Theoretical Investigations of the BaRh2Ge4X6 (X = S, Se, Te) Compounds


Abstract
:
1. Introduction
2. Bader’s Quantum Theory of Atoms in Molecules and Computational Details
2.1. Bader’s Quantum Theory of Atoms in Molecules
2.2. Computational Details
3. Structural, Electronic and Transport Results
3.1. BaRh2Ge4X6 (X = S, Se, Te)
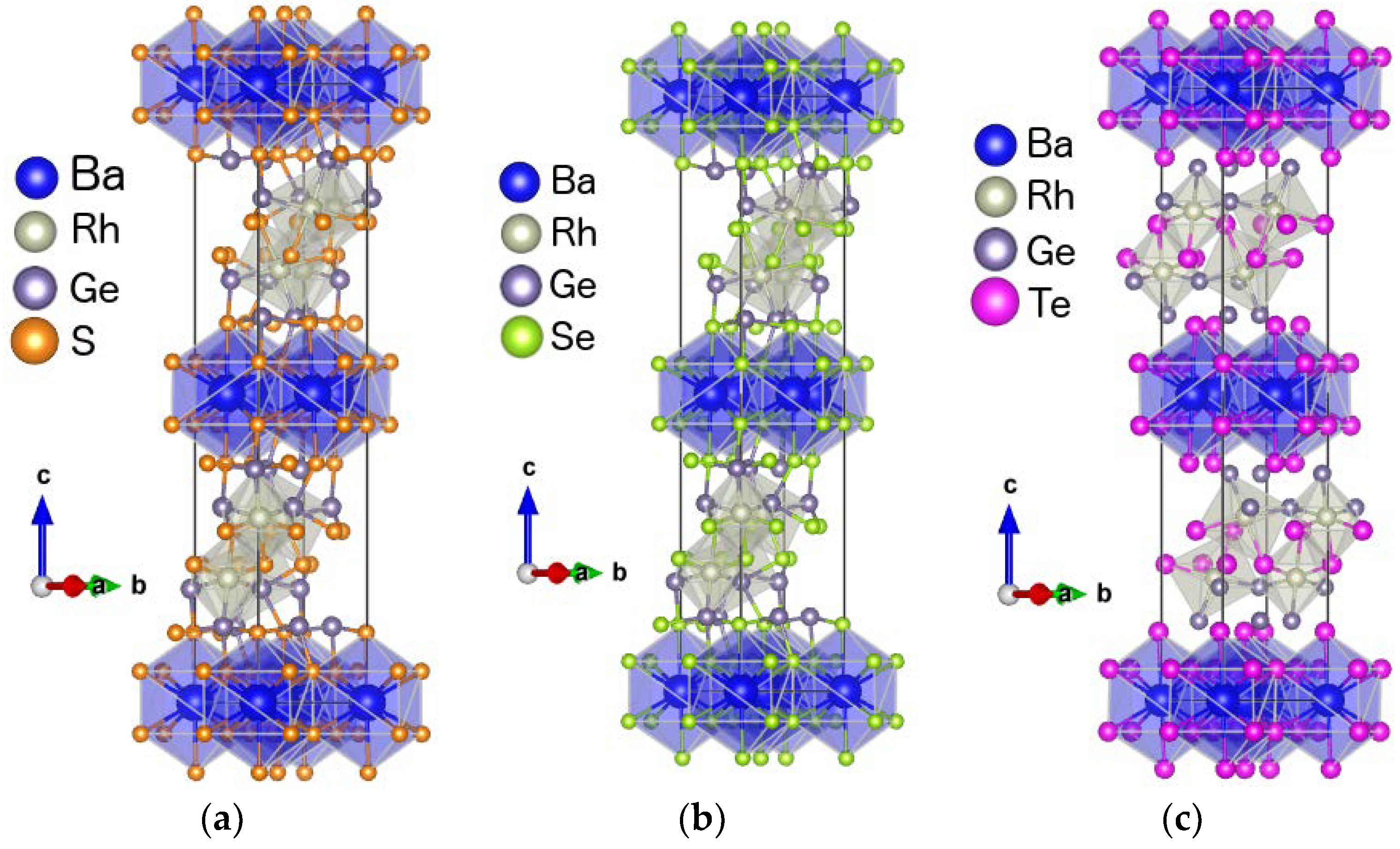
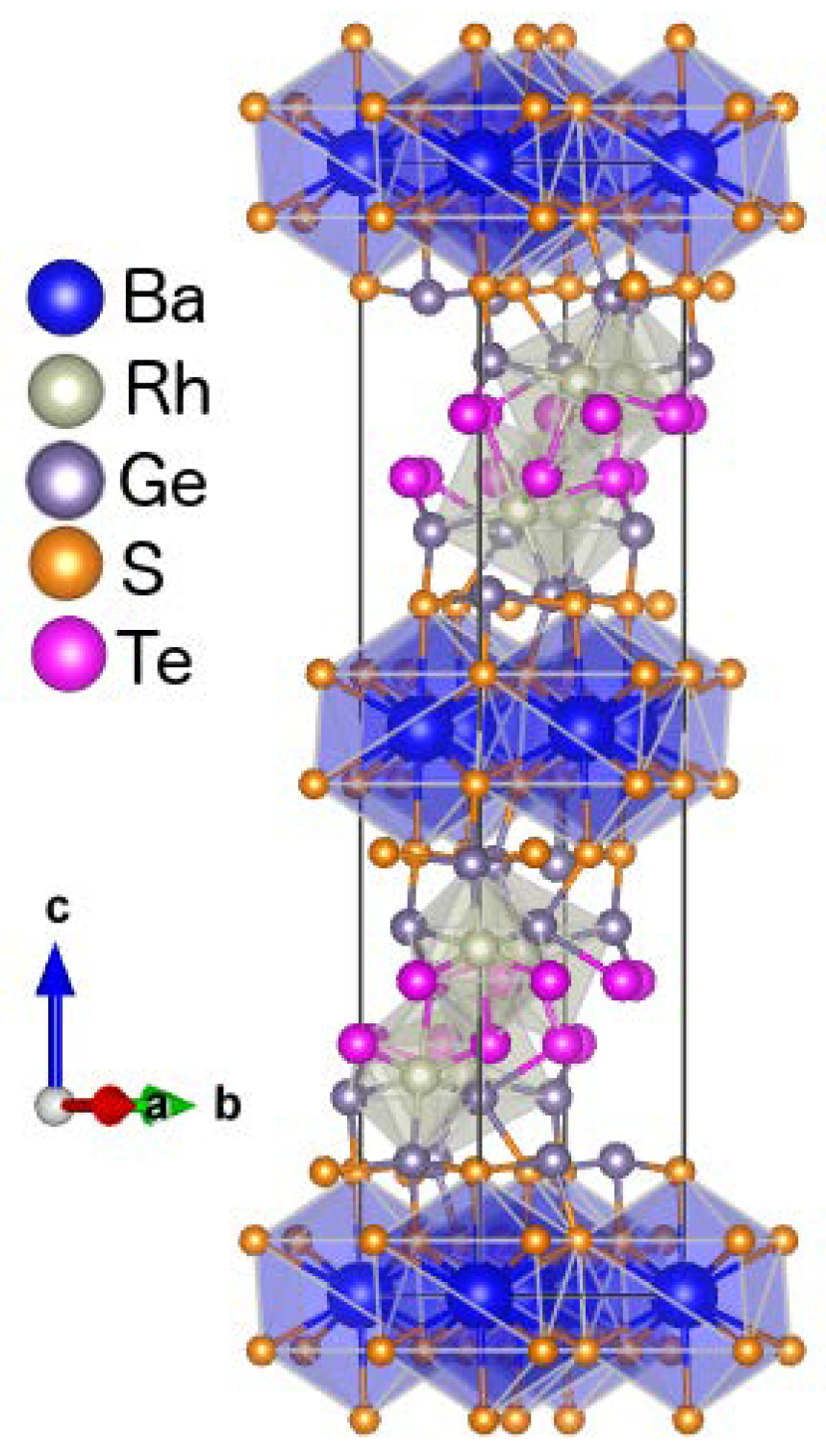
3.1.1. Structural Characteristics of the Compounds
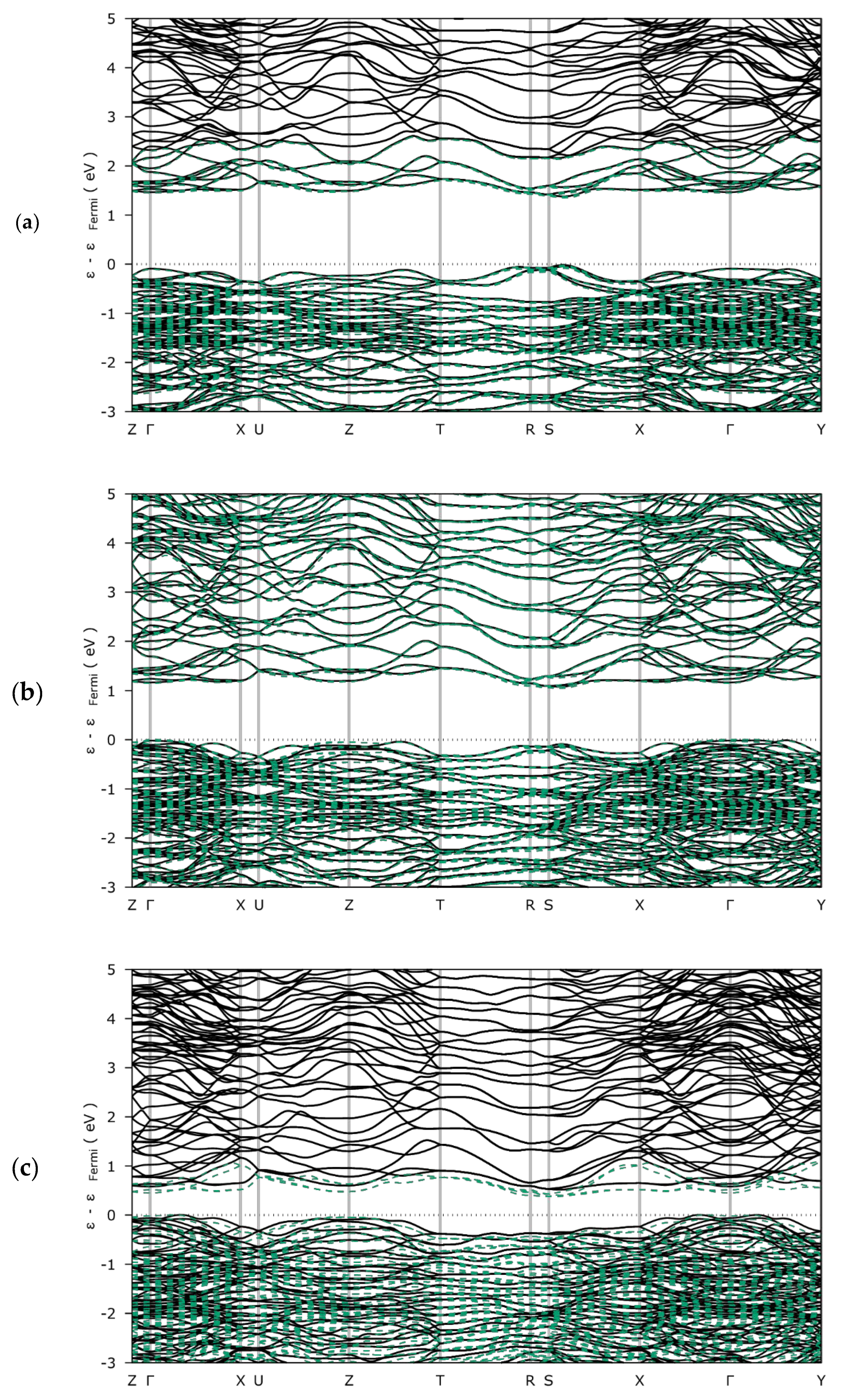
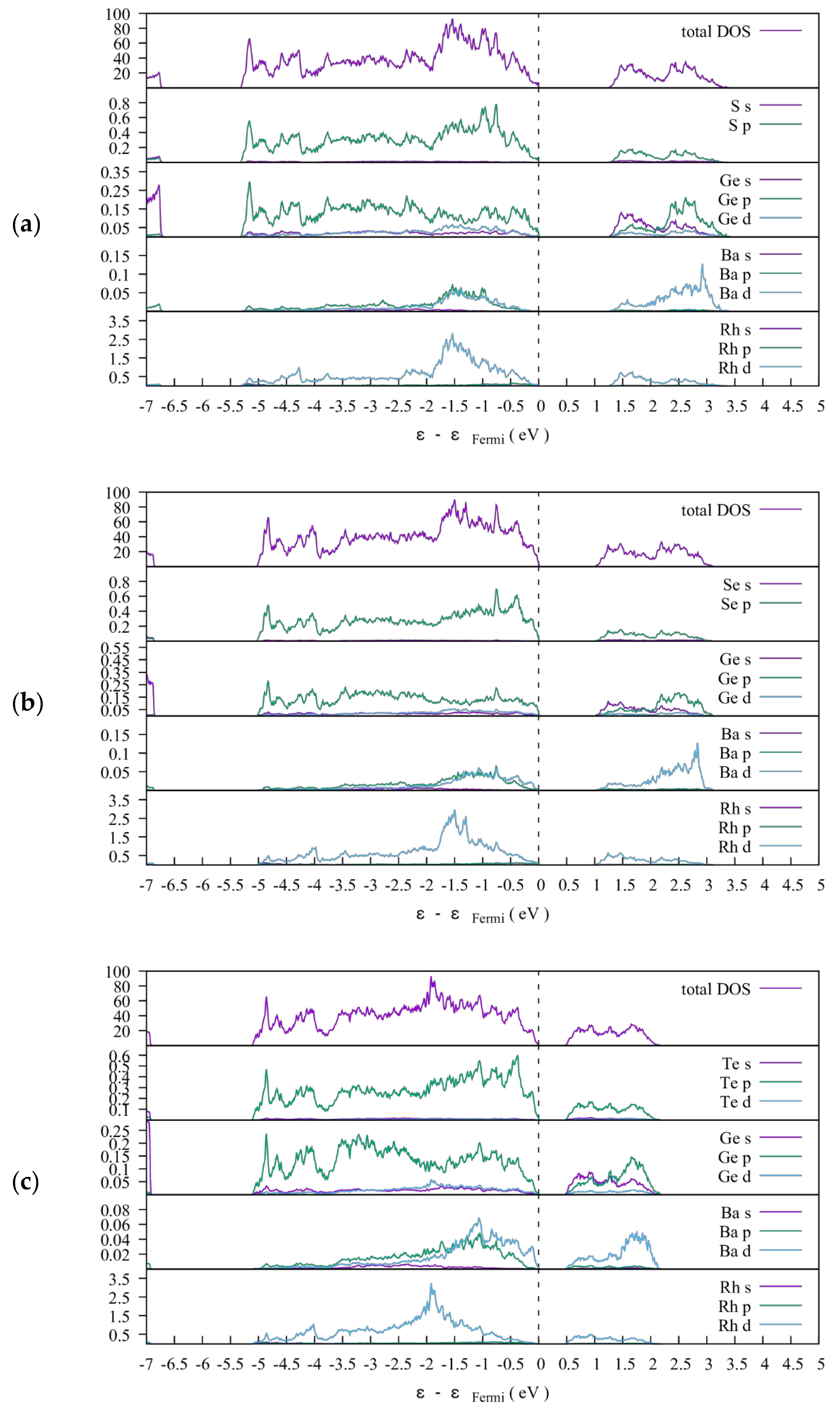
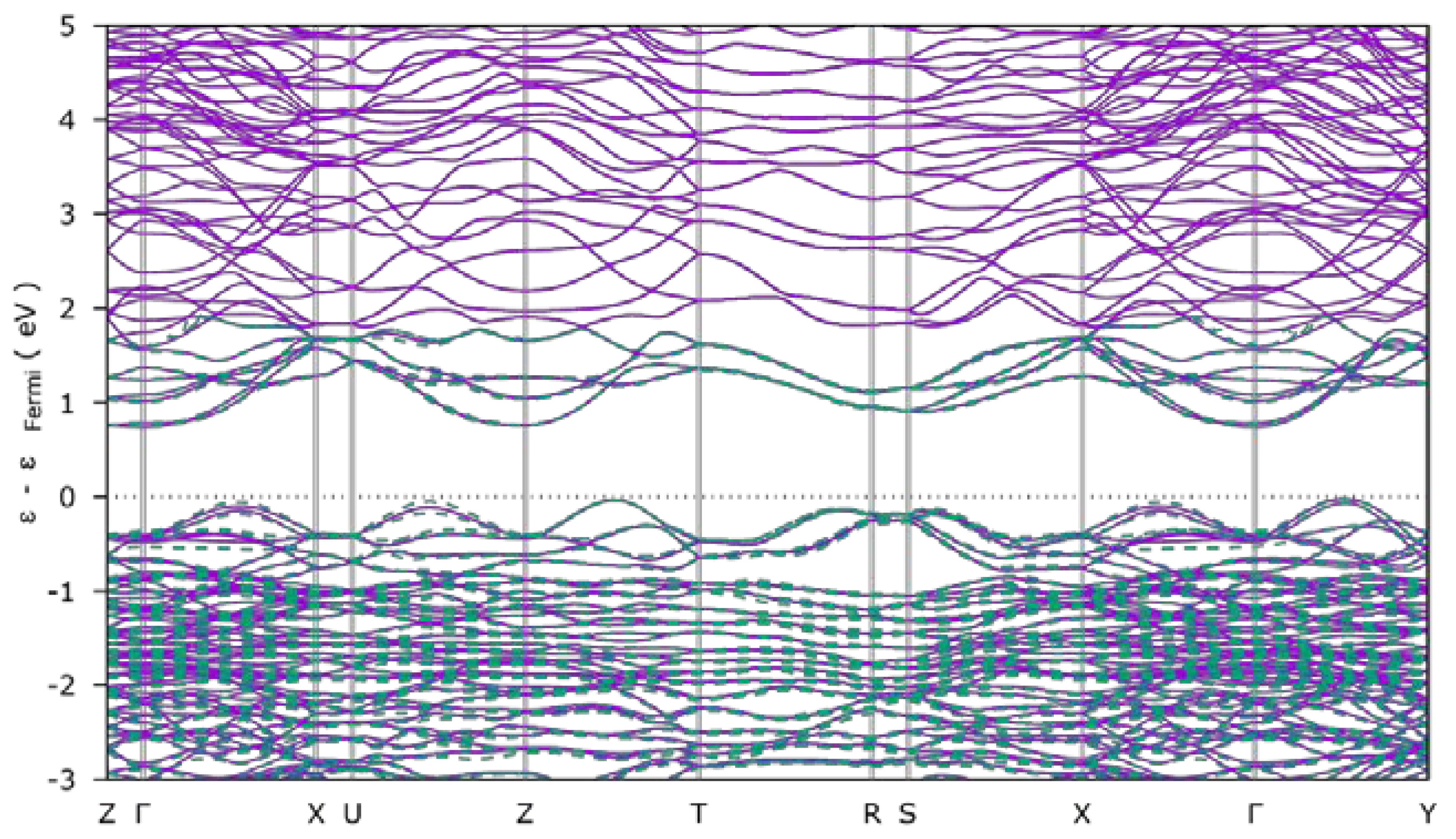
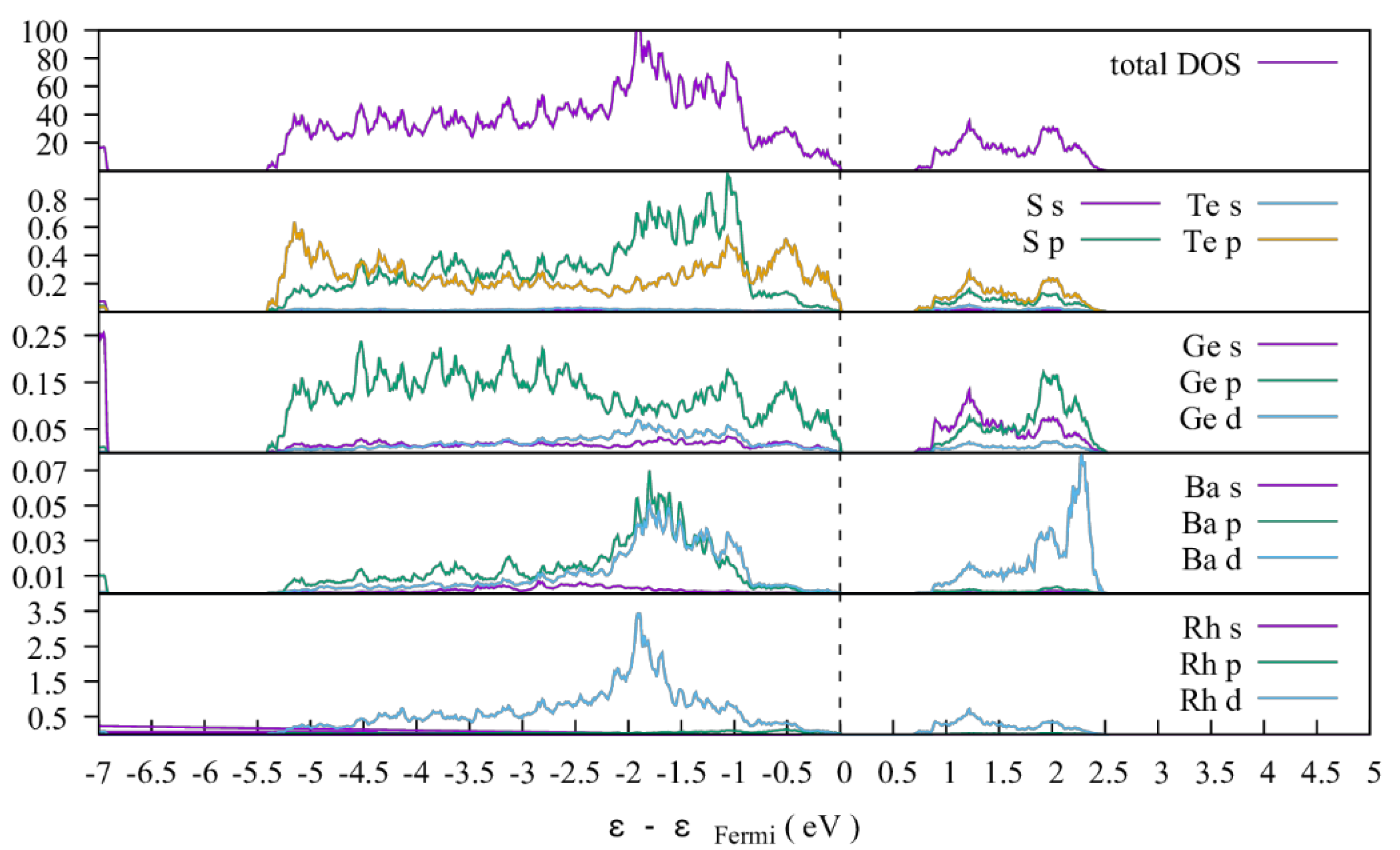
3.1.2. Electronic Structures
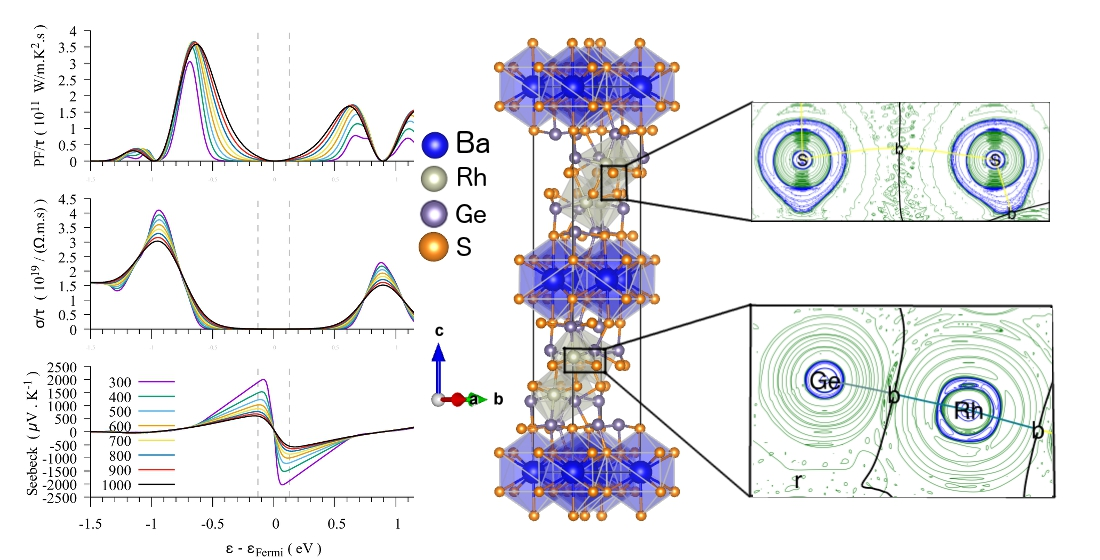
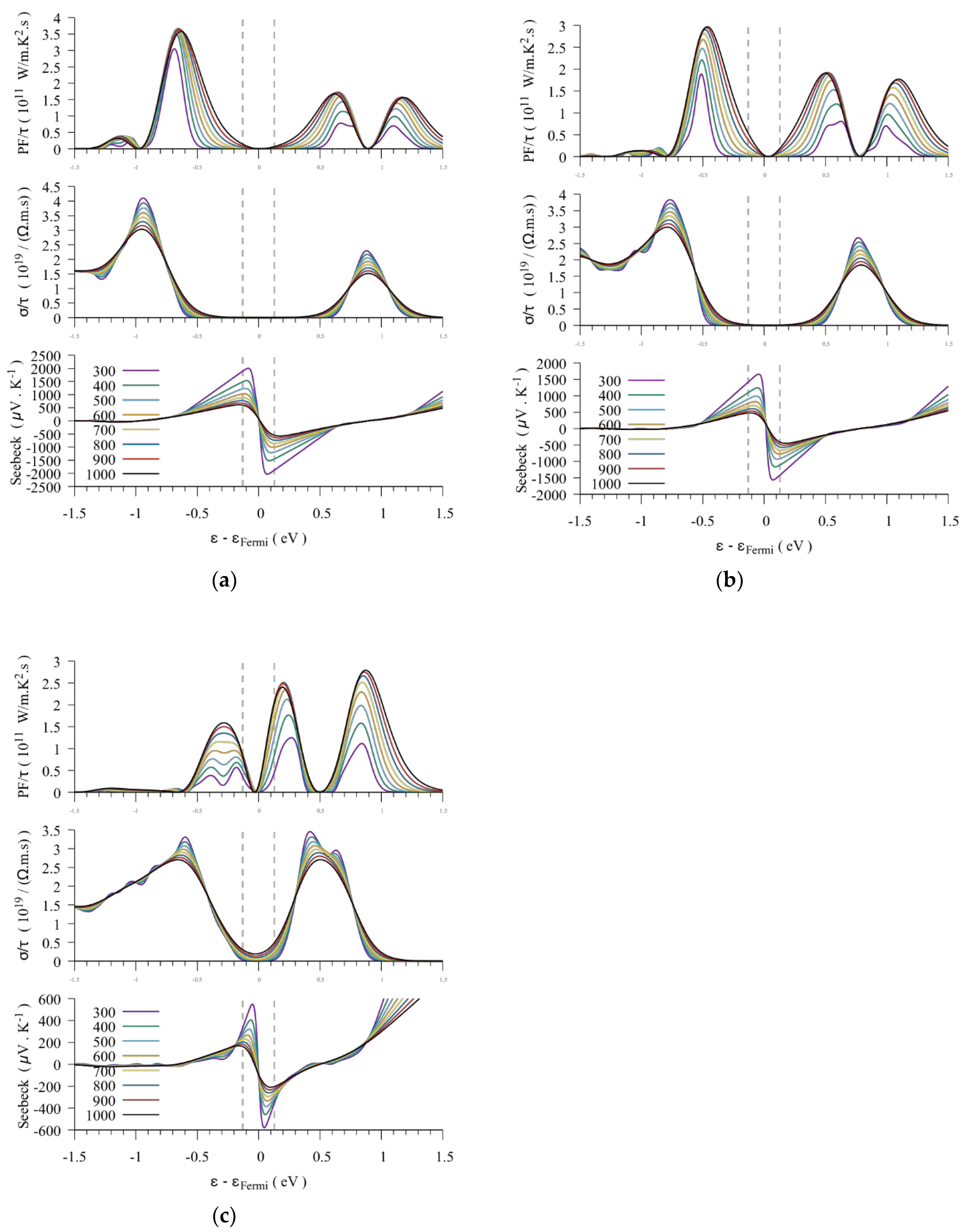
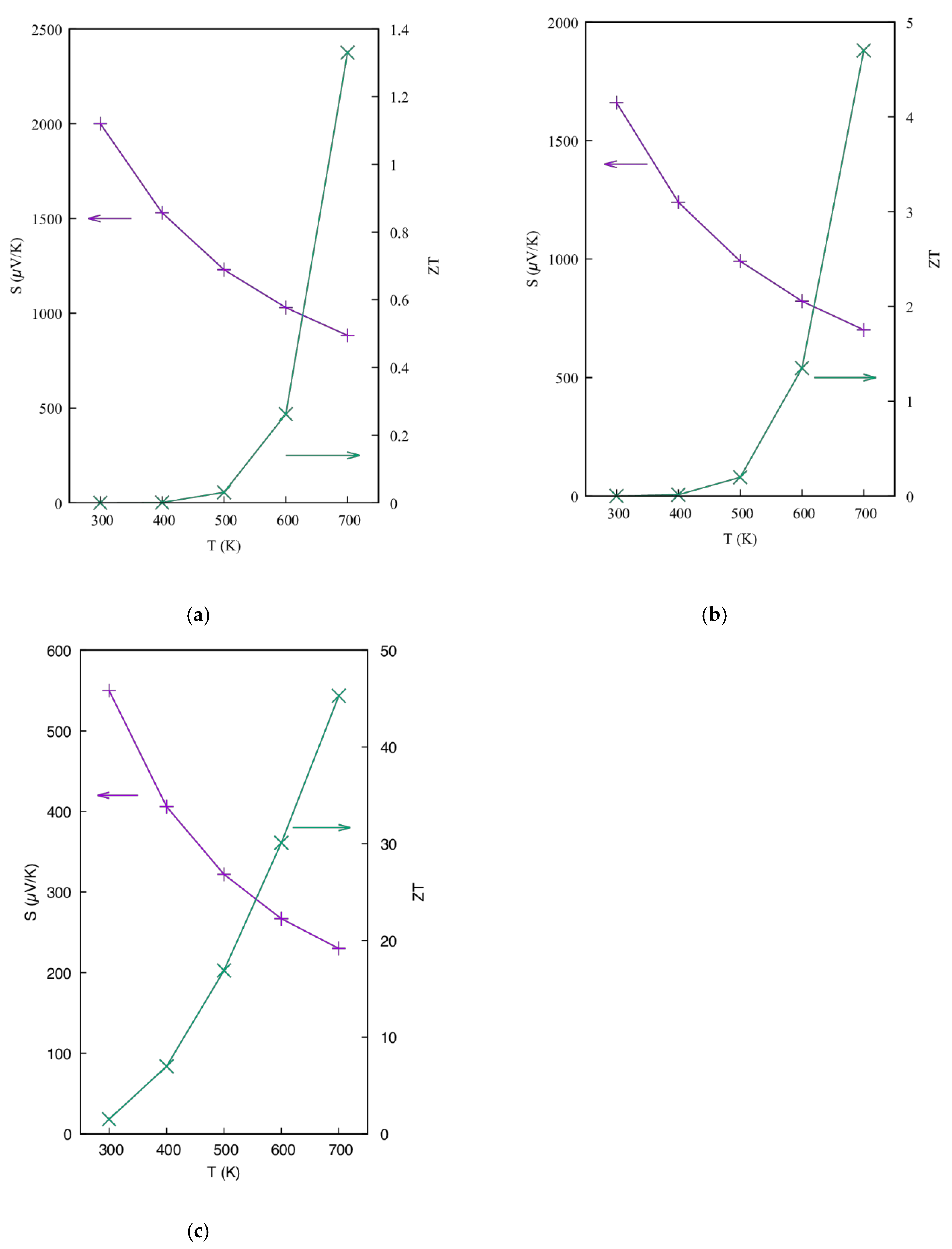
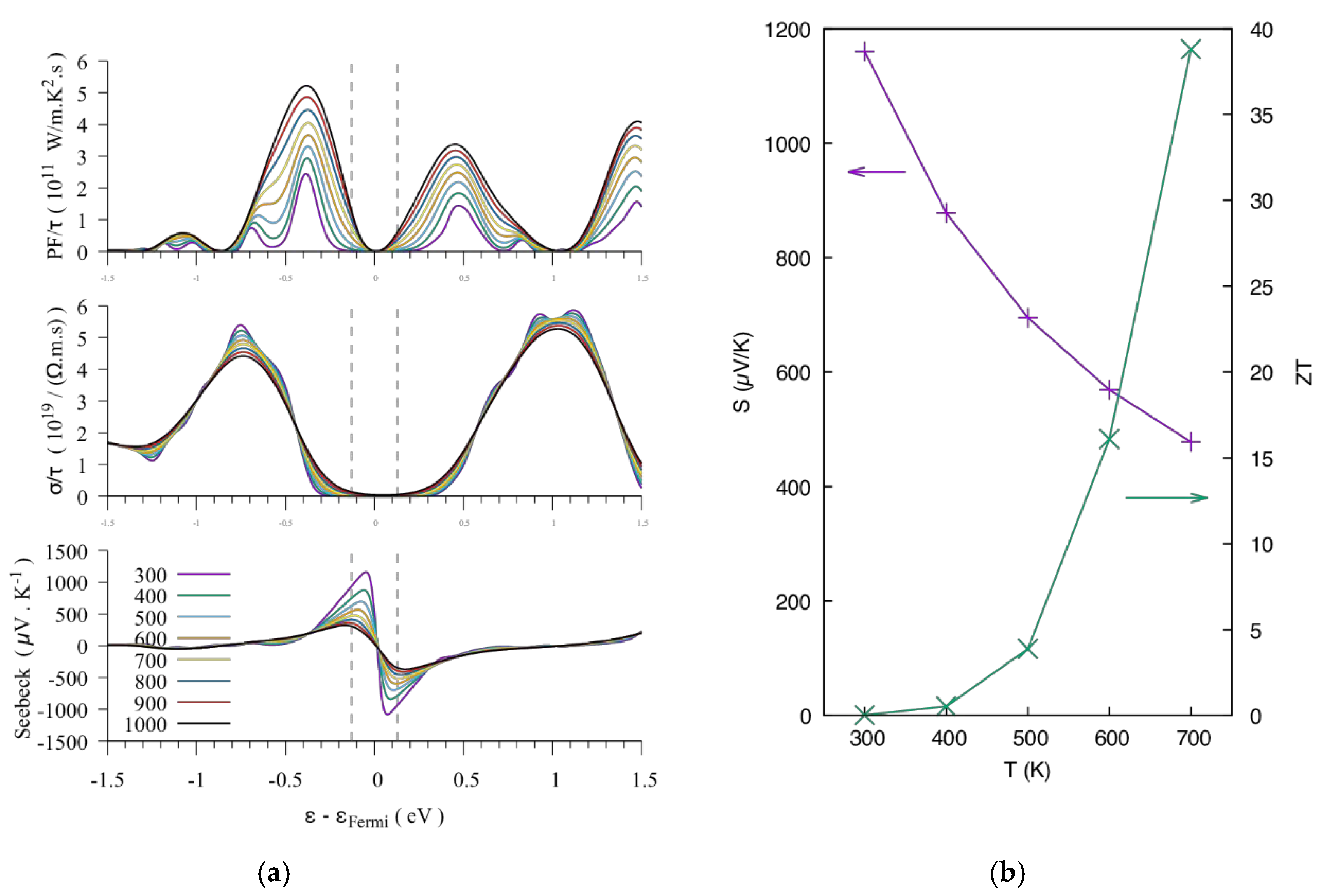
3.1.3. Thermoelectric Properties
3.2. The BaRh2Ge4S4Te2 Compound
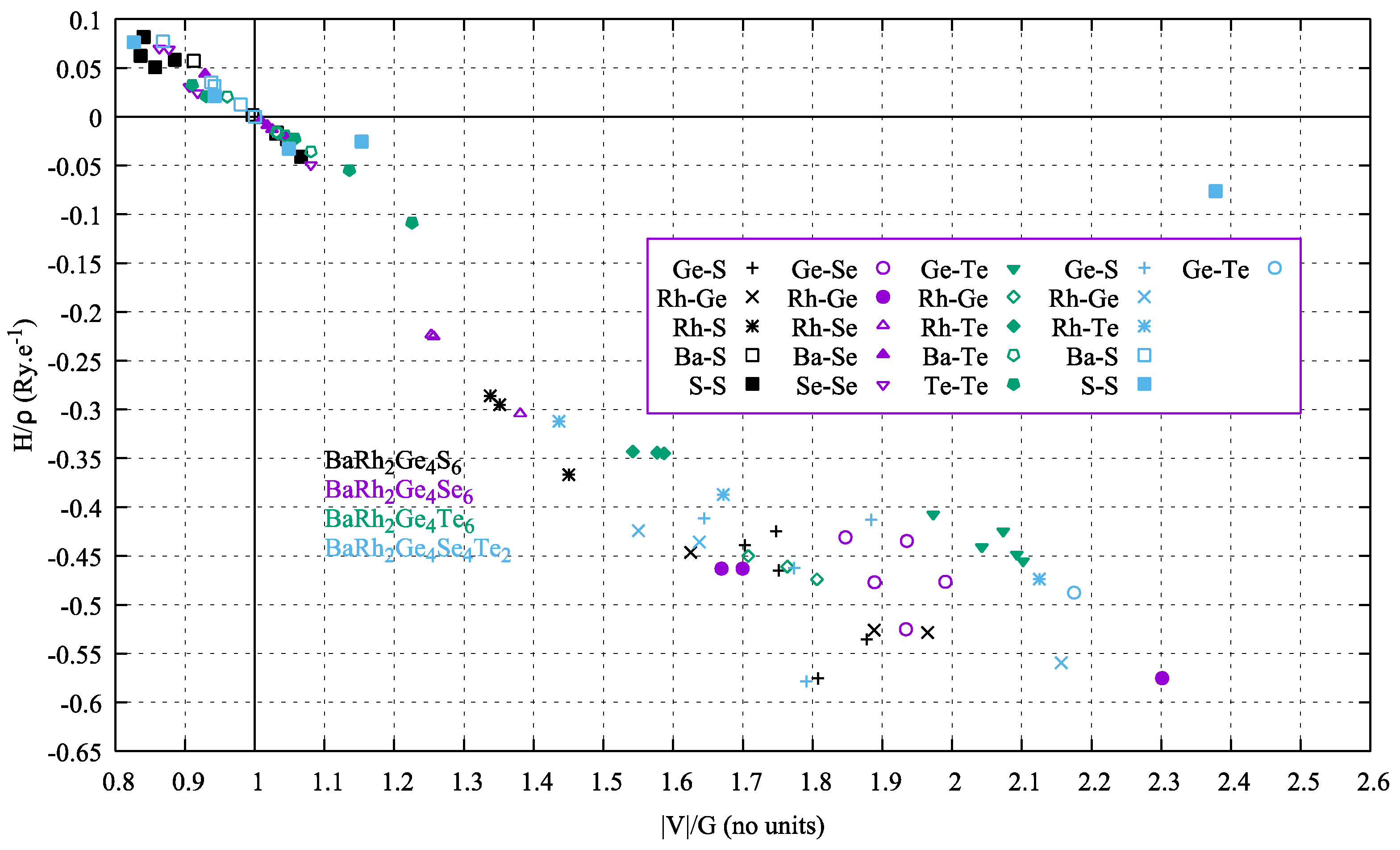
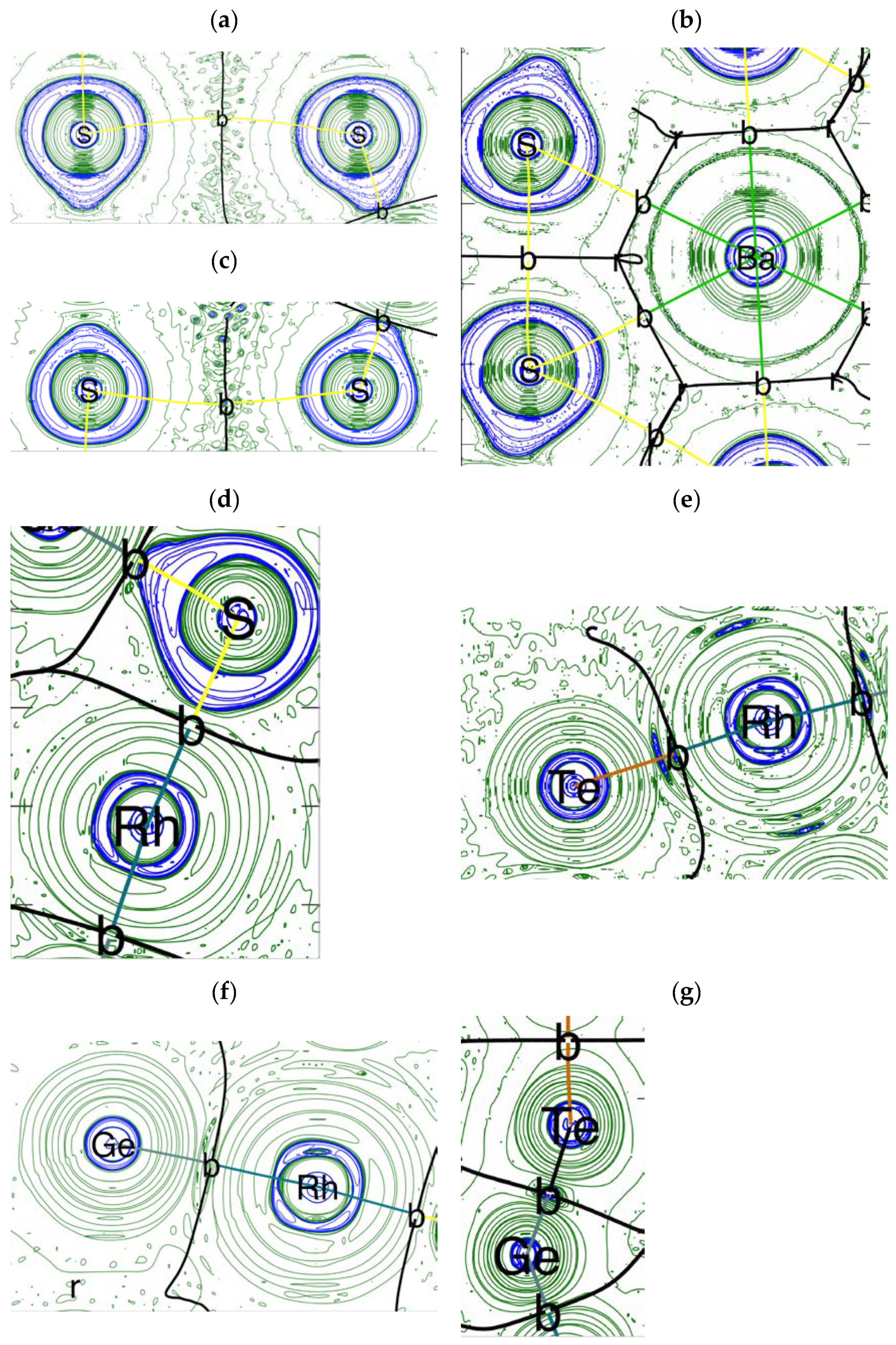
4. Electron Density Topology Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biswas, K.; He, J.; Blum, I.D.; Wu, C.I.; Hogan, T.P.; Seidman, D.N.; Dravid, V.P.; Kanatzidis, M.G. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 2012, 489, 414–418. [Google Scholar] [CrossRef]
- Hsu, K.F.; Loo, S.; Guo, F.; Chen, W.; Dyck, J.S.; Uher, C.; Hogan, T.; Polychroniadis, E.K.; Kanatzidis, M.G. Cubic AgPb(m)SbTe(2+m): Bulk thermoelectric materials with high figure of merit. Science 2004, 303, 818–821. [Google Scholar] [CrossRef]
- Poudeu, P.F.R.; D’Angelo, J.; Downey, A.D.; Short, J.L.; Hogan, T.P.; Kanatzidis, M.G. High thermoelectric figure of merit and nanostructuring in bulk p-type Na1-xPbmSbyTem+2. Angew. Chem. Int. Ed. 2006, 45, 3835–3839. [Google Scholar] [CrossRef] [PubMed]
- Nolas, G.S.; Cohn, J.L.; Slack, G.A.; Schujman, S.B. Semiconducting Ge clathrates: Promising candidates for thermoelectric applications. Appl. Phys. Lett. 1998, 73, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Keppens, V.; Mandrus, D.; Sales, B.C.; Chakoumakos, B.C.; Dai, P.; Coldea, R.; Maple, M.B.; Gajewski, D.A.; Freeman, E.J.; Bennington, S. Localized vibrational modes in metallic solids. Nature 1998, 395, 876–878. [Google Scholar] [CrossRef]
- Sales, B.C.; Chakoumakos, B.C.; Mandrus, D. Thermoelectric properties of thallium-filled skutterudites. Phys. Rev. B 2000, 61, 2475–2481. [Google Scholar] [CrossRef]
- Christensen, M.; Abrahamsen, A.B.; Christensen, N.B.; Juranyi, F.; Andersen, N.H.; Lefmann, K.; Andreasson, J.; Bahl, C.R.H.; Iversen, B.B. Avoided crossing of rattler modes in thermoelectric materials. Nat. Mater. 2008, 7, 811–815. [Google Scholar] [CrossRef]
- Zhang, J.; Song, L.; Madsen, G.K.H.; Fischer, K.F.F.; Zhang, W.; Shi, X.; Iversen, B.B. Designing high-performance layered thermoelectric materials through orbital engineering. Nat. Commun. 2016, 7, 10892. [Google Scholar] [CrossRef]
- Heremans, J.P.; Wiendlocha, B.; Chamoire, A.M. Resonant levels in bulk thermoelectric semiconductors. Energy Environ. Sci. 2012, 5, 5510–5530. [Google Scholar] [CrossRef]
- Balout, H.; Boulet, P.; Record, M.C. Electronic and transport properties of Mg2Si under isotropic strains. Intermetallics 2014, 50, 8–13. [Google Scholar] [CrossRef]
- Balout, H.; Boulet, P.; Record, M.C. Strain-induced electronic band convergence: Effect on the Seebeck coefficient of Mg2Si for thermoelectric applications. J. Mol. Mod. 2017, 23, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauling, L. The nature of the chemical bond. IV The energy of single bonds and the relative electronegativity of atoms. J. Am. Chem. Soc. 1932, 54, 3570–3582. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Pearson, R.G. Recent advances in the concept of hard and soft acids and bases. J. Chem. Educ. 1987, 64, 561–567. [Google Scholar] [CrossRef]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comput. Chem. 1999, 20, 129. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Gatti, C. Chemical bonding in crystals: New directions. Z. Kristallogr. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Evangeli, C.; Spiece, J.; Sangtarash, S.; Molina-Mendoza, A.J.; Mucientes, M.; Mueller, T.; Lambert, C.; Sadeghi, H.; Kolosov, O. Nanoscale Thermal Transport in 2D Nanostructures from Cryogenic to Room Temperature. Adv. Electron. Mater. 2019, 5, 1900331. [Google Scholar] [CrossRef]
- Oh, J.; Kim, Y.; Chung, S.; Kim, H.; Son, J.G. Fabrication of a MoS2/Graphene Nanoribbon Heterojunction Network for Improved Thermoelectric Properties. Adv. Mater. Interf. 2019, 6, 1901333. [Google Scholar] [CrossRef]
- Uematsu, Y.; Terada, T.; Sato, K.; Ishibe, T.; Nakamura, Y. Low thermal conductivity in single crystalline epitaxial germanane films. Appl. Phys. Express 2020, 13, 55503. [Google Scholar] [CrossRef]
- Lei, H.; Yamaura, J.I.; Guo, J.; Qi, Y.; Toda, Y.; Hosono, H. Layered Compounds BaM2Ge4Ch6 (M = Rh, Ir and Ch = S, Se) with Pyrite-Type Building Blocks and Ge–Ch Heteromolecule-Like Anions. Inorg. Chem. 2014, 53, 5684–5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, R.F.W.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [Green Version]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Silvi, B.; Gatti, C. Direct Space Representation of the Metallic Bond. J. Chem. Phys. A 2000, 104, 947–953. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Kirzhnits, D.A. Quantum Corrections to the Thomas-Fermi Equation. Sov. Phys. JETP 1957, 5, 64. [Google Scholar]
- Hodges, C.H. Quantum Corrections to the Thomas–Fermi Approximation—The Kirzhnits Method. Can. J. Phys. 1973, 51, 1428–1437. [Google Scholar] [CrossRef]
- Abramov, Y.A. Secondary Interactions and Bond Critical Points in Ionic Crystals. Acta Cryst. Sect. A Found. Cryst. 1997, 53, 264–272. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [Green Version]
- Medasani, B.; Haranczyk, M.; Canning, A.; Asta, M. Vacancy formation energies in metals: A comparison of MetaGGA with LDA and GGA exchange-correlation functionnals. Comput. Mater. Sci. 2015, 101, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.; Wang, Y.; Guan, P.; Wang, W.Y.; Fang, H.; Anderson, T.; Liu, Z.K. Insight into structural, elastic, phonon, and thermodynamic properties of α-sulfur and energy-related sulfides: A comprehensive first-principles study. J. Mater. Chem. A 2015, 3, 8002–8014. [Google Scholar] [CrossRef]
- Tran, F.; Koller, D.; Blaha, P. Application of screened hybrid functionals to the bulk transition metals Rh, Pd, and Pt. Phys. Rev. B 2012, 86, 134406. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.J. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Otero-de-la-Roza, A.; Blanco, M.A.; Martín Pendás, A.; Luaña, V. Critic: A new program for the topological analysis of solid-state electron densities. Comput. Phys. Commun. 2009, 180, 157–166. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Available online: http://www.gnuplot.info (accessed on 3 December 2020).
- Lin, S.; Li, W.; Bu, Z.; Gao, B.; Li, J.; Pei, Y. Thermoelectric properties of Ag9GaS6 with ultralow lattice thermal conductivity. Mater. Today Phys. 2018, 6, 60–67. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Zhang, C.; Yang, D.; Liu, T.; Yan, Y.; Liu, W.; Tan, G.; Su, X.; Uher, C.; et al. Ultralow thermal conductivity of BaAg2SnSe4 and the effect of doping by Ga and In. Mater. Today Phys. 2019, 9, 100098. [Google Scholar] [CrossRef]
- Yang, H.; Boulet, P.; Record, M.C. A rapid method for analyzing the chemical bond from energy densities calculations at the bond critical point. Comput. Theor. Chem. 2020, 1178, 112784. [Google Scholar] [CrossRef]
- Yang, H.; Boulet, P.; Record, M.C. New insight into the structure-property relationships from chemical bonding analysis: Application to thermoelectric materials. J. Solid State Mater. 2020, 286, 121266. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Gonzalez, J.; Kessens, R.; Schuster, H.U. Darstellung und Kristallstruktur neuer AM2X2-Verbindungen in den Systemen Erdalkalimetall-Platinmetall-Germanium. Z. Anorg. Allg. Chem. 1993, 619, 13–16. [Google Scholar] [CrossRef]
- Salma, M.U.; Rahman, M.A. Physical properties of ThCr2Si2-type Rh-based compounds ARh2Ge2 (A = Ca, Sr, Y and Ba): DFT based first-principles investigation. Int. J. Mod. Phys. B 2018, 32, 1850357. [Google Scholar] [CrossRef]
- Bu, K.; Huang, J.; Luo, M.; Guan, M.; Zheng, C.; Pan, J.; Zhang, X.; Wang, S.; Zhao, W.; Shi, X.; et al. Observation of High Seebeck Coefficient and Low Thermal Conductivity in [SrO]-Intercalated CuSbSe2 Compound. Chem. Mater. 2018, 30, 5539–5543. [Google Scholar] [CrossRef]
- Coppens, P. X-Ray Charge Densities and Chemical Bonding, IUCr Texts on Crystallography; Oxford University Press: Oxford, UK, 1997; Volume 4. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
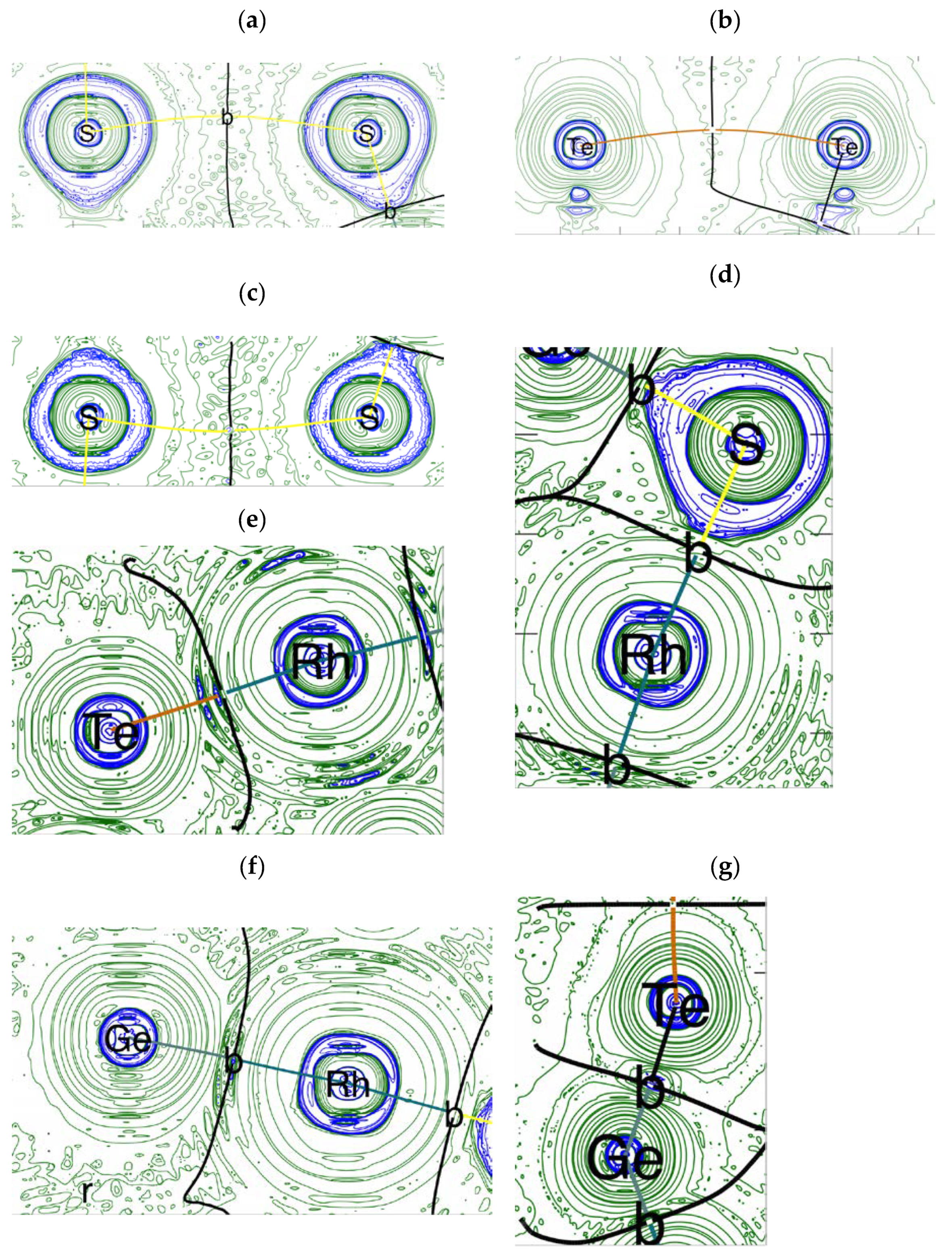{kind=link}
| Parameter (Å) | BaRh2Ge4S6 | BaRh2Ge4Se6 | BaRh2Ge4Te6 |
|---|---|---|---|
| a | 6.022 (5.9473(2)) | 6.219 (6.1318(3)) | 6.506 |
| b | 5.967 (5.8891(2)) | 6.156 (6.0700(3)) | 6.458 |
| c | 29.424 (29.1781(9)) | 30.626 (30.3144(9)) | 32.580 |
| Gap Energy | Band Gap Type | Edge Valence State Features | Edge Conduction State Features | |
|---|---|---|---|---|
| BaRh2Ge4S6 | ||||
| NR | 1.26 | Ge-p, S-p | Rh-d, Ge-s, S-p | |
| FR | 1.27 | – | – | |
| BaRh2Ge4Se6 | ||||
| NR | 1.01 | Ge-p, Se-p | Rh-d, Ge-s, Se-p | |
| FR | 1.00 | – | – | |
| BaRh2Ge4Te6 | ||||
| NR | 0.48 | Ba-d, Ge-p, Te-p | Rh-d, Ge-s, Te-p | |
| FR | 0.35 | – | – | |
| Compound | Atom | Charge | Volume Change |
|---|---|---|---|
| BaRh2Ge4S6 | Ba | 1.47 | −8.3 |
| Rh | −0.09 | 4.4 | |
| Ge | 0.78 | −9.4 | |
| 0.95 | −12.7 | ||
| S | −0.58 | 3.7 | |
| −0.81 | 7.0 | ||
| −0.99 | 7.7 | ||
| BaRh2Ge4Se6 | Ba | 1.41 | −8.3 |
| Rh | −0.21 | 5.2 | |
| Ge | 0.62 | −8.0 | |
| 0.72 | −10.1 | ||
| Se | −0.36 | 2.0 | |
| −0.60 | 4.6 | ||
| −0.87 | 6.7 | ||
| BaRh2Ge4Te6 | Ba | 1.32 | −6.4 |
| Rh | −0.40 | 7.1 | |
| Ge | 0.41 | 5.4 | |
| 0.40 | 5.8 | ||
| Te | −0.03 | −0.8 | |
| −0.31 | 0.6 | ||
| −0.72 | 5.1 | ||
| BaRh2Ge4S4Te2 | Ba | 1.50 | −9.6 |
| Rh | −0.43 | 7.3 | |
| Ge | 0.56 | −7.1 | |
| 0.93 | −13.4 | ||
| Te | −0.01 | −0.8 | |
| S | −0.82 | 6.8 | |
| −0.98 | 9.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulet, P.; Record, M.-C. Theoretical Investigations of the BaRh2Ge4X6 (X = S, Se, Te) Compounds. Energies 2020, 13, 6434. https://doi.org/10.3390/en13236434
Boulet P, Record M-C. Theoretical Investigations of the BaRh2Ge4X6 (X = S, Se, Te) Compounds. Energies. 2020; 13(23):6434. https://doi.org/10.3390/en13236434
Chicago/Turabian StyleBoulet, Pascal, and Marie-Christine Record. 2020. "Theoretical Investigations of the BaRh2Ge4X6 (X = S, Se, Te) Compounds" Energies 13, no. 23: 6434. https://doi.org/10.3390/en13236434