
Dendritic Iron(III) Carbazole Complexes: Structural, Optical, and Magnetic Characteristics

 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physical Measurements
2.3. DFT Calculations
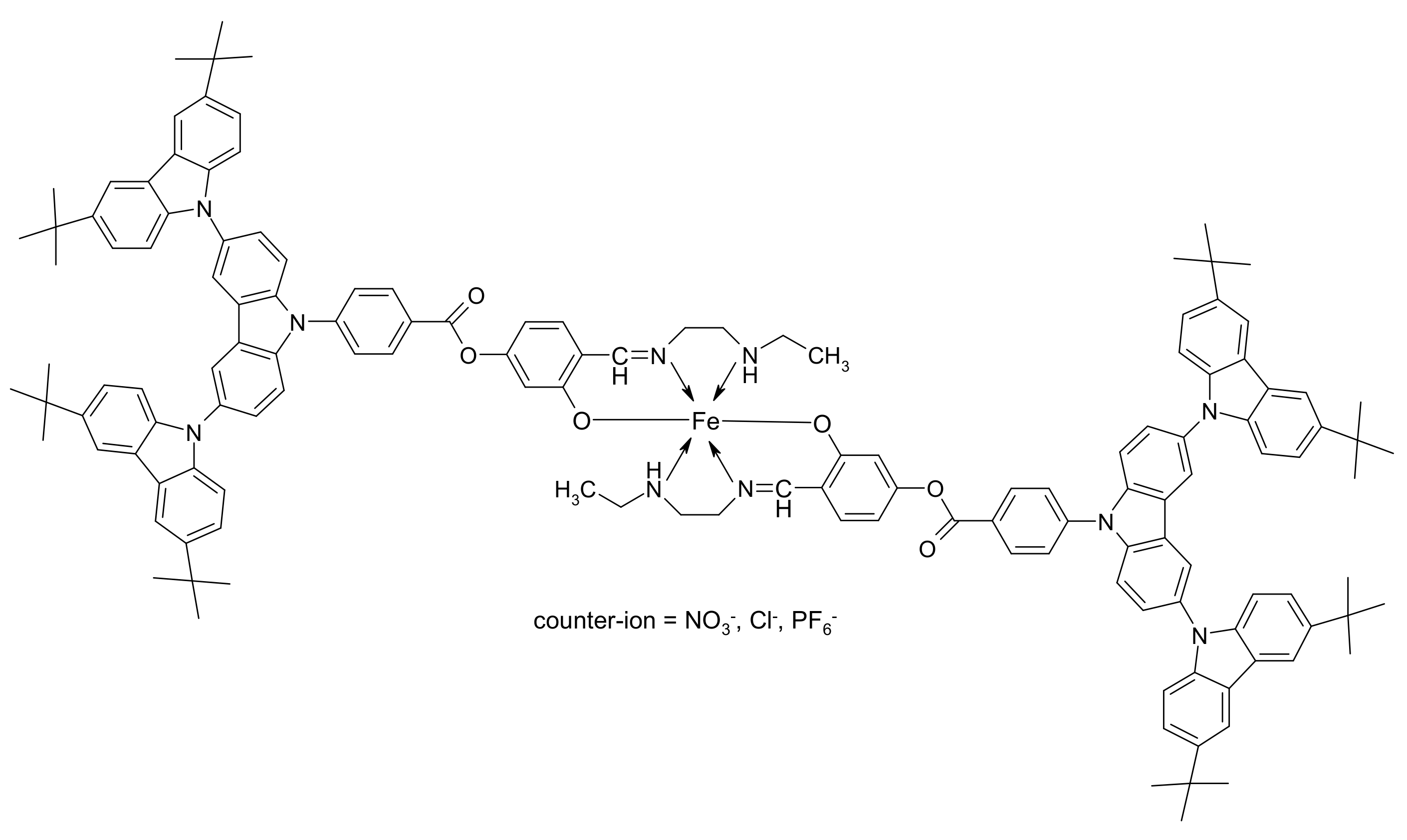
2.4. Synthesis: General Procedure
- [Fe(L)2]NO3 (1). Product is a light-brown fine-dispersed powder. Yield is 85.78% (0.39 g). FT-IR (KBr, ν, cm–1): 3424 (m, NH), 3045 (m, Ph–H), 2960–2865 (s, CH2, CH3), 1722 (s, –COO–), 1607 (s, HC=N), 1388 (s, NO3−). Anal.: Calculated (%): C, 77.08; H, 6.65; N, 7.07; O, 6.11; Fe, 2.59. C140H144N10O6Fe·NO3. Found (%): C, 77.16; H, 6.69; N, 7.02; O, 6.59; Fe, 2.54. MS (m/z): Calculated 1032.84; Found 1034.45 [L]+.
- [Fe(L)2]Cl (2). Product is a grey fine-dispersed powder. Yield is 70.61% (0.32 g). FT-IR (KBr, ν, cm–1): 3421 (m, NH), 3044 (m, Ph–H), 2959–2864 (s, CH2, CH3), 1720 (s, –COO–), 1609 (s, HC=N). Anal.: Calculated (%): C, 78.03; H, 6.74; N, 6.5; O, 4.45; Fe, 2.63; Cl, 1.65. C140H144N10O6Fe·Cl. Found (%): C, 77.98; H, 6.78; N, 6.43; O, 4.67; Fe, 2.56; Cl, 1.58. MS (m/z): Calculated 1032.84; Found 1034.81 [L]+.
- [Fe(L)2]PF6 (3). Product is a grey fine-dispersed powder. Yield is 70.83% (0.34 g). FT-IR (KBr, ν, cm–1): 3419 (m, NH), 3050 (s, Ph–H), 2960–2865 (s, CH2, CH3), 1721 (s, –COO–), 1605 (s, HC=N), 839 (m, PF6−), 559 (m, PF6−). Anal.: Calculated (%): C, 74.26; H, 6.41; N, 6.18; O, 4.24; Fe, 2.51; P, 1.37; F, 5.03. C140H144N10O6Fe·PF6. Found (%): C, 74.28; H, 6.44; N, 6.08; O, 4.11; Fe, 2.63; P, 1.48; F, 4.98. MS (m/z): Calculated 1032.84; Found 1034.16 [L]+.
3. Results
3.1. Infrared Spectroscopy
3.2. 1H NMR Spectroscopy
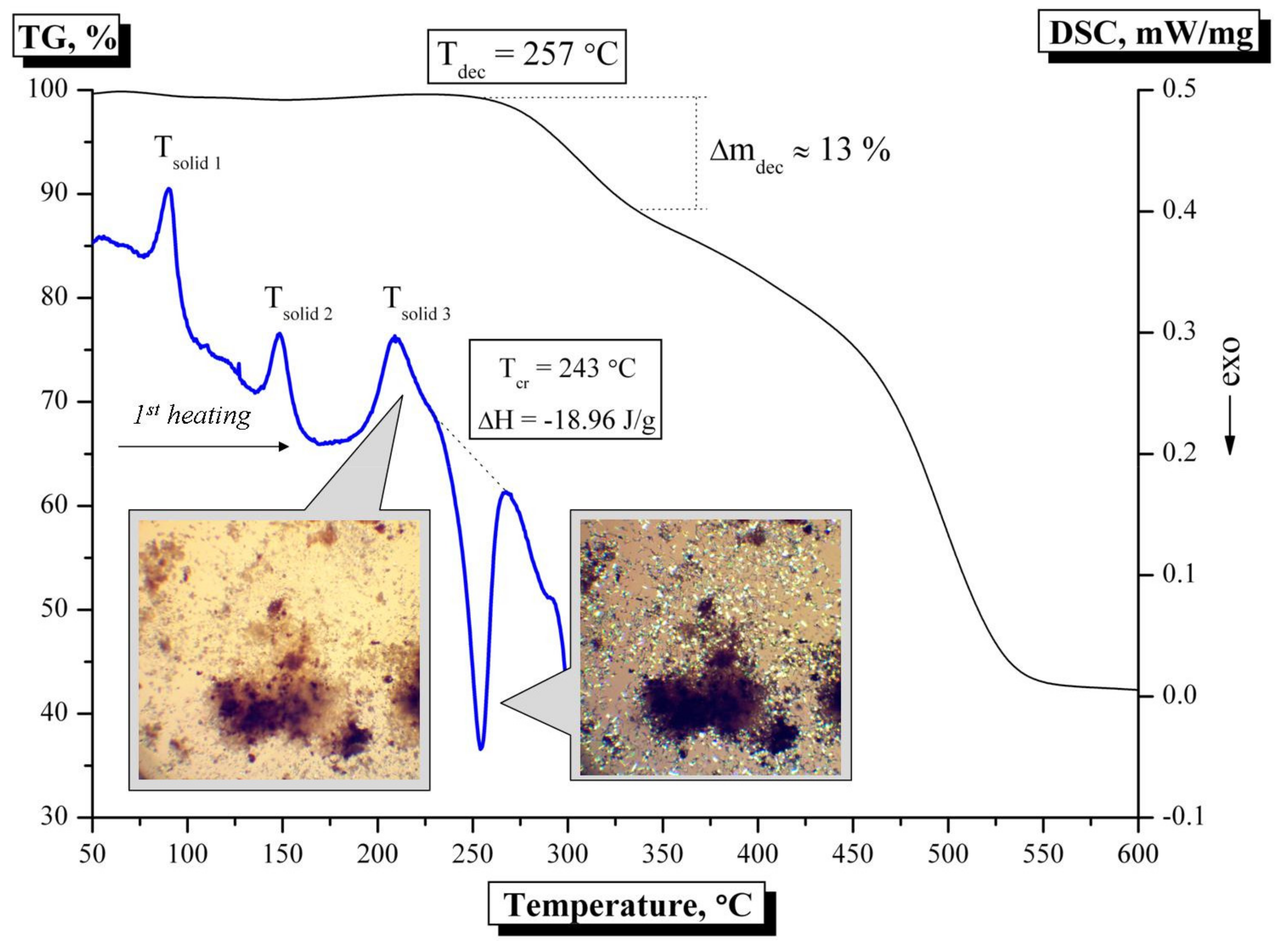
3.3. Thermal Analysis
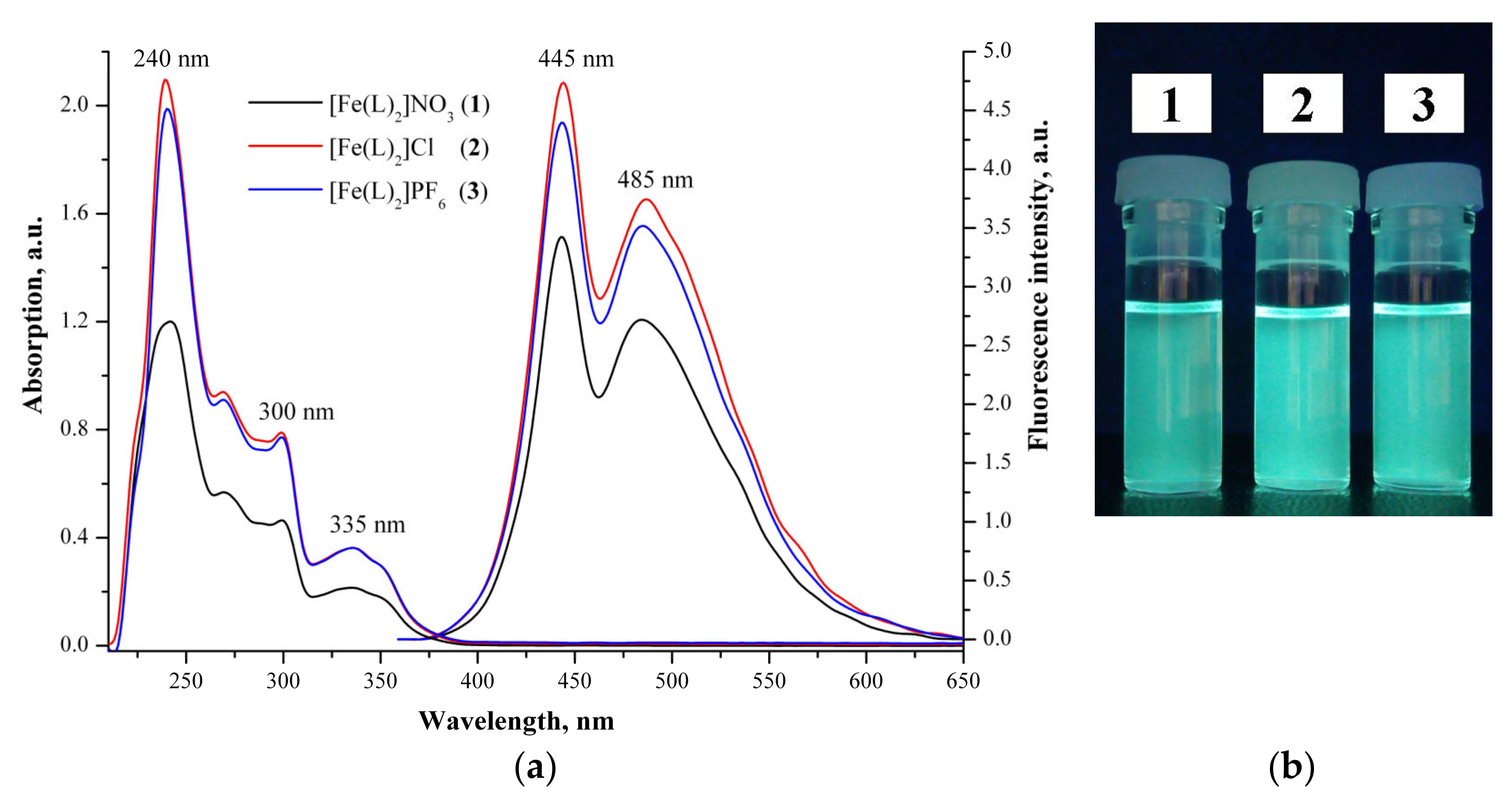
3.4. Optical Properties
3.5. TD-DFT Calculations
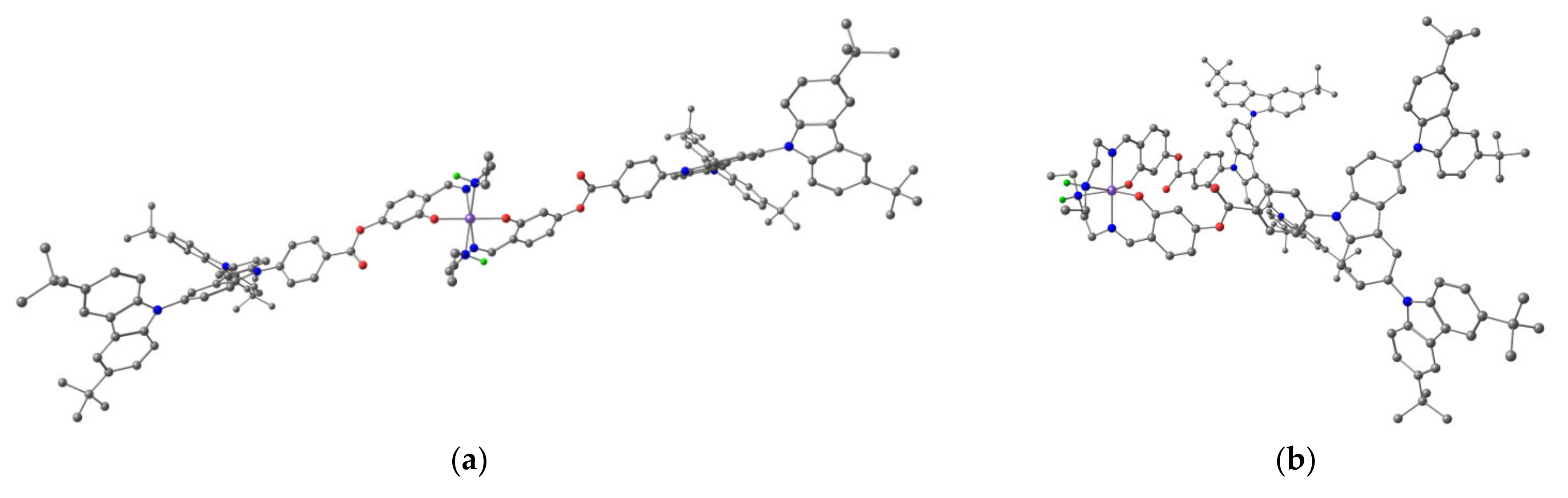
3.6. X-ray Powder Diffraction Analysis
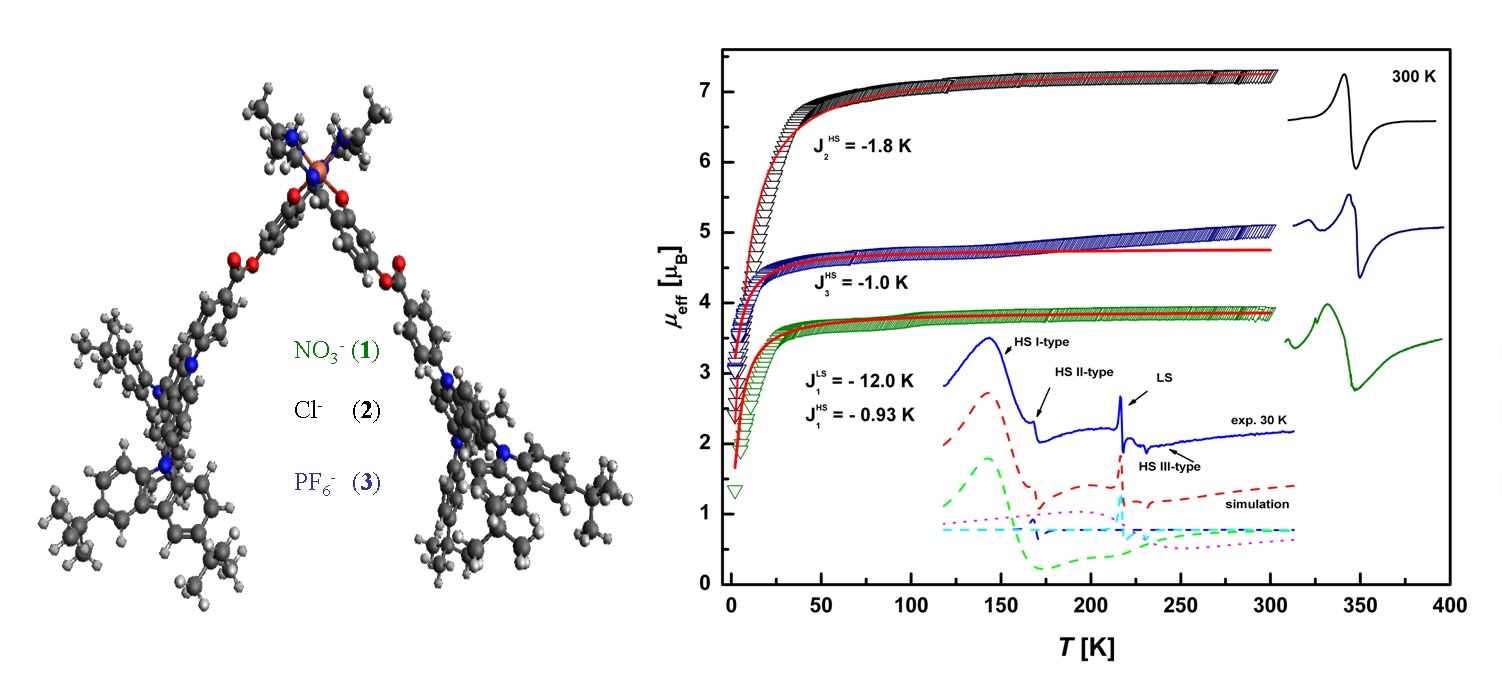
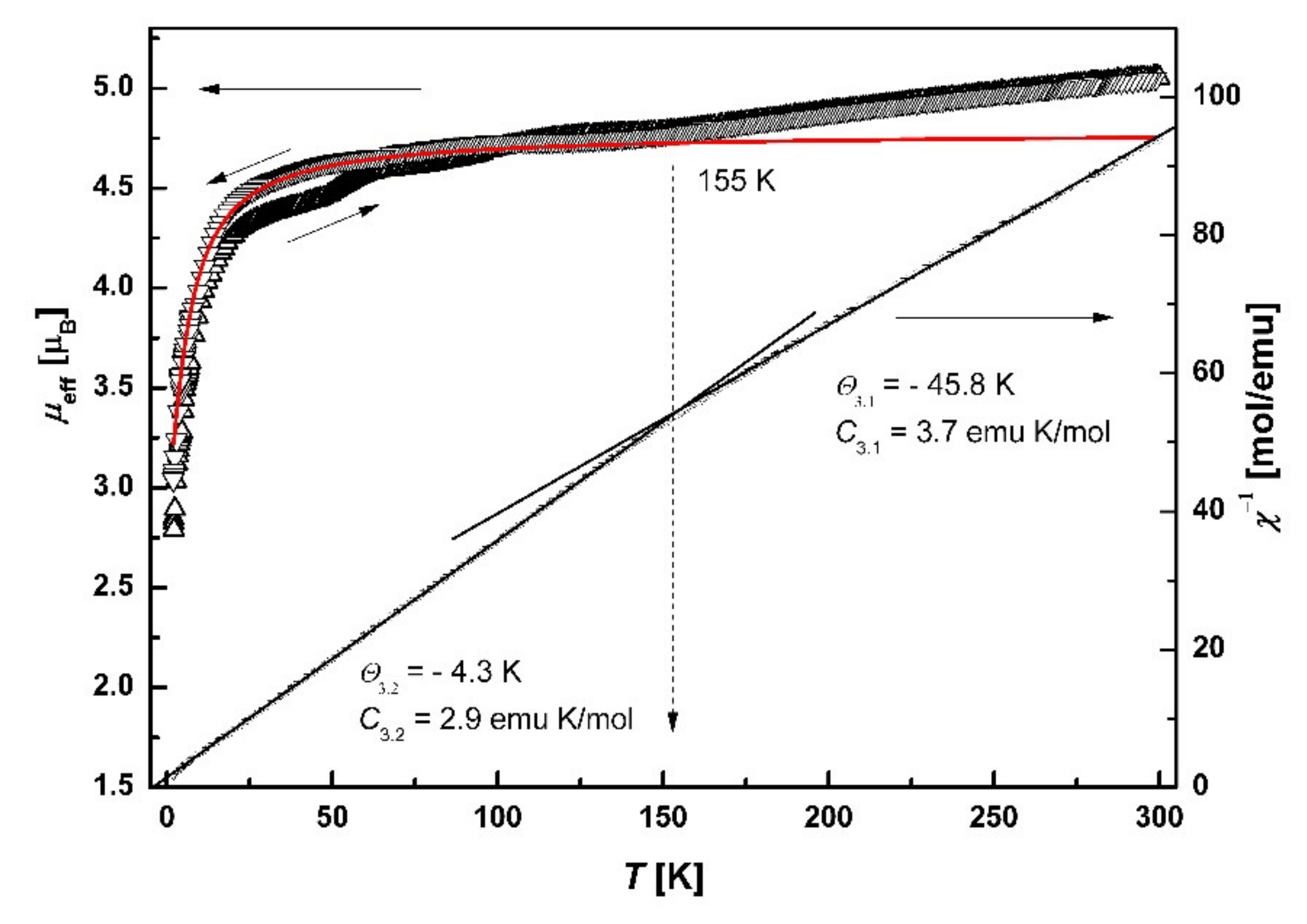
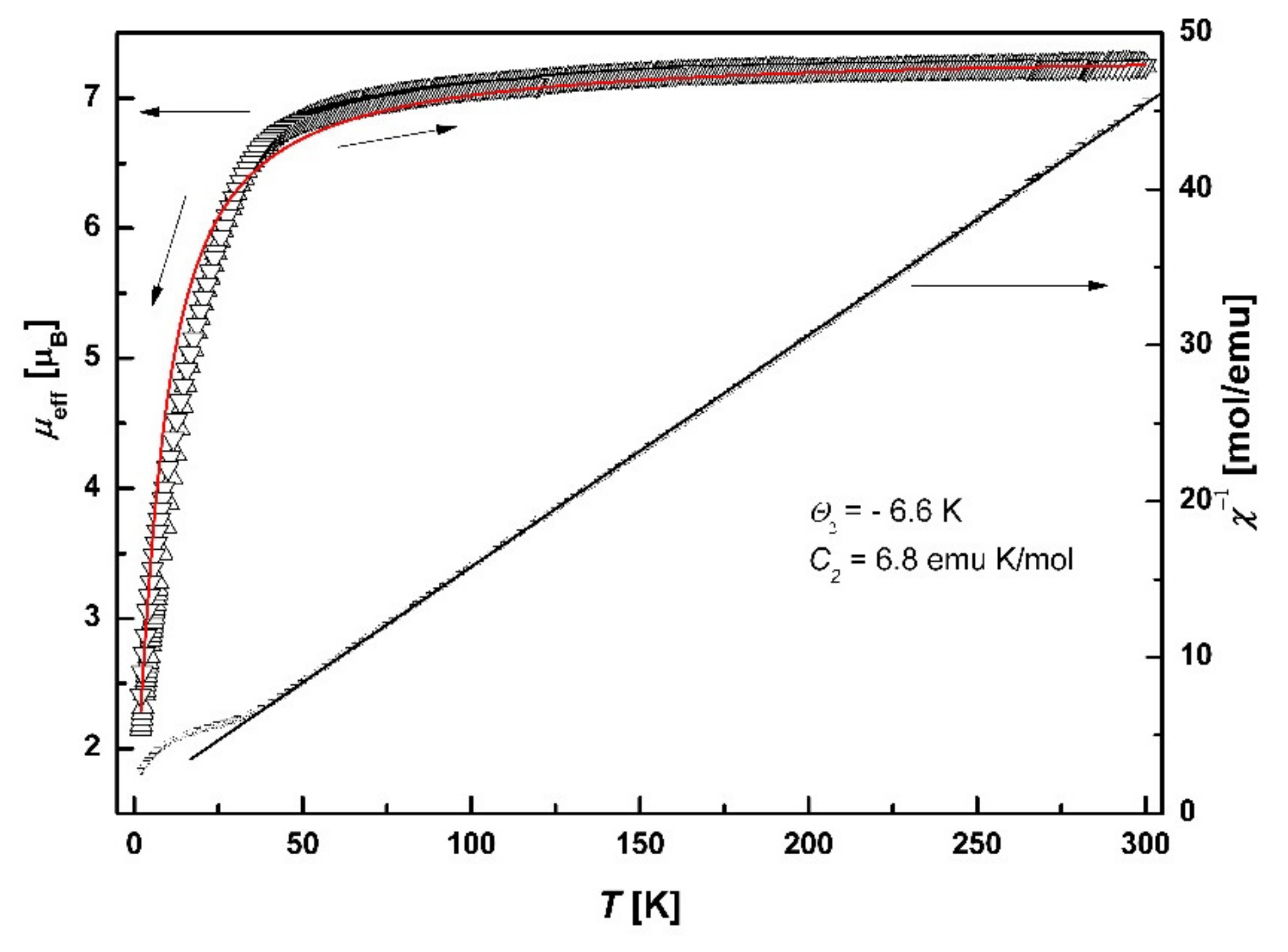
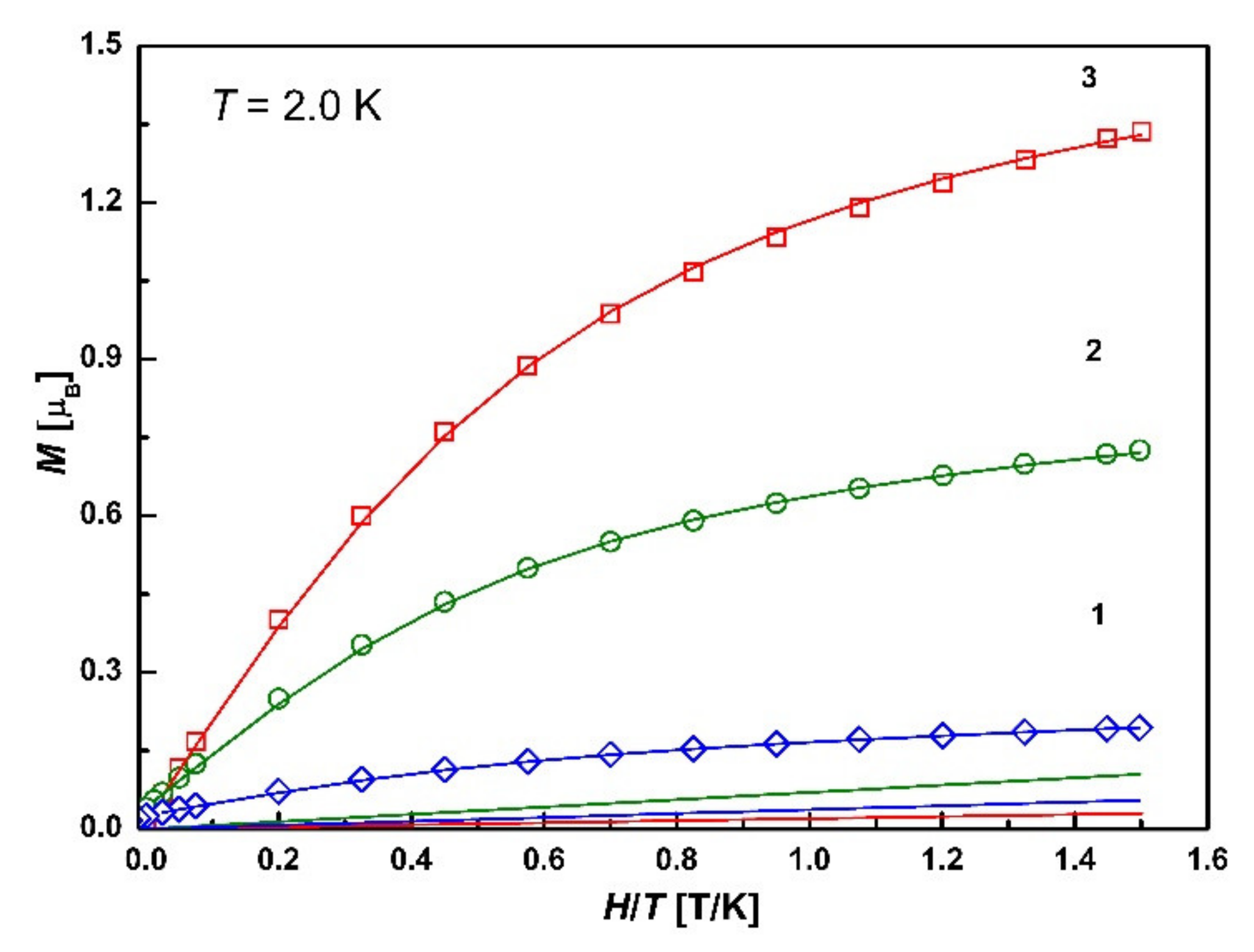
3.7. Magnetic Properties
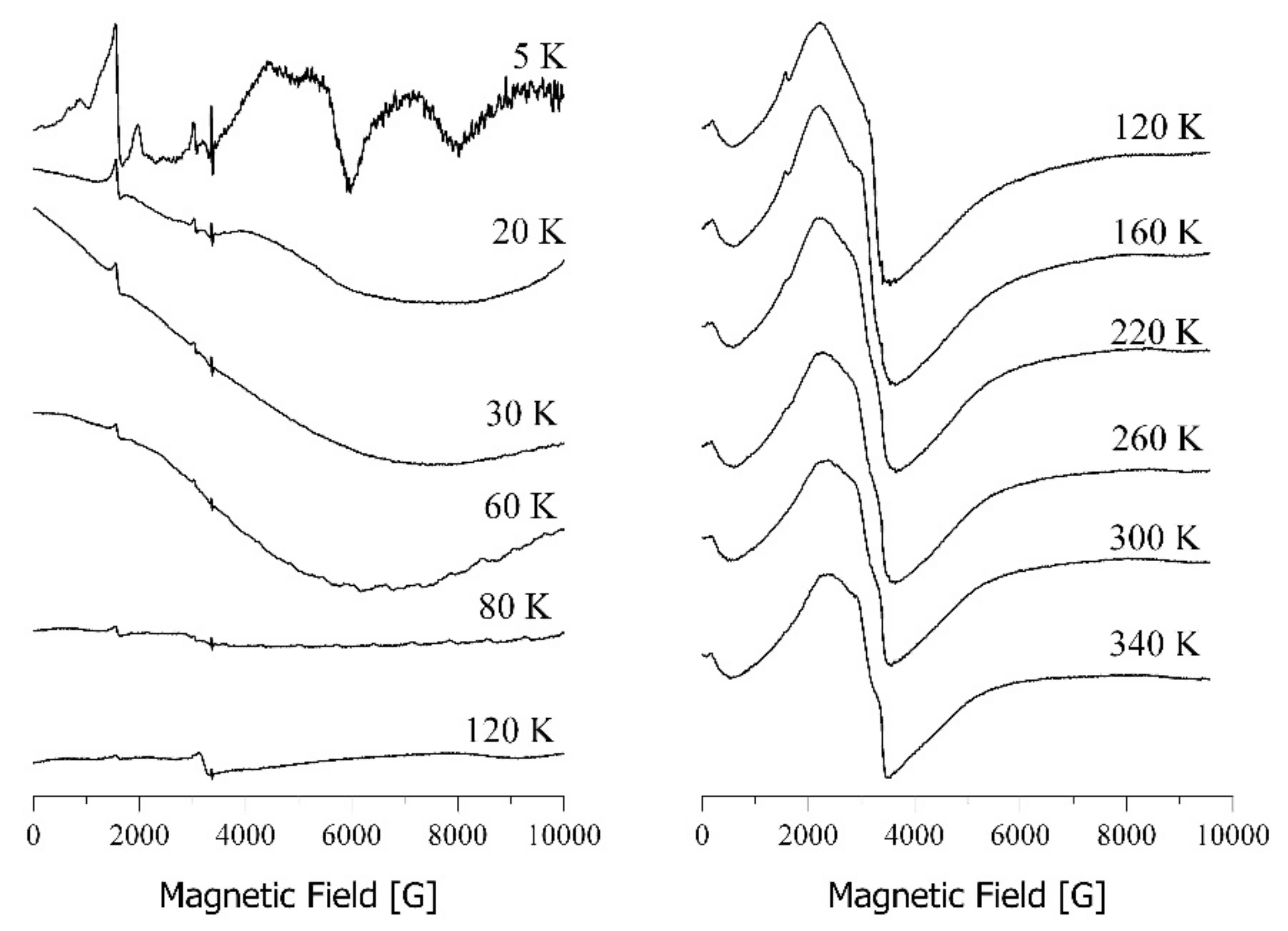
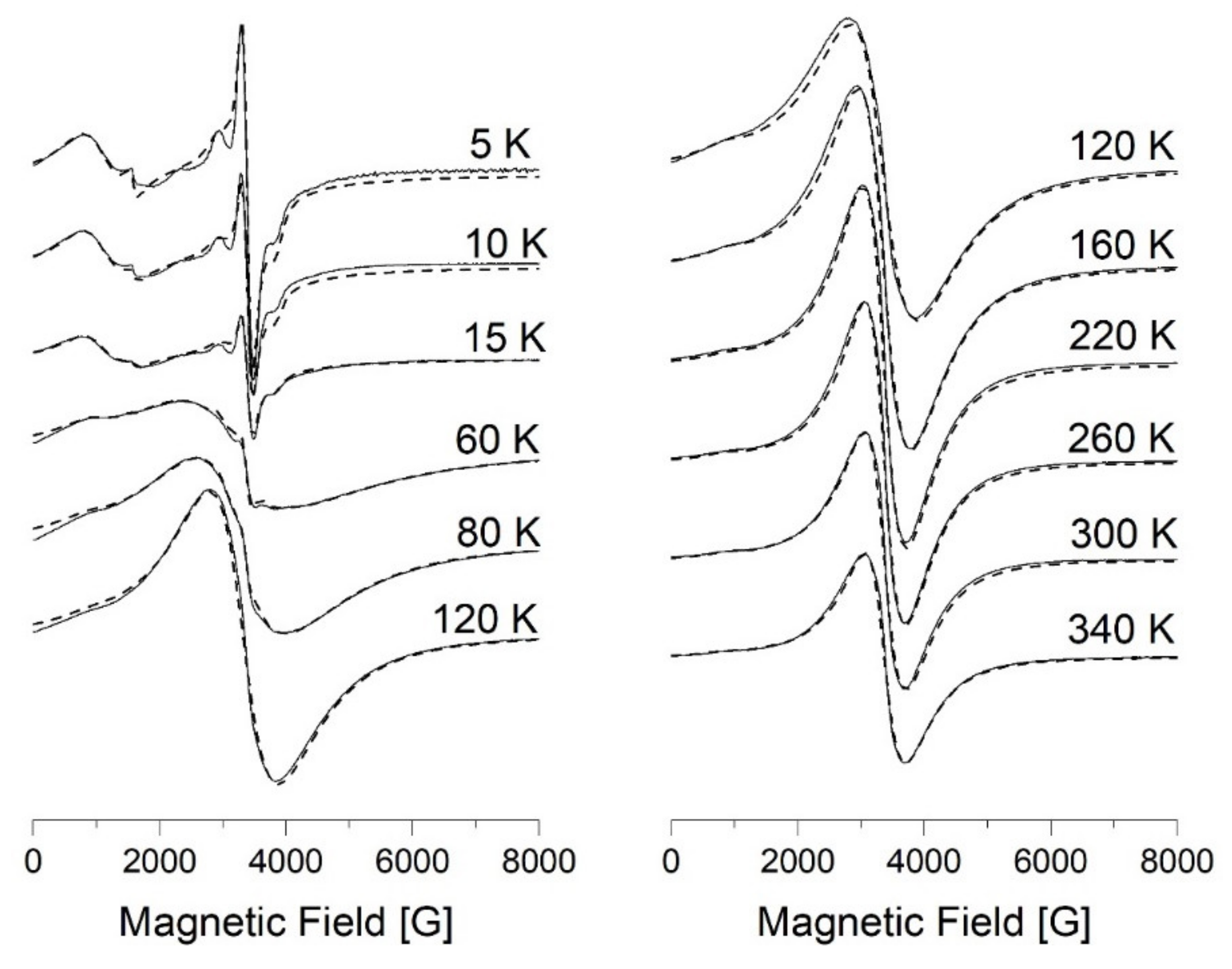
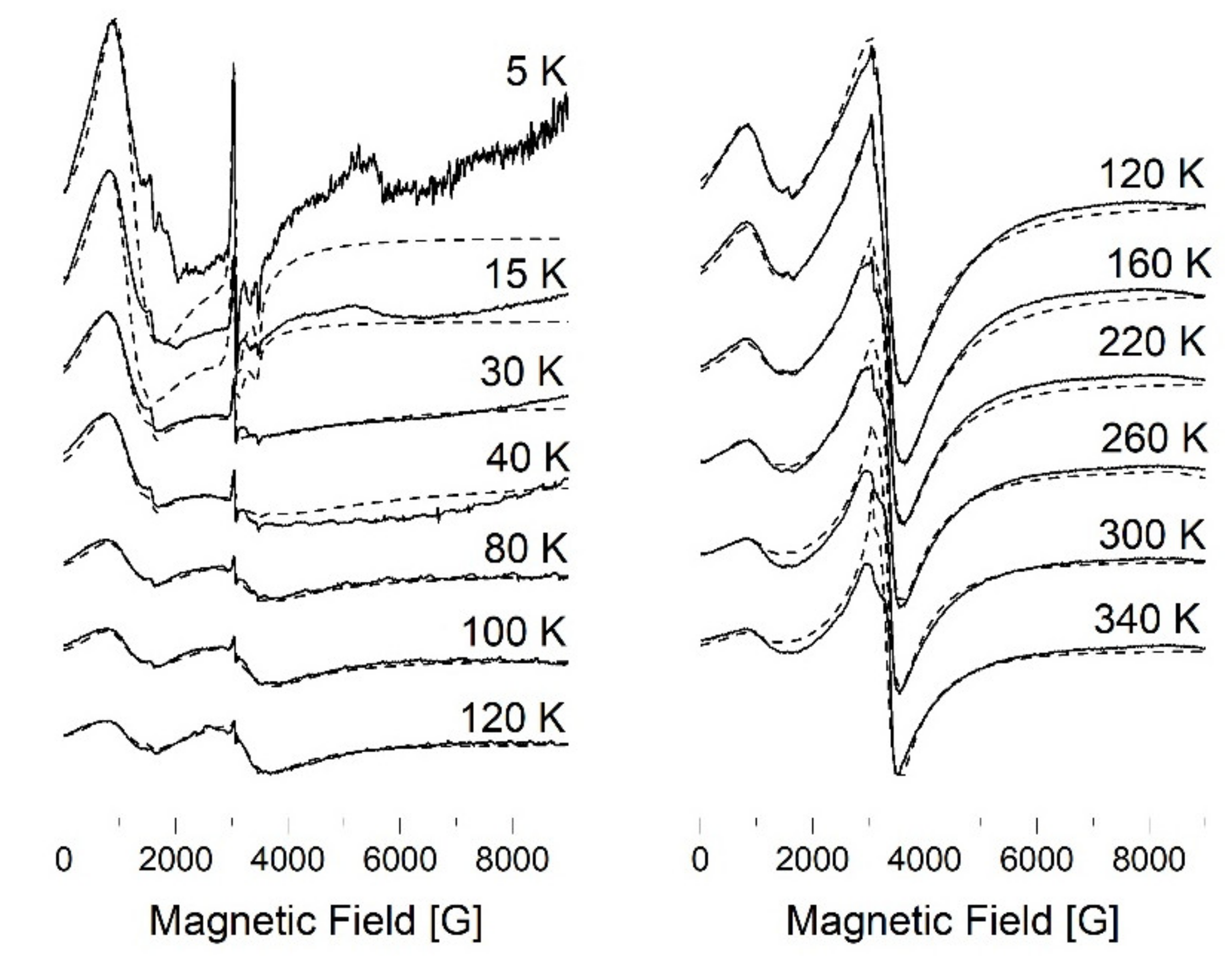
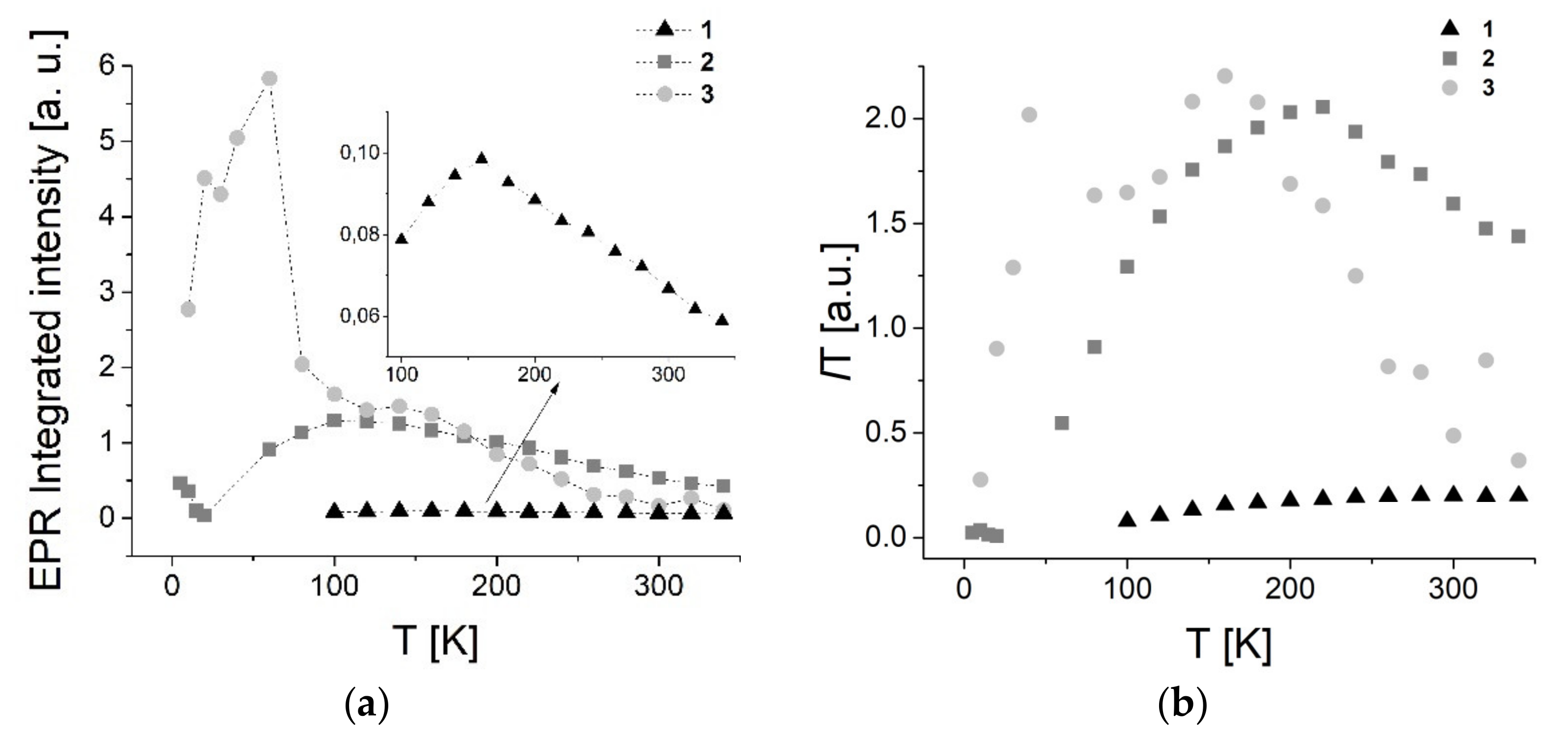
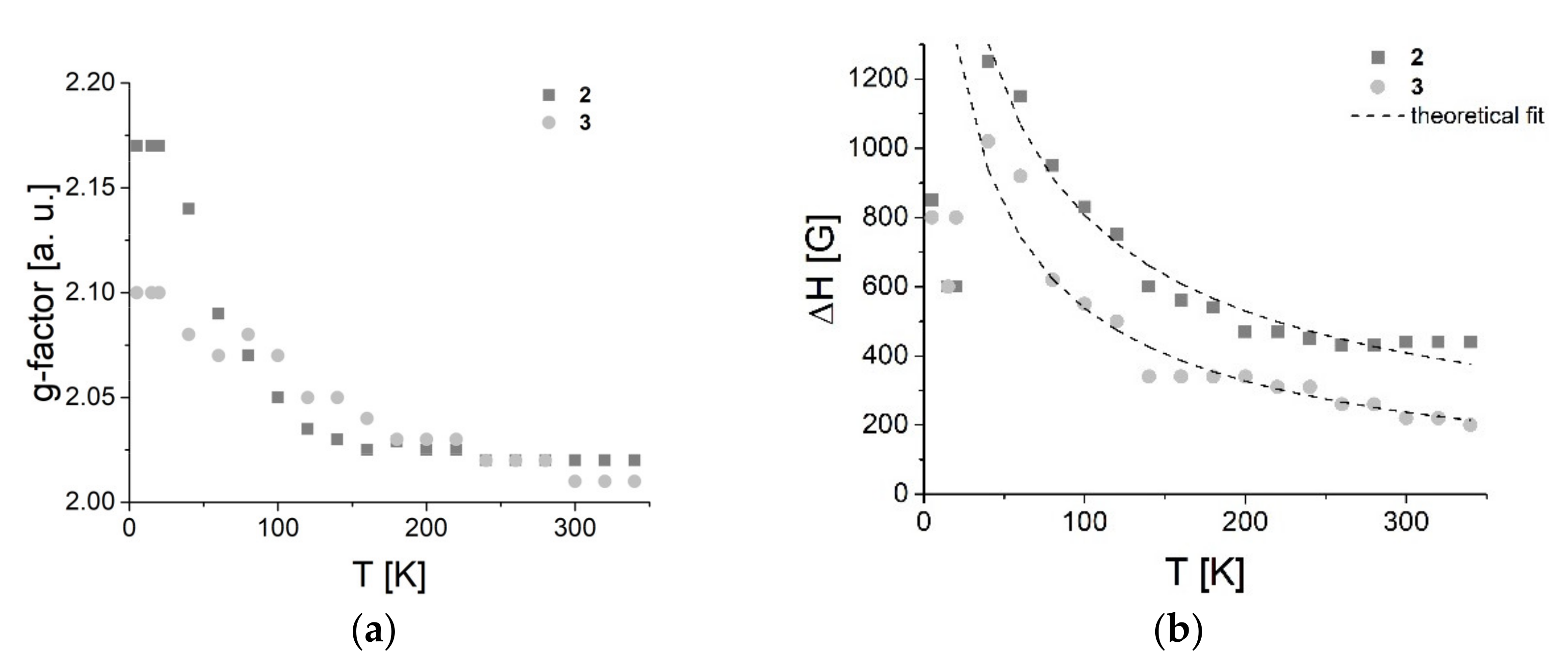
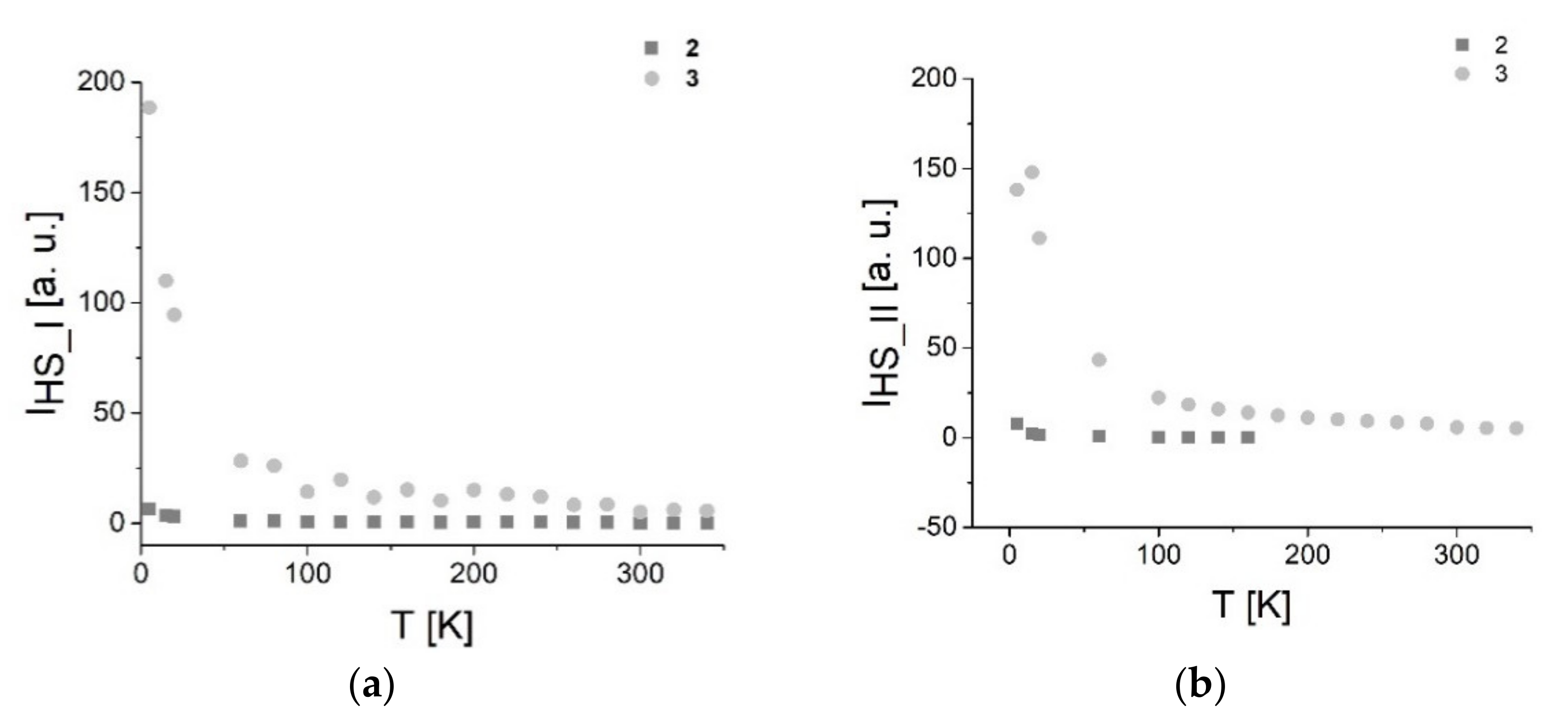
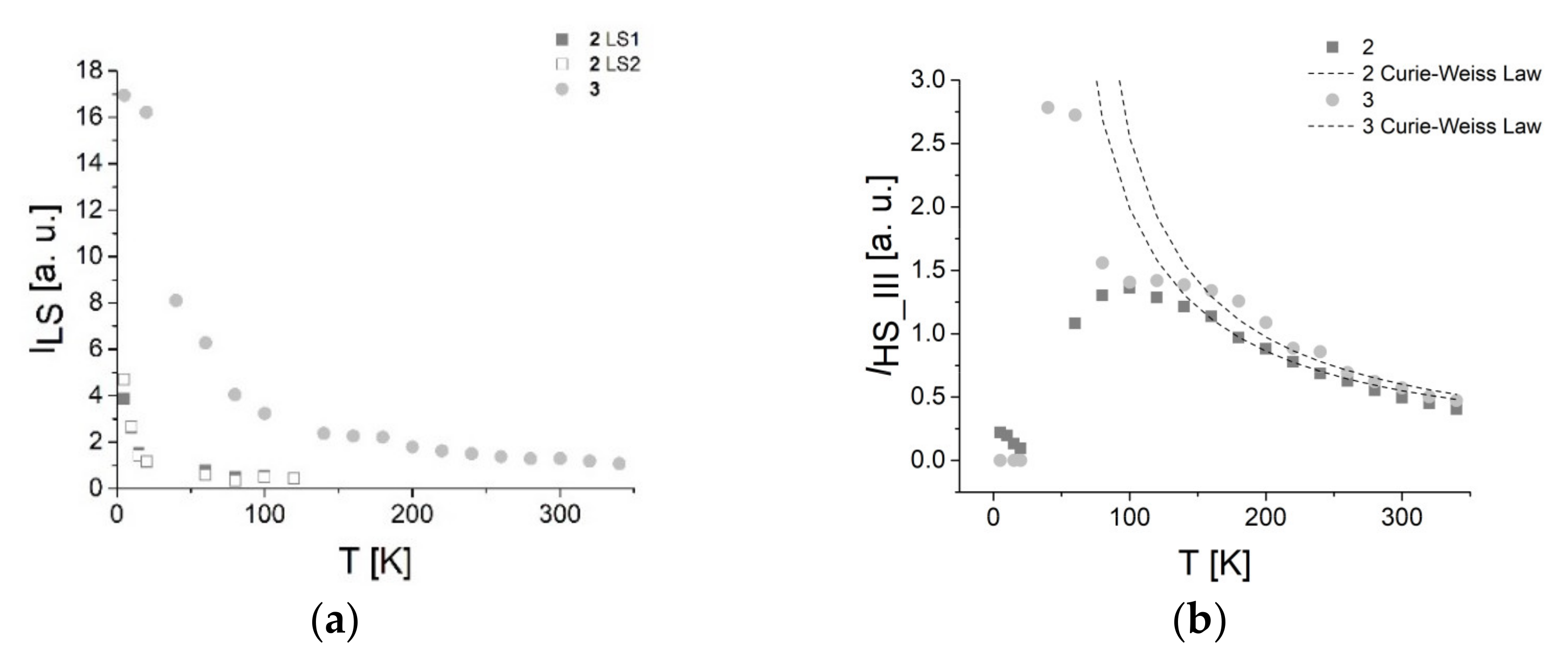
3.7.1. EPR Spectroscopy
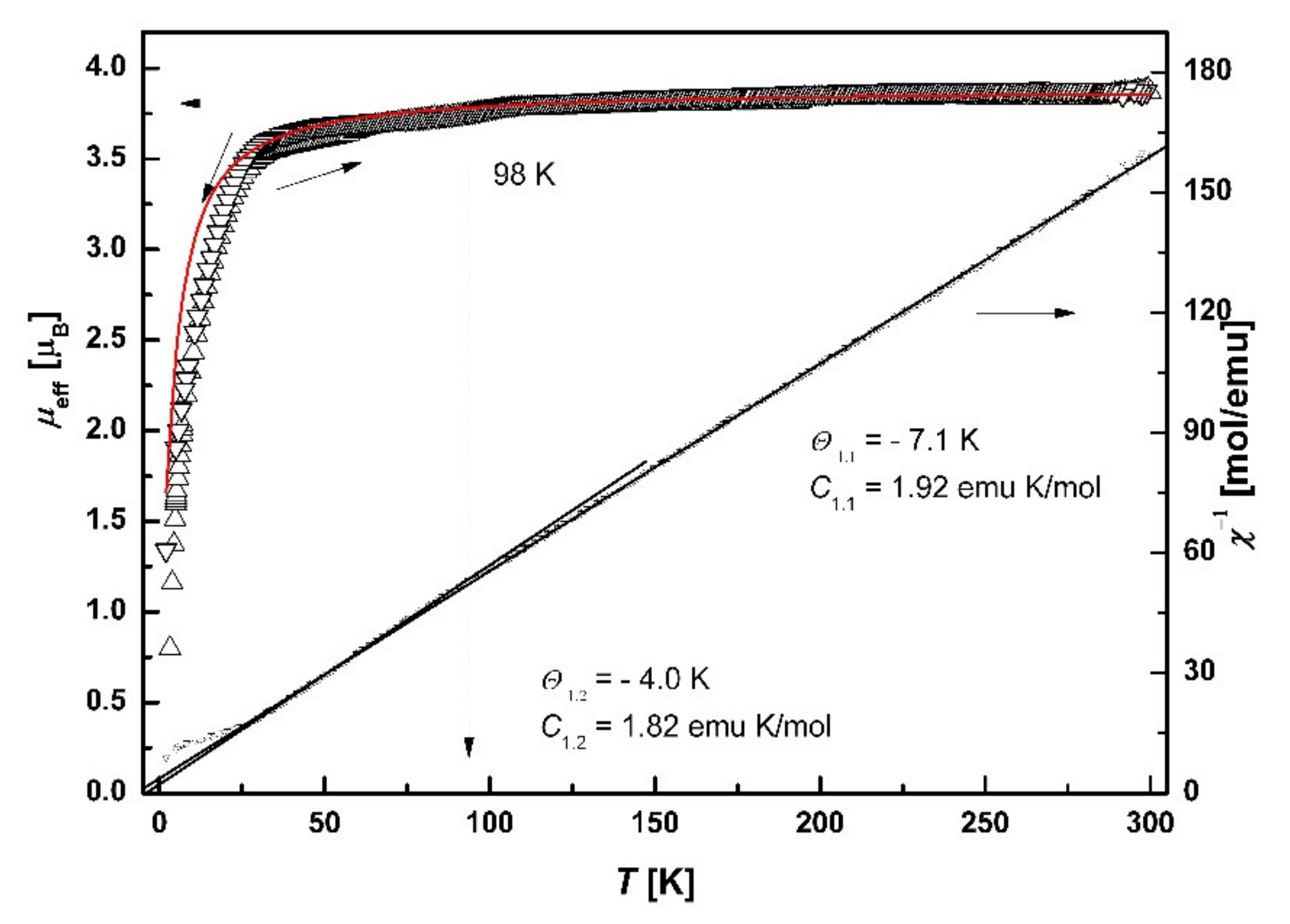
3.7.2. SQUID Magnetometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newkome, G.R.; Moorefield, C.N.; Vögtle, F. Dendritic Molecules: Concepts, Syntheses, Perspectives; VCH: Weinheim, Germany, 1996; 272p. [Google Scholar]
- Tomalia, D.A.; Naylor, A.M.; Goddard, W.A. Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew. Chem. Int. Ed. Engl. 1990, 29, 138–175. [Google Scholar] [CrossRef]
- Mangione, M.I.; Spanevello, R.A.; Minudri, D.; Cavallo, P.; Otero, L.; Fungo, F. Electrochemical films deposition and electro-optical properties of bis-carbazol-triphenylamine end-caped dendrimeric polymers. Electrochim. Acta 2018, 263, 585–595. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Z.; Ye, S.; Jim, C.K.W.; Yu, G.; Liu, Y.; Qin, J.; Tang, B.Z.; Li, Z. Solution-processable π-conjugated dendrimers with hole-transporting, electroluminescent and fluorescent pattern properties. J. Mater. Chem. 2011, 21, 14663–14671. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Xin, D.; Bai, S.-D.; Fang, L.; Duan, X.-E.; Roose, J.; Peng, H.; Chen, S.; Tang, B.Z. The synthesis, crystal structures, aggregation-induced emission and electroluminescence properties of two novel green-yellow emitters based on carbazole-substituted diphenylethene and dimesitylboron. Org. Electron. 2016, 33, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Zhu, Y.-Z.; Chang, W.-Y.; Song, J.; Pan, B.; Lu, L.; Gao, H.-H.; Zheng, J.-Y. Benzo[a]carbazole-Based Donor−π−Acceptor Type Organic Dyes for Highly Efficient Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 9015–9022. [Google Scholar] [CrossRef]
- Campbell, N.; Barclay, B.M. Recent Advances in the Chemistry of Carbazole. Chem. Rev. 1947, 40, 359–380. [Google Scholar] [CrossRef]
- McClenaghan, N.D.; Passalacqua, R.; Loiseau, F.; Campagna, S.; Verheyde, B.; Hameurlaine, A.; Dehaen, W. Ruthenium(II) Dendrimers Containing Carbazole-Based Chromophores as Branches. J. Am. Chem. Soc. 2003, 125, 5356–5365. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Shiga, T.; Maeda, Y.; Oshio, H. Spin crossover iron(III) complexes. Coord. Chem. Rev. 2007, 251, 2606–2621. [Google Scholar] [CrossRef]
- Gruzdev, M.S.; Domracheva, N.E.; Chervonova, U.V.; Kolker, A.M.; Golubeva, A.S. Bis-chelate Fe(III) complex of an azomethine at the focal point of a branched ester functionalized with cyclohexylbenzoic acid. J. Coord. Chem. 2012, 65, 1812–1820. [Google Scholar] [CrossRef]
- Fan, W.-J.; Sun, B.; Ma, J.; Li, X.; Tan, H.; Xu, L. Coordination-Driven Self-Assembly of Carbazole-Based Metallodendrimers with Generation-Dependent Aggregation-Induced Emission Behavior. Chem. Eur. J. 2015, 21, 12947–12959. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.; Connolly, J.D. Synthesis, characterization, thermal and catalytic properties of a novel carbazole derived Azo ligand and its metal complexes. Arab. J. Chem. 2017, 10, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Rajamouli, B.; Dwaraka Viswanath, C.S.; Giri, S.; Jayasankar, C.K.; Sivakumar, V. Carbazole functionalized new bipolar ligand for monochromatic red light-emitting europium(III) complex: Combined experimental and theoretical study. New J. Chem. 2017, 41, 3112–3123. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Pan, Y.-L.; Zhang, X.-J.; Sun, T.; Tian, Y.-P.; Yang, J.-X.; Chen, Z.-N. Synthesis, photoluminescence and electrochemical properties of a series of carbazole-functionalized ligands and their silver(I) complexes. Inorg. Chim. Acta 2007, 360, 2083–2091. [Google Scholar] [CrossRef]
- Gee, H.-C.; Lee, C.-H.; Jeong, Y.-H.; Jang, W.-D. Highly sensitive and selective cyanide detection via Cu2+ complex ligand exchange. Chem. Commun. 2011, 47, 11963–11965. [Google Scholar] [CrossRef] [PubMed]
- Hwang, A.R.; Han, W.-S.; Wee, K.-R.; Kim, H.Y.; Cho, D.W.; Min, B.K.; Nam, S.W.; Pac, C.; Kang, S.O. Photodynamic Behavior of Heteroleptic Ir(III) Complexes with Carbazole-Functionalized Dendrons Associated with Efficient Electron Transfer Processes. J. Phys. Chem. C 2012, 116, 1973–1986. [Google Scholar] [CrossRef]
- Gruzdev, M.S.; Chervonova, U.V.; Ksenofontov, A.A.; Bichan, N.G.; Kolker, A.M. Chemical design of carbazole dendrons: Optical properties and DFT analysis. Submitted to press.
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, J.F.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory; Schaefer, H.F., III, Ed.; Springer US: New York, NY, USA, 1977; Volume 2, pp. 1–462. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Casida, M.E. Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, 1995; pp. 155–192. [Google Scholar]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Kazitsyna, L.A.; Kupletskaya, N.B. Application of UV, IR, and NMR Spectroscopy in Organic Chemistry; Vysshaya Shkola: Moscow, Russia, 1971; 263p. (In Russian) [Google Scholar]
- Pretsch, E.; Buhlman, P.; Affolter, C. Structure Determination of Organic Compounds—Tables of Spectra Data; Springer: Berlin/Heidelberg, Germany, 2000; 421p. [Google Scholar]
- Nakanishi, K. Infrared Absorption Spectroscopy; Holden-Day, Inc., San Francisco und Nankodo Co.: Tokyo, Japan, 1962; 133p. [Google Scholar]
- Cheremisina, I.M. Frequencies of predominantly stretching vibrations and the nature of the metal-ligand bond. J. Struct. Chem. 1978, 19, 286–300. [Google Scholar] [CrossRef]
- Nawar, N.; Hosny, N.M. Synthesis, spectral and antimicrobial activity studies of o-aminoacetophenone o-hydroxybenzoylhydrazone complexes. Transit. Met. Chem. 2000, 25, 1–8. [Google Scholar] [CrossRef]
- James, D.W.; Nolan, M.J. Vibrational Spectra of Transition Metal Complexes and the Nature of the Metal-Ligand Bond. In Progress in Inorganic Chemistry; Cotton, F.A., Ed.; Interscience Publishers: New York, NY, USA, 1968; Volume 9, pp. 195–275. [Google Scholar]
- Chervonova, U.V.; Gruzdev, M.S.; Zueva, E.M.; Vorobeva, V.E.; Ksenofontov, A.A.; Alexandrov, A.I.; Pashkova, T.V.; Kolker, A.M. Synthesis, EPR study and photophysical properties of a mononuclear Fe(III) Schiff base complex functionalized by 3,6-di-tert-butyl-carbazole moieties. J. Mol. Struct. 2020, 1200, 127090. [Google Scholar] [CrossRef]
- Gruzdev, M.S.; Vorobeva, V.E.; Zueva, E.M.; Chervonova, U.V.; Petrova, M.M.; Domracheva, N.E. High-spin Fe(III) Schiff based complexes with photoactive ligands. Synthesis, EPR study and magnetic properties. Polyhedron 2018, 155, 415–424. [Google Scholar] [CrossRef]
- Hagen, W.R. Biomolecular EPR Spectroscopy; CRC Press, Tailor & Francis Group: New York, NY, USA, 2009; 248p. [Google Scholar]
- Wickman, H.; Klein, M.; Shirley, D. Paramagnetic Resonance of Fe3+ in Polycrystalline Ferrichrome A. J. Chem. Phys. 1965, 42, 2113–2117. [Google Scholar] [CrossRef] [Green Version]
- Aasa, R. Powder Line Shapes in the Electron Paramagnetic Resonance Spectra of High-Spin Ferric Complexes. J. Chem. Phys. 1970, 52, 3919–3930. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S. CW-EPR Spectral Simulations: Solid State. In Electron Paramagnetic Resonance Investigations of Biological Systems by Using Spin Labels, Spin Probes, and Intrinsic Metal Ions, Part A; Qin, P.Z., Warncke, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 563, pp. 121–142. [Google Scholar] [CrossRef]
- Ivanova, T.A.; Ovchinnikov, I.V.; Gil’mutdinov, I.F.; Mingalieva, L.V.; Turanova, O.A.; Ivanova, G.I. Specific features of spin-variable properties of [Fe(acen)pic2]BPh4·nH2O. Phys. Solid State 2016, 58, 280–283. [Google Scholar] [CrossRef]
- Altshuler, T.S.; Bresler, M.S.; Goryunov, Y.V. Ferromagnetic ordering of iron impurities in the Kondo semiconductor SmB6. JETP Lett. 2005, 81, 475–478. [Google Scholar] [CrossRef]
- Brooker, S. Spin crossover with thermal hysteresis: Practicalities and lessons learnt. Chem. Soc. Rev. 2014, 44, 2880–2892. [Google Scholar] [CrossRef] [Green Version]
- Halcrow, M.A. Spin-Crossover Materials: Properties and Applications; John Wiley & Sons, Ltd.: Chicheser, UK, 2013. [Google Scholar] [CrossRef]
- Domracheva, N.; Vorobeva, V.; Pyataev, A.; Ivanova, A. Magnetic properties of novel dendrimeric iron (III) complexes of the first generation: EPR and mössbauer study. Appl. Magn. Reson. 2016, 47, 903–913. [Google Scholar] [CrossRef]
- Fisher, M. Magnetism in One-Dimensional Systems-The Heisenberg Model for Infinite Spin. Am. J. Phys. 1964, 32, 343–346. [Google Scholar] [CrossRef]
- Bonner, J.C.; Fisher, M.E. Linear Magnetic Chains with Anisotropic Coupling. Phys. Rev. 1964, 135, A640. [Google Scholar] [CrossRef] [Green Version]
- Carlin, R.L. Magnetochemistry; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Fe(L)2]NO3 | [Fe(L)2]Cl | [Fe(L)2]PF6 | |
|---|---|---|---|
| Symmetry | P1 | P1 | P1 |
| a, Å | 14.58 | 11.78 | 8.48 |
| b, Å | 11.93 | 15.49 | 6.12 |
| c, Å | 20.80 | 24.90 | 23.06 |
| α, ° | 78.64 | 46.41 | 53.83 |
| β, ° | 70.84 | 127.8 | 97.40 |
| γ, ° | 24.86 | 136.6 | 105.1 |
| № | Ms, μB | Weight Factor C1 for the Brillouin Function S = 5/2 | Weight Factor C2 for the Brillouin Function S = 1/2 | Linear Factor k, T/K |
|---|---|---|---|---|
| 1 | 0.19 | 0.025 | 0 | 0.034 |
| 2 | 0.72 | 0.1 | 0 | 0.07 |
| 3 | 1.33 | 0.21 | 0.39 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruzdev, M.; Chervonova, U.; Kolker, A.; Fomina, N.; Zueva, E.; Vorobeva, V.; Starichenko, D.; Korolev, A. Dendritic Iron(III) Carbazole Complexes: Structural, Optical, and Magnetic Characteristics. Materials 2021, 14, 5445. https://doi.org/10.3390/ma14185445
Gruzdev M, Chervonova U, Kolker A, Fomina N, Zueva E, Vorobeva V, Starichenko D, Korolev A. Dendritic Iron(III) Carbazole Complexes: Structural, Optical, and Magnetic Characteristics. Materials. 2021; 14(18):5445. https://doi.org/10.3390/ma14185445
Chicago/Turabian StyleGruzdev, Matvey, Ulyana Chervonova, Arkadiy Kolker, Nadezhda Fomina, Ekaterina Zueva, Valerya Vorobeva, Denis Starichenko, and Alexander Korolev. 2021. "Dendritic Iron(III) Carbazole Complexes: Structural, Optical, and Magnetic Characteristics" Materials 14, no. 18: 5445. https://doi.org/10.3390/ma14185445