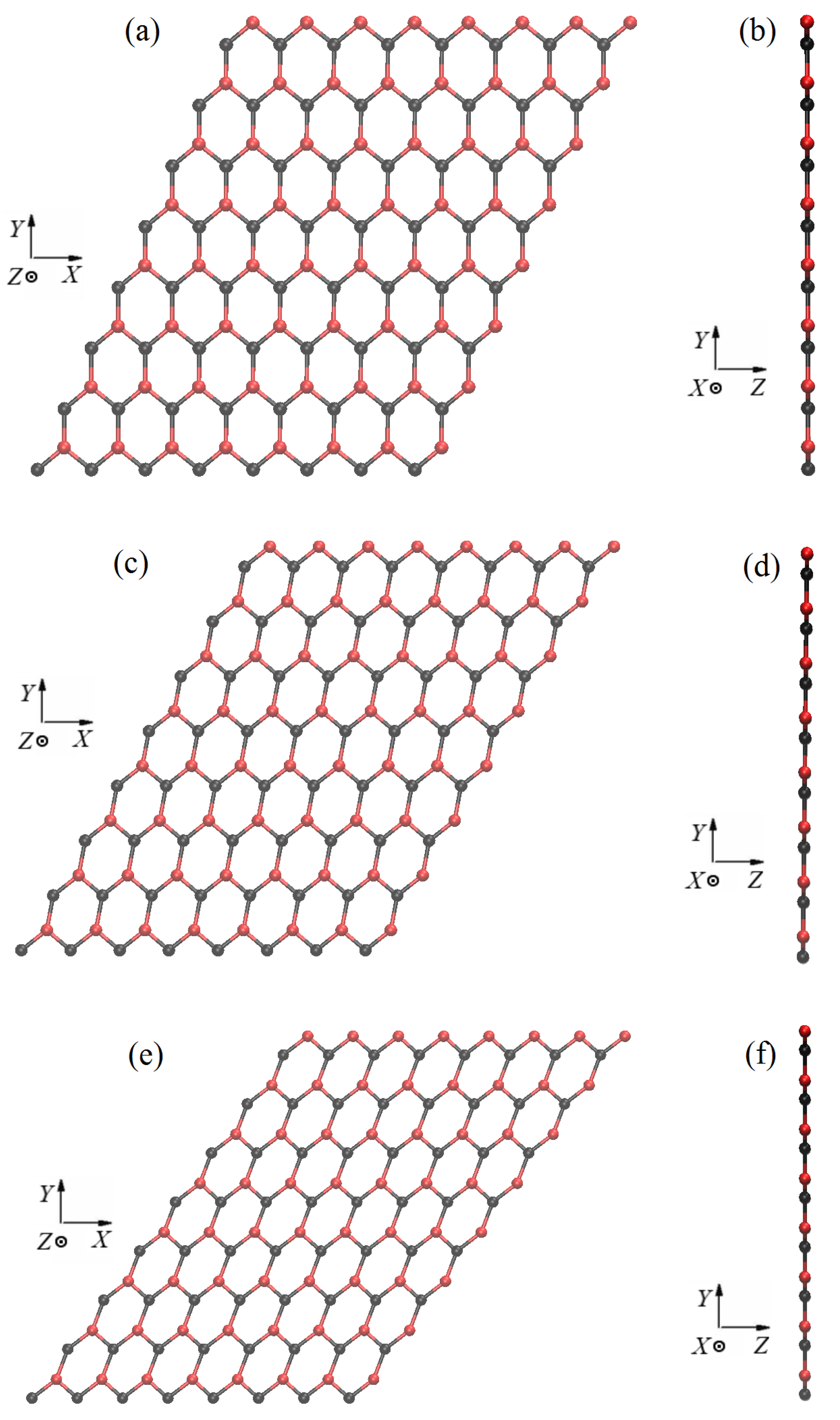
2.1. Structure of Stanene
2.2. Structure Generation and Homogeneous Deformation Application
2.3. Tersoff Potential
2.4. Check for Stability of Strained Stanene
3. Results and Discussion
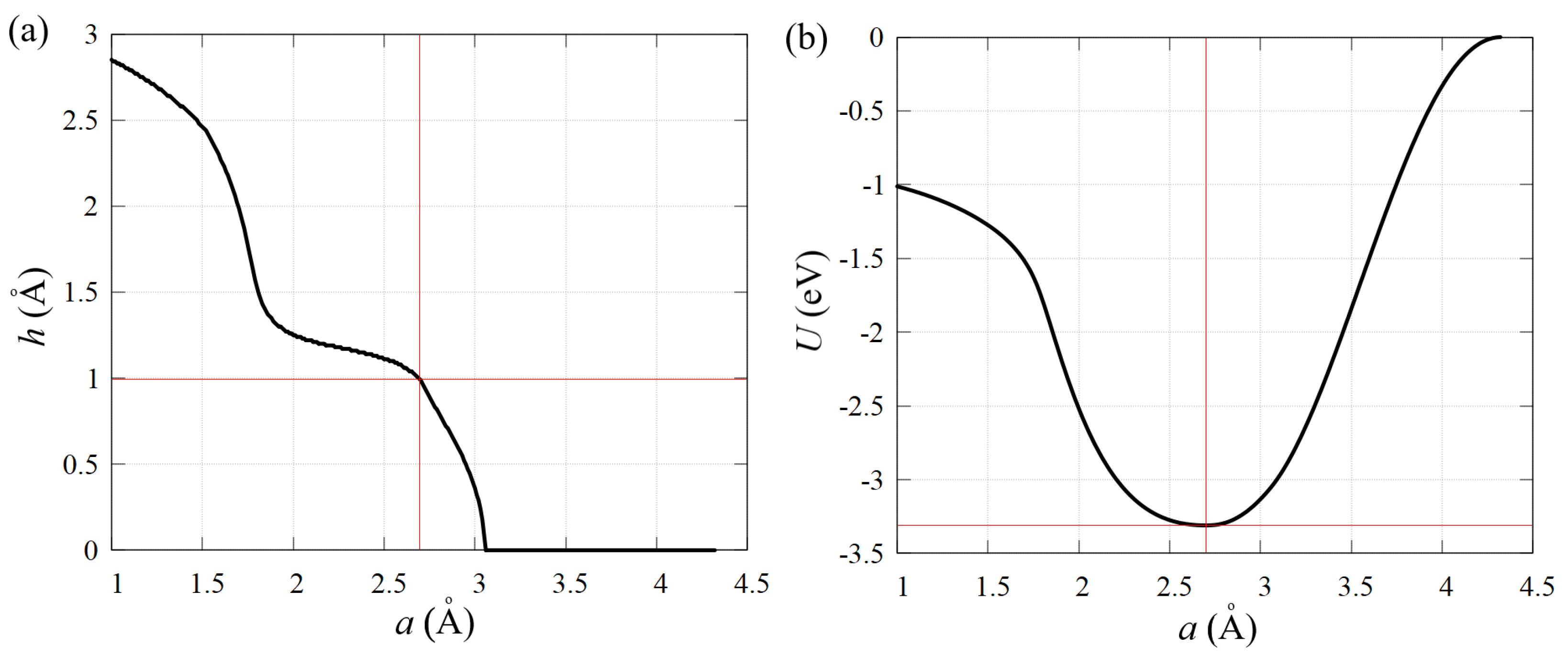
3.1. Equilibrium State of Stanene
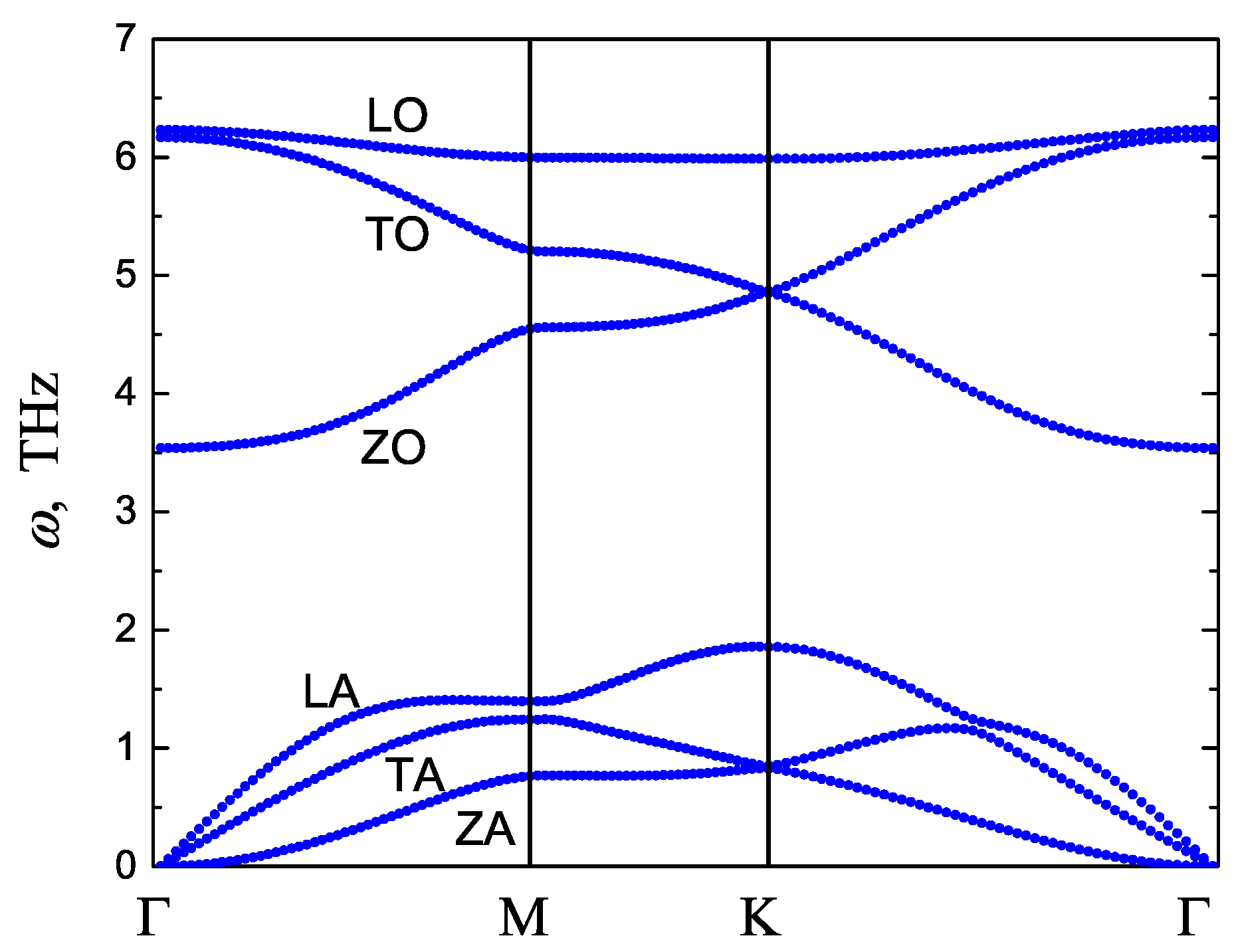
3.2. Phonon Dispersion Curves
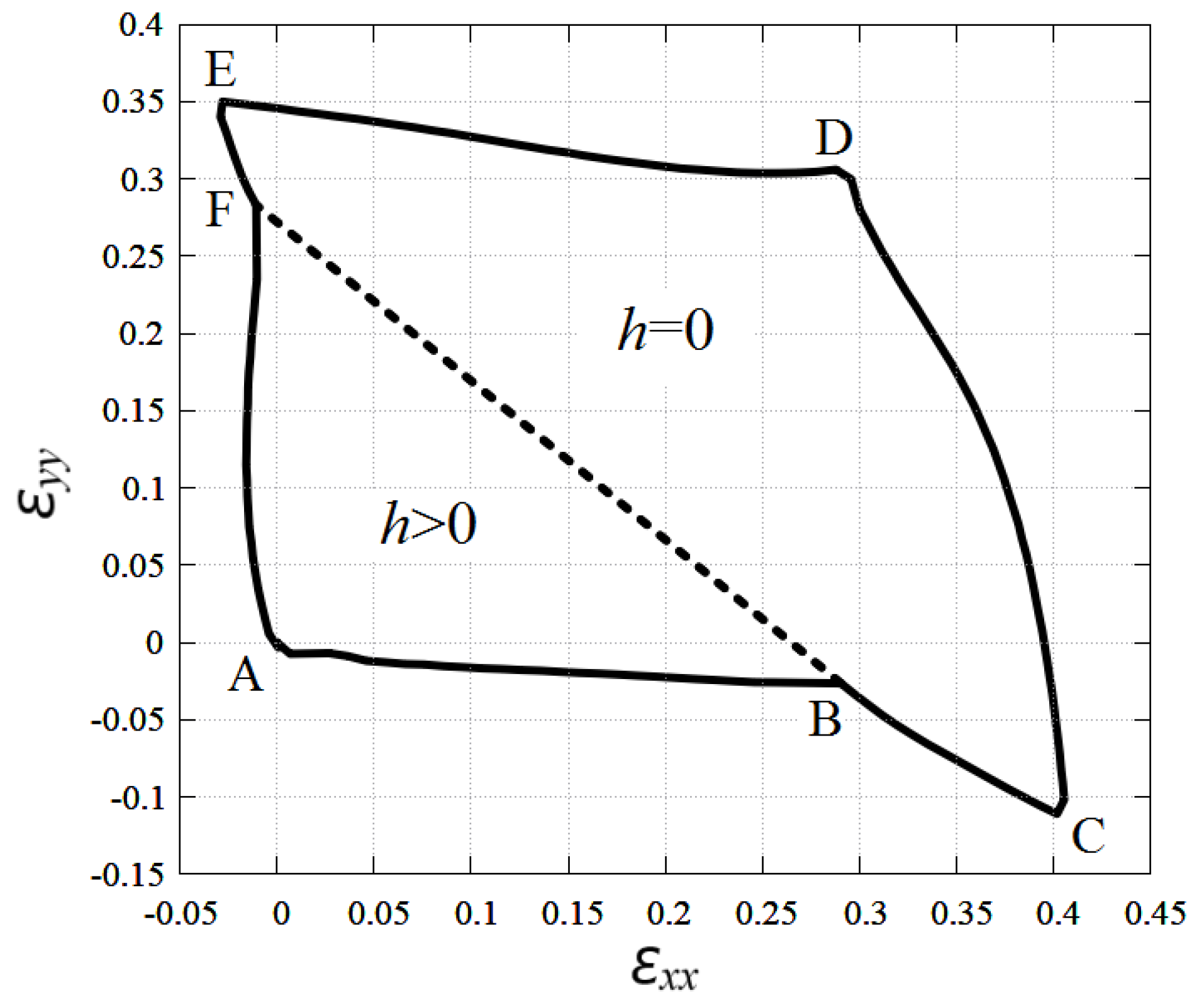
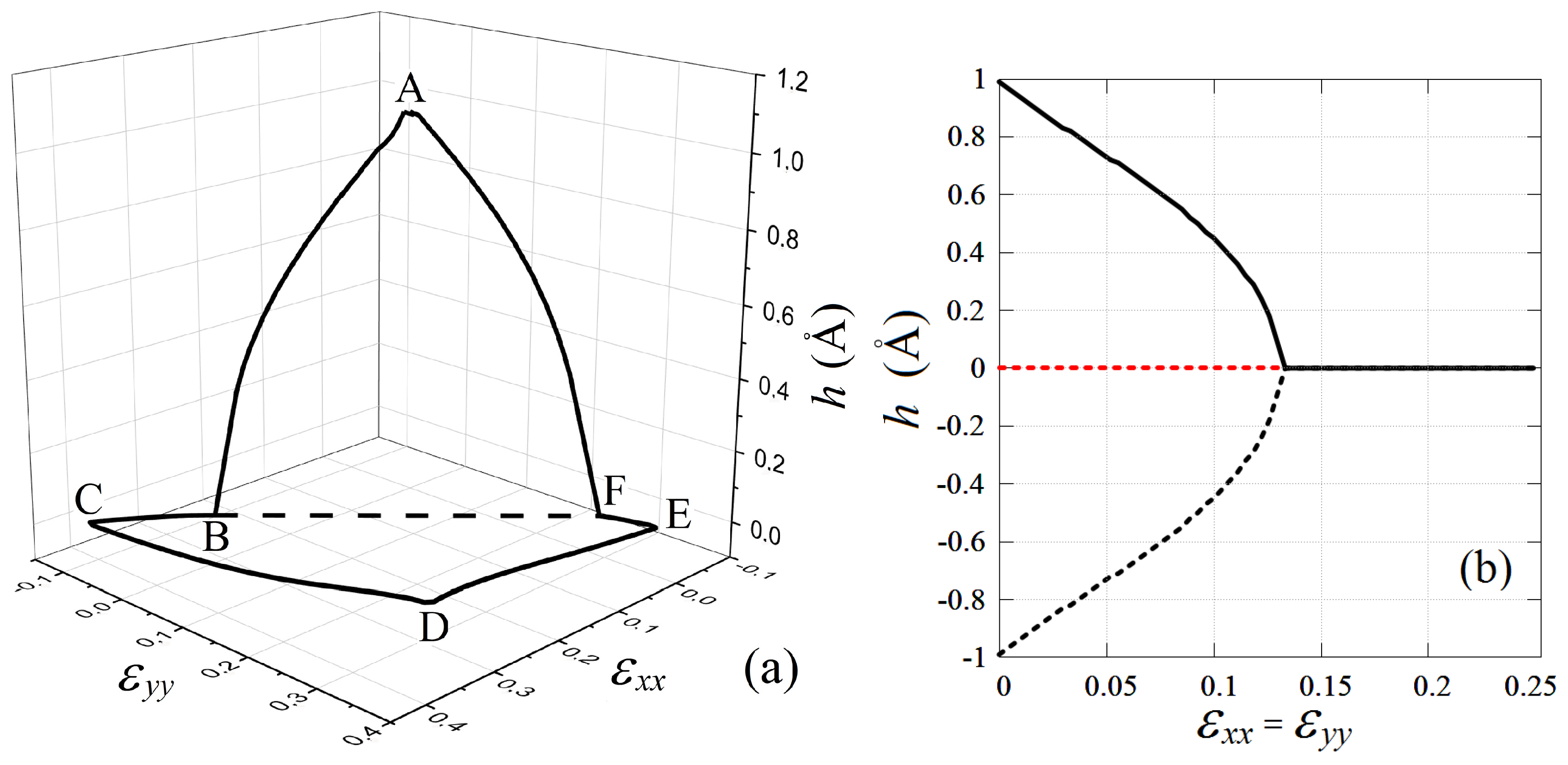
3.3. Stability Region of Planar Stanene under In-Plane Tensile and Compressive Strain
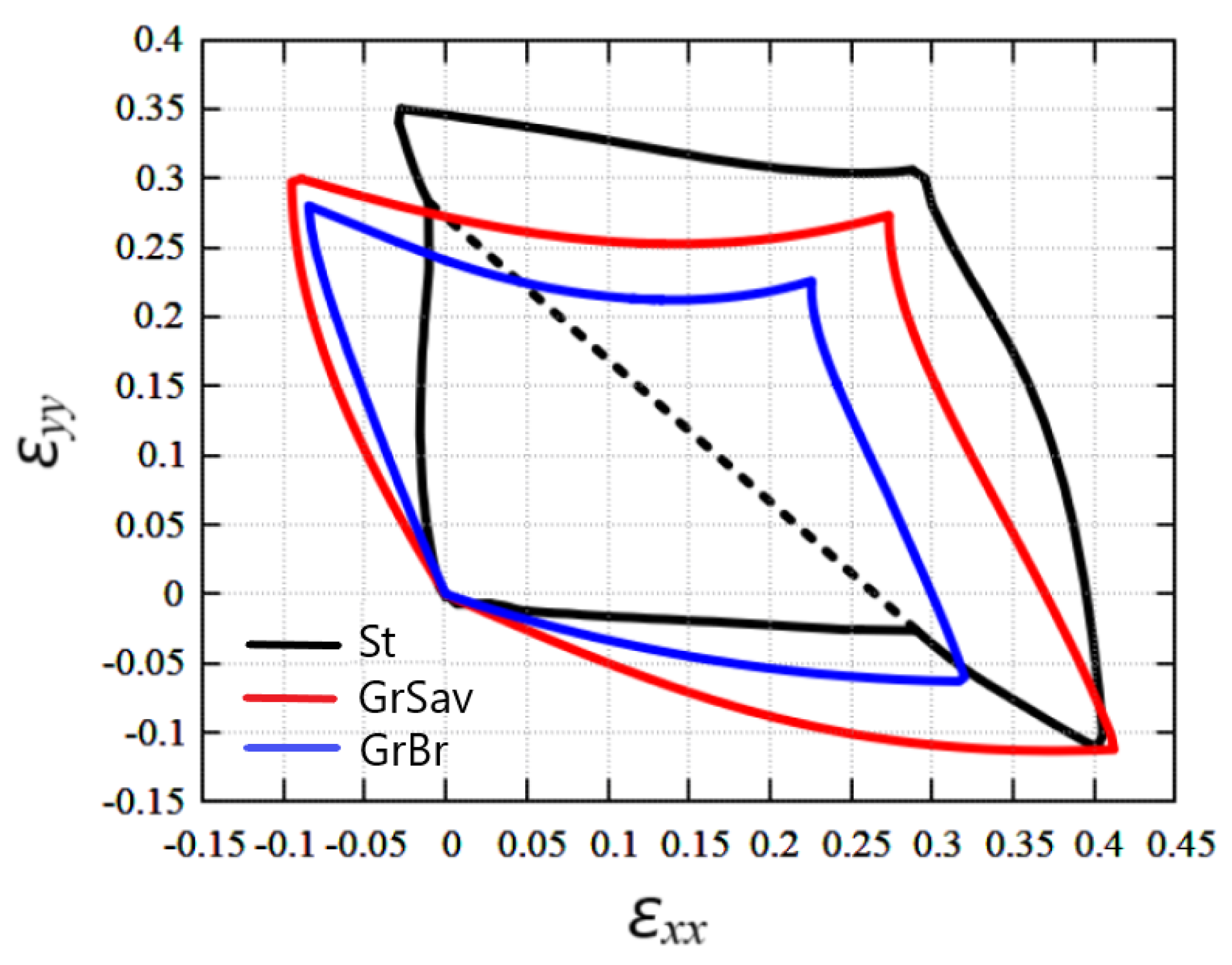
3.4. Comparison of the Stability Regions of Stanene and Graphene
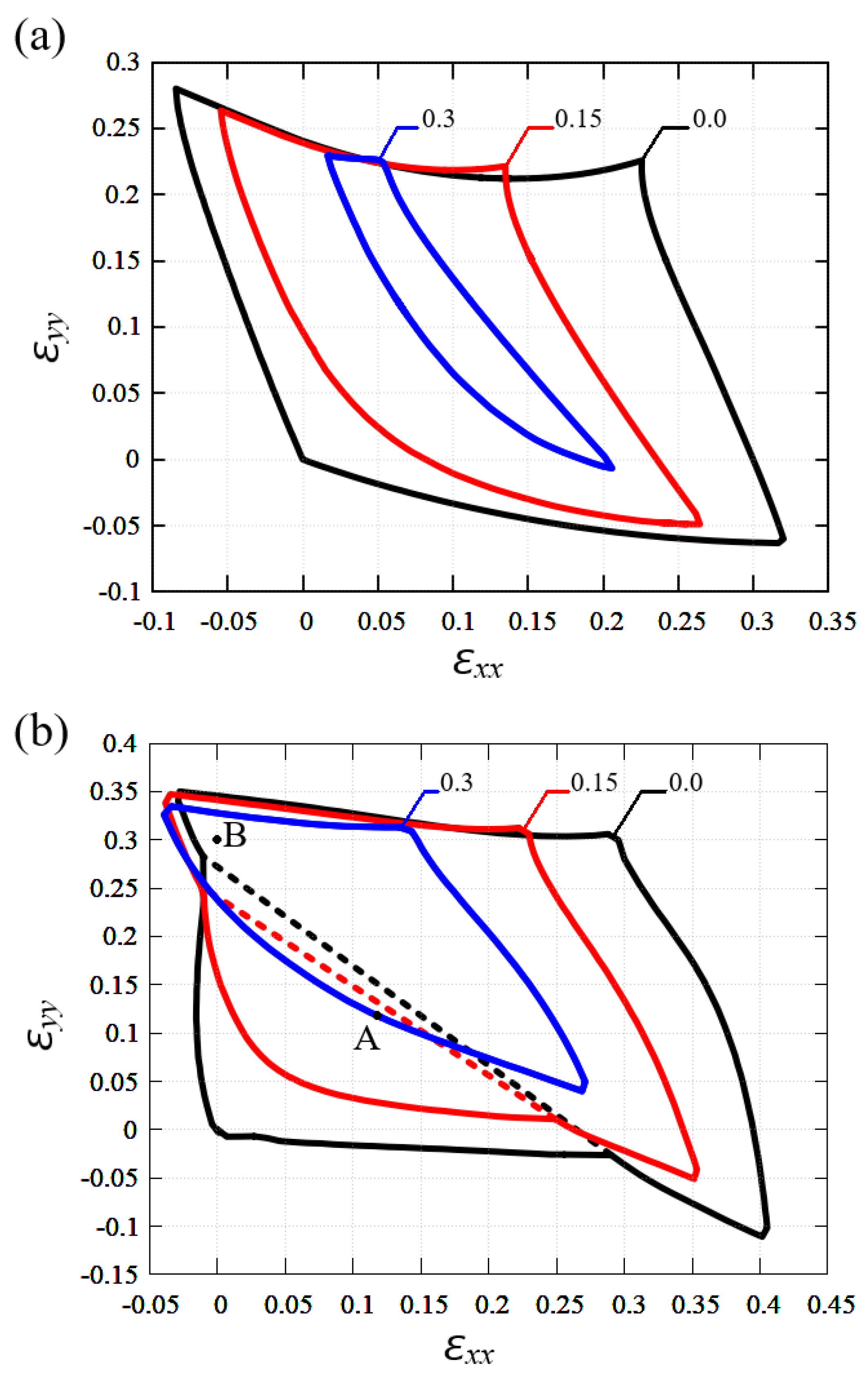
3.5. Region of Stability of Planar Stanene and Graphene in the Presence of Shear Strain
4. Conclusions and Future Work
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Matusalem, F.; Koda, D.S.; Bechstedt, F.; Marques, M.; Teles, L.K. Deposition of topological silicene, germanene and stanene on graphene-covered SiC substrates. Sci. Rep. 2017, 7, 15700. [Google Scholar] [CrossRef]
- Izhnin, I.I.; Kurbanov, K.R.; Lozovoy, K.A.; Kokhanenko, A.P.; Dirko, V.V.; Voitsekhovskii, A.V. Epitaxial fabrication of 2D materials of group IV elements. Appl. Nanosci. 2020, 10, 4375–4383. [Google Scholar] [CrossRef]
- Lozovoy, K.A.; Izhnin, I.I.; Kokhanenko, A.P.; Dirko, V.V.; Vinarskiy, V.P.; Voitsekhovskii, A.V.; Fitsych, O.I.; Akimenko, N.Y. Single-element 2D materials beyond graphene: Methods of epitaxial synthesis. Nanomaterials 2022, 12, 2221. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Wei, K.-H. A perspective on recent advances in 2D stanene nanosheets. Adv. Mater. Interfaces 2019, 6, 1900752. [Google Scholar] [CrossRef]
- Shodja, H.M.; Ojaghnezhad, F.; Etehadieh, A.; Tabatabaei, M. Elastic moduli tensors, ideal strength, and morphology of stanene based on an enhanced continuum model and first principles. Mech. Mater. 2017, 110, 1–15. [Google Scholar] [CrossRef]
- Hess, P. Thickness of elemental and binary single atomic monolayers. Nanoscale Horiz. 2020, 5, 385–399. [Google Scholar] [CrossRef]
- Matthes, L.; Pulci, O.; Bechstedt, F. Massive Dirac quasiparticles in the optical absorbance of graphene, silicene, germanene, and tinene. J. Phys. Condens. Matt. 2013, 25, 395305. [Google Scholar] [CrossRef]
- John, R.; Merlin, B. Theoretical investigation of structural, electronic, and mechanical properties of two-dimensional C, Si, Ge, Sn. Cryst. Struct. Theory Appl. 2016, 5, 43–55. [Google Scholar] [CrossRef]
- Zhu, F.-F.; Chen, W.-J.; Xu, Y.; Gao, C.-L.; Guan, D.-D.; Liu, C.-H.; Qian, D.; Zhang, S.-C.; Jia, J.-F. Epitaxial growth of two-dimensional stanene. Nat. Mater. 2015, 14, 1020–1025. [Google Scholar] [CrossRef]
- Lozovoy, K.A.; Dirko, V.V.; Vinarskiy, V.P.; Kokhanenko, A.P.; Voitsekhovskii, A.V.; Akimenko, N.Y. Two-dimensional materials of group IVA: Latest advances in epitaxial methods of growth. Russ. Phys. J. 2022, 64, 1583–1591. [Google Scholar] [CrossRef]
- Tang, P.; Chen, P.; Cao, W.; Huang, H.; Cahangirov, S.; Xian, L.; Xu, Y.; Zhang, S.-C.; Duan, W.; Rubio, A. Stable two-dimensional dumbbell stanene: A quantum spin Hall insulator. Phys. Rev. B 2014, 90, 121408. [Google Scholar] [CrossRef]
- Yuhara, J.; Fujii, Y.; Nishino, K.; Isobe, N.; Nakatake, M.; Xian, L.; Rubio, A.; Le Lay, G. Large area planar stanene epitaxially grown on Ag(111). 2D Mater. 2018, 5, 025002. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, G.; Zhang, Y.-W. Exploring Ag(111) substrate for epitaxially growing monolayer stanene: A first-principles study. Sci. Rep. 2016, 6, 29107. [Google Scholar] [CrossRef]
- Pang, W.; Nishino, K.; Ogikubo, T.; Araidai, M.; Nakatake, M.; Le Lay, G.; Yuhara, J. Epitaxial growth of honeycomb-like stanene on Au(111). Appl. Surf. Sci. 2020, 517, 146224. [Google Scholar] [CrossRef]
- Deng, J.; Xia, B.; Ma, X.; Chen, H.; Shan, H.; Zhai, X.; Li, B.; Zhao, A.; Xu, Y.; Duan, W.; et al. Epitaxial growth of ultraflat stanene with topological band inversion. Nat. Mater. 2018, 17, 1081–1086. [Google Scholar] [CrossRef]
- Yuhara, J.; Ogikubo, T.; Araidai, M.; Takakura, S.-I.; Nakatake, M.; Le Lay, G. In-plane strain-free stanene on a Pd2Sn(111) surface alloy. Phys. Rev. Mater. 2021, 5, 053403. [Google Scholar] [CrossRef]
- Tsai, H.-S.; Wang, Y.; Liu, C.; Wang, T.; Huo, M. The elemental 2D materials beyond graphene potentially used as hazardous gas sensors for environmental protection. J. Hazard. Mater. 2022, 423, 127148. [Google Scholar] [CrossRef]
- Chen, X.; Tan, C.; Yang, Q.; Meng, R.; Liang, Q.; Cai, M.; Zhang, S.; Jiang, J. Ab initio study of the adsorption of small molecules on stanene. J. Phys. Chem. C 2016, 120, 13987–13994. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, R.; Zhao, M.; Zhao, X.; An, Y.; Dai, X.; Xia, C. Effects of applied strain and electric field on small-molecule sensing by stanene monolayers. J. Mater. Sci. 2017, 52, 5083–5096. [Google Scholar] [CrossRef]
- Vovusha, H.; Hussain, T.; Sajjad, M.; Lee, H.; Karton, A.; Ahuja, R.; Schwingenschlögl, U. Sensitivity enhancement of stanene towards toxic SO2 and H2S. Appl. Surf. Sci. 2019, 495, 143622. [Google Scholar] [CrossRef]
- Abbasi, A.; Sardroodi, J.J. The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: Applications to gas sensor devices. Phys. E 2019, 108, 382–390. [Google Scholar] [CrossRef]
- Kumar, V.; Roy, D.R. Single-layer stanane as potential gas sensor for NO2, SO2, CO2 and NH3 under DFT investigation. Phys. E 2019, 110, 100–106. [Google Scholar] [CrossRef]
- Zhu, T.; Li, J. Ultra-strength materials. Prog. Mater. Sci. 2010, 55, 710–757. [Google Scholar] [CrossRef]
- Han, Y.; Gao, L.; Zhou, J.; Hou, Y.; Jia, Y.; Cao, K.; Duan, K.; Lu, Y. Deep elastic strain engineering of 2D materials and their twisted bilayers. ACS Appl. Mater. Interfaces 2022, 14, 8655–8663. [Google Scholar] [CrossRef]
- Baimova, J.A. Property control by elastic strain engineering: Application to graphene. J. Micromech. Mol. Phys. 2017, 2, 17500011. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, P.; Wang, Q.; Chen, X.; Lu, P. Strain-induced energetic and electronic properties of stanene nanomeshes. J. Comput. Electron. 2020, 19, 1357–1364. [Google Scholar] [CrossRef]
- Kalosakas, G.; Lathiotakis, N.N.; Papagelis, K. Uniaxially strained graphene: Structural characteristics and G-mode splitting. Materials 2022, 15, 67. [Google Scholar] [CrossRef]
- Wang, H.; Qian, X. Two-dimensional multiferroics in monolayer group IV monochalcogenides. 2D Mater. 2017, 4, 015042. [Google Scholar] [CrossRef]
- Qian, X.; Fu, L.; Li, J. Topological crystalline insulator nanomembrane with strain-tunable band gap. Nano Res. 2015, 8, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Iff, O.; Tedeschi, D.; Martín-Sánchez, J.; Moczała-Dusanowska, M.; Tongay, S.; Yumigeta, K.; Taboada-Gutiérrez, J.; Savaresi, M.; Rastelli, A.; Alonso-González, P.; et al. Strain-tunable single photon sources in WSe2 monolayers. Nano Lett. 2019, 19, 6931–6936. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.D.; Lindsay, L.; Shi, S.Q.; Zheng, G.P. Tensile strains give rise to strong size effects for thermal conductivities of silicene, germanene and stanene. Nanoscale 2016, 8, 3760–3767. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.V.; Korznikova, E.A.; Krivtsov, A.M.; Dmitriev, S.V. Longitudinal stiffness and thermal conductivity of twisted carbon nanoribbons. Eur. J. Mech. A/Solids 2020, 80, 103920. [Google Scholar] [CrossRef]
- Zhou, K.; Liu, B.; Cai, Y.; Dmitriev, S.V.; Li, S. Modelling of low-dimensional functional nanomaterials. Phys. Status Solidi RRL 2022, 16, 2100654. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, K. Recent progress on graphene-analogous 2D nanomaterials: Properties, modeling and applications. Prog. Mater. Sci. 2019, 100, 99–169. [Google Scholar] [CrossRef]
- Savin, A.V.; Korznikova, E.A.; Dmitriev, S.V. Dynamics of surface graphene ripplocations on a flat graphite substrate. Phys. Rev. B 2019, 99, 235411. [Google Scholar] [CrossRef]
- Modarresi, M.; Kakoee, A.; Mogulkoc, Y.; Roknabadi, M.R. Effect of external strain on electronic structure of stanene. Comput. Mater. Sci. 2015, 101, 164–167. [Google Scholar] [CrossRef]
- Iskandarov, A.M.; Dmitriev, S.V.; Umeno, Y. Temperature effect on ideal shear strength of Al and Cu. Phys. Rev. B 2011, 84, 224118. [Google Scholar] [CrossRef]
- Shi, Z.; Singh, C.V. Ideal strength of two-dimensional stanene could possibly exceed Griffith theoretical strength estimate. Nanoscale 2017, 9, 7055–7062. [Google Scholar] [CrossRef]
- Mojumder, S.; Amin, A.A.; Islam, M.M. Mechanical properties of stanene under uniaxial and biaxial loading: A molecular dynamics study. J. Appl. Phys. 2015, 118, 124305. [Google Scholar] [CrossRef] [Green Version]
- Mahata, A.; Mukhopadhyay, T. Probing the chirality-dependent elastic properties and crack propagation behavior of single and bilayer stanene. Phys. Chem. Chem. Phys. 2018, 20, 22768–22782. [Google Scholar] [CrossRef] [PubMed]
- Hess, P. Predictive modeling of intrinsic strengths for several groups of chemically related monolayers by a reference model. Phys. Chem. Chem. Phys. 2018, 20, 7604–7611. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, T.; Mahata, A.; Adhikari, S.; Asle Zaeem, M. Probing the shear modulus of two-dimensional multiplanar nanostructures and heterostructures. Nanoscale 2018, 10, 5280–5294. [Google Scholar] [CrossRef] [PubMed]
- Eugster, S.R.; Dell’Isola, F.; Steigmann, D.J. Continuum theory for mechanical metamaterials with a cubic lattice substructure. Math. Mech. Complex Syst. 2019, 7, 75–98. [Google Scholar] [CrossRef]
- Pavlov, I.S.; Vasiliev, A.A.; Porubov, A.V. Dispersion properties of the phononic crystal consisting of ellipse-shaped particles. J. Sound Vib. 2016, 384, 163–176. [Google Scholar] [CrossRef]
- Vasiliev, A.A.; Pavlov, I.S. Auxetic properties of chiral hexagonal Cosserat lattices composed of finite-sized particles. Phys. Status Solidi B 2020, 257, 1900389. [Google Scholar] [CrossRef]
- Ghadiyali, M.; Chacko, S. Band splitting in bilayer stanene electronic structure scrutinized via first principle DFT calculations. Comput. Condens. Matter 2018, 17, e00341. [Google Scholar] [CrossRef]
- Baimova, J.A.; Dmitriev, S.V.; Zhou, K.; Savin, A.V. Unidirectional ripples in strained graphene nanoribbons with clamped edges at zero and finite temperatures. Phys. Rev. B 2012, 86, 035427. [Google Scholar] [CrossRef]
- Savin, A.V.; Kivshar, Y.S.; Hu, B. Suppression of thermal conductivity in graphene nanoribbons with rough edges. Phys. Rev. B 2010, 82, 195422. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, A.S.; Suryanarayana, P. Symmetry-adapted real-space density functional theory for cylindrical geometries: Application to large group-IV nanotubes. Phys. Rev. B 2019, 100, 125143. [Google Scholar] [CrossRef] [Green Version]
- Sadki, K.; Kourra, M.H.; Drissi, L.B. Non linear and thermoelastic behaviors of group-IV hybrid 2D nanosheets. Superlattice. Microstructur. 2019, 132, 106172. [Google Scholar] [CrossRef]
- Tu, Z.; Wu, M. Ultrahigh-strain ferroelasticity in two-dimensional honeycomb monolayers: From covalent to metallic bonding. Sci. Bull. 2020, 65, 147–152. [Google Scholar] [CrossRef]
- Si, C.; Wang, X.-D.; Fan, Z.; Feng, Z.-H.; Cao, B.-Y. Impacts of potential models on calculating the thermal conductivity of graphene using non-equilibrium molecular dynamics simulations. Int. J. Heat Mass Transf. 2017, 107, 450–460. [Google Scholar] [CrossRef]
- Rowe, P.; Csányi, G.; Alfè, D.; Michaelides, A. Development of a machine learning potential for graphene. Phys. Rev. B 2018, 97, 054303. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater. Horiz. 2020, 7, 2359–2367. [Google Scholar] [CrossRef]
- Rowe, P.; Deringer, V.L.; Gasparotto, P.; Csányi, G.; Michaelides, A. An accurate and transferable machine learning potential for carbon. J. Chem. Phys. 2020, 153, 034702. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yan, B.; Zhang, H.-J.; Wang, J.; Xu, G.; Tang, P.; Duan, W.; Zhang, S.-C. Large-Gap Quantum Spin Hall Insulators in Tin Films. Phys. Rev. Lett. 2013, 111, 136804. [Google Scholar] [CrossRef]
- Lyu, J.-K.; Zhang, S.-F.; Zhang, C.-W.; Wang, P.-J. Stanene: A Promising Material for New Electronic and Spintronic Applications. Ann. Phys. 2019, 531, 1900017. [Google Scholar] [CrossRef]
- van den Broek, B.; Houssa, M.; Scalise, E.; Pourtois, G.; Afanasev, V.V.; Stesmans, A. Two-dimensional hexagonal tin: Ab initio geometry, stability, electronic structure and functionalization. 2D Mater. 2014, 1, 021004. [Google Scholar] [CrossRef]
- van den Broek, B.; Houssa, M.; Scalise, E.; Pourtois, G.; Afanasev, V.V. Stability and electronic structure of two-dimensional allotropes of group-IV materials. Phys. Rev. B 2015, 92, 045436. [Google Scholar]
- Rivero, P.; Yan, J.A.; Garc’ia-Su’arez, V.M.; Ferrer, J.; Barraza-Lopez, S. Stability and properties of high-buckled two-dimensional tin and lead. Phys. Rev. B 2014, 90, 241408(R). [Google Scholar] [CrossRef] [Green Version]
- Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 1988, 37, 6991–7000. [Google Scholar] [CrossRef] [PubMed]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Narayanan, B.; Sasikumar, K.; Mei, Z.-G.; Kinaci, A.; Sen, F.G.; Davis, M.J.; Gray, S.K.; Chan, M.K.Y.; Sankaranarayanan, S.K.R.S. Development of a Modified Embedded Atom Force Field for Zirconium Nitride Using Multi-Objective Evolutionary Optimization. J. Phys. Chem. C 2016, 120, 17475–17483. [Google Scholar] [CrossRef]
- Lindsay, L.; Broido, D.A. Optimized Tersoff and Brenner empirical potential parameters for lattice dynamics and phonon thermal transport in carbon nanotubes and graphene. Phys. Rev. B 2010, 81, 205441. [Google Scholar] [CrossRef]
- Cherukara, M.J.; Narayanan, B.; Kinaci, A.; Sasikumar, K.; Gray, S.K.; Chan, M.K.Y.; Sankaranarayanan, S.K.R.S. Ab-initio based bond order potential to investigate low thermal conductivity of stanene Nanostructures. J. Chem. Phys. 2016, 7, 3752–3759. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rahaman, O.; Makaremi, M.; Dianat, A.; Cuniberti, G.; Rabczuk, T. First-principles investigation of mechanical properties of silicene, germanene and stanene. Phys. E 2017, 87, 228–232. [Google Scholar] [CrossRef]
- Tao, L.; Yang, C.; Wu, L.; Han, L.; Song, Y.; Wang, S.; Lu, P. Tension-induced mechanical properties of stanene. Mod. Phys. Lett. B 2016, 30, 1650146. [Google Scholar] [CrossRef]
- Cao, G. Atomistic studies of mechanical properties of graphene. Polymers 2014, 6, 2404–2432. [Google Scholar] [CrossRef]
- Dmitriev, S.V.; Baimova, J.A.; Savin, A.V.; Kivshar, Y.S. Ultimate strength, ripples, sound velocities, and density of phonon states of strained graphene. Comput. Mater. Sci. 2012, 53, 194–203. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458–9471. [Google Scholar] [CrossRef] [PubMed]
- Savin, A.V.; Kivshar, Y.S. Modeling of second sound in carbon nanostructures. Phys. Rev. B 2022, 105, 205414. [Google Scholar] [CrossRef]
- Savin, A.V.; Korznikova, E.A.; Dmitriev, S.V. Plane vibrational modes and localized nonlinear excitations in carbon nanotube bundle. J. Sound Vib. 2022, 520, 116627. [Google Scholar] [CrossRef]
- Savin, A.V. Multistability of carbon nanotube packings on flat substrate. Phys. Status Solidi-RRL 2022, 16, 2100437. [Google Scholar] [CrossRef]
- Savin, A.V.; Dmitriev, S.V. The frequency spectrum of rotobreathers with many degrees of freedom. Europhys. Lett. 2022, 137, 36005. [Google Scholar] [CrossRef]
- Shcherbinin, S.A.; Zhou, K.; Dmitriev, S.V.; Korznikova, E.A.; Davletshin, A.R.; Kistanov, A.A. Two-dimensional black phosphorus carbide: Rippling and formation of nanotubes. J. Phys. Chem. C 2020, 124, 10235–10243. [Google Scholar] [CrossRef]
- Rysaeva, L.K.; Bachurin, D.V.; Murzaev, R.T.; Abdullina, D.U.; Korznikova, E.A.; Mulyukov, R.R.; Dmitriev, S.V. Evolution of the carbon nanotube bundle structure under biaxial and shear strains. Facta Univ. Ser. Mech. Eng. 2020, 18, 525–536. [Google Scholar] [CrossRef]
- Rysaeva, L.K.; Korznikova, E.A.; Murzaev, R.T.; Abdullina, D.U.; Kudreyko, A.A.; Baimova, J.A.; Lisovenko, D.S.; Dmitriev, S.V. Elastic damper based on the carbon nanotube bundle. Facta Univ. Ser. Mech. Eng. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Galiakhmetova, L.K.; Pavlov, I.S.; Bayazitov, A.M.; Kosarev, I.V.; Dmitriev, S.V. Mechanical properties of cubene crystals. Materials 2022, 15, 4871. [Google Scholar] [CrossRef]
- Shepelev, I.A.; Chetverikov, A.P.; Dmitriev, S.V.; Korznikova, E.A. Shock waves in graphene and boron nitride. Comput. Mater. Sci. 2020, 177, 109549. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | 3.0 |
| 1.0 | |
| (Å) | 0.198543 |
| c | 501643.0 |
| d | 155.4496 |
| −0.321475 | |
| N | 1.423982 |
| 0.006901 | |
| (Å) | 0.221467 |
| B (eV) | 4.534091 |
| R (Å) | 3.59983 |
| D (Å) | 0.723836 |
| (Å) | 2.824898 |
| A (eV) | 638.6396 |
| Point | Angles in Degrees | Bond Length in Å | U (eV) | h (Å) | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | |||||||
| A | 0.0 | 119.20 | 119.20 | 119.20 | 3.027 | 3.027 | 3.027 | −3.11 | 0.32 |
| 0.15 | 119.01 | 128.44 | 110.12 | 3.108 | 3.038 | 2.973 | −3.06 | 0.27 | |
| 0.3 | 119.02 | 138.71 | 102.27 | 3.208 | 3.039 | 2.911 | −2.88 | 0.0 | |
| B | 0.0 | 101.66 | 130.19 | 112.15 | 3.043 | 3.360 | 2.989 | −2.73 | 0.0 |
| 0.15 | 101.37 | 138.98 | 119.65 | 3.090 | 3.366 | 2.970 | −2.72 | 0.0 | |
| 0.3 | 100.66 | 148.08 | 111.26 | 3.215 | 3.381 | 2.924 | −2.60 | 0.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosarev, I.V.; Dmitriev, S.V.; Semenov, A.S.; Korznikova, E.A. Stability of Strained Stanene Compared to That of Graphene. Materials 2022, 15, 5900. https://doi.org/10.3390/ma15175900
Kosarev IV, Dmitriev SV, Semenov AS, Korznikova EA. Stability of Strained Stanene Compared to That of Graphene. Materials. 2022; 15(17):5900. https://doi.org/10.3390/ma15175900
Chicago/Turabian StyleKosarev, Igor V., Sergey V. Dmitriev, Alexander S. Semenov, and Elena A. Korznikova. 2022. "Stability of Strained Stanene Compared to That of Graphene" Materials 15, no. 17: 5900. https://doi.org/10.3390/ma15175900