
Expanding the Chemical Space of Arsenicin A-C Related Polyarsenicals and Evaluation of Some Analogs as Inhibitors of Glioblastoma Stem Cell Growth
 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
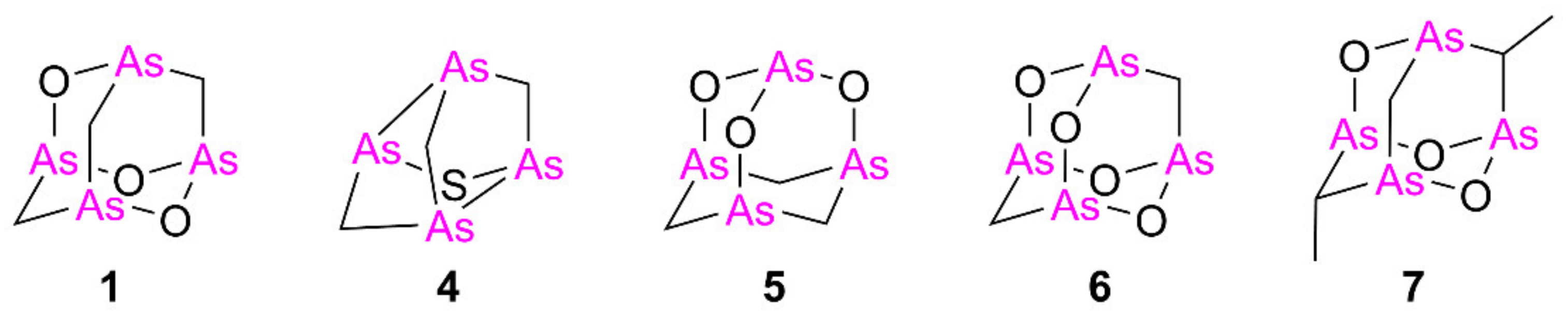
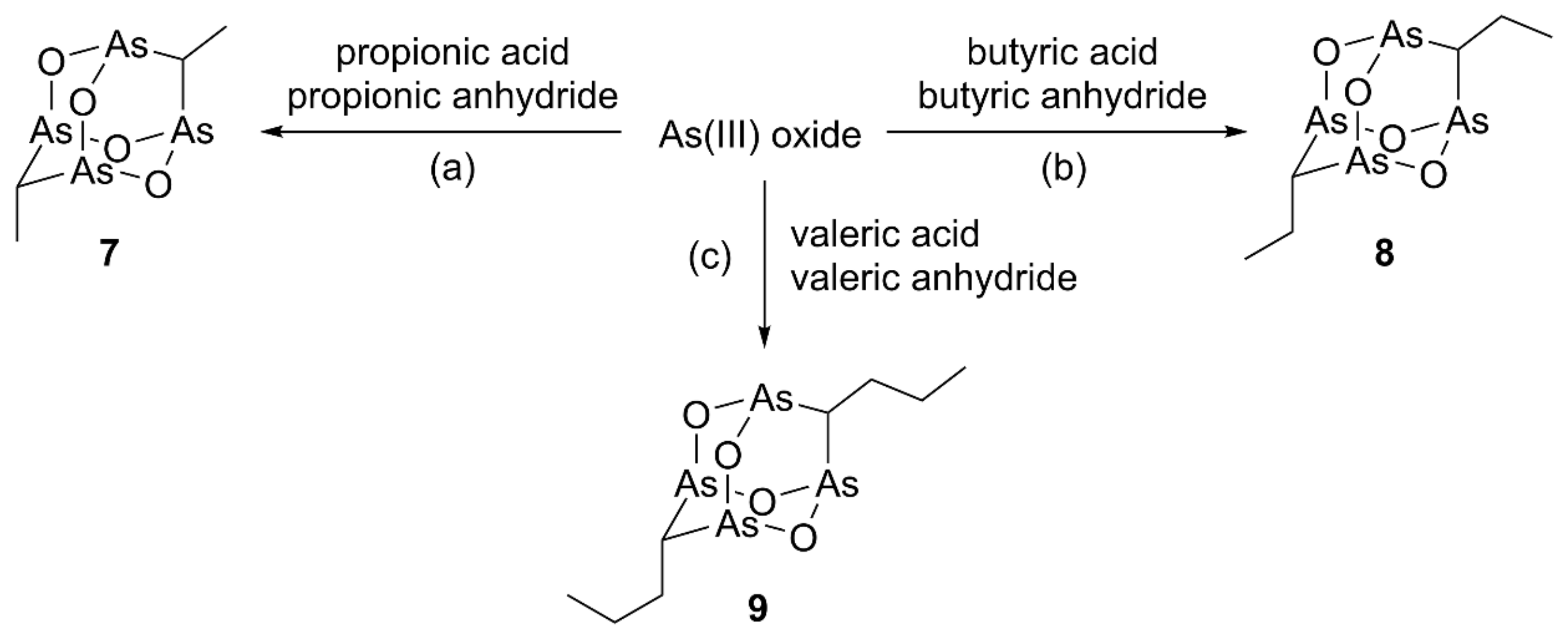
2.1.1. Synthesis of Alkyl Polyarsenical Analogs
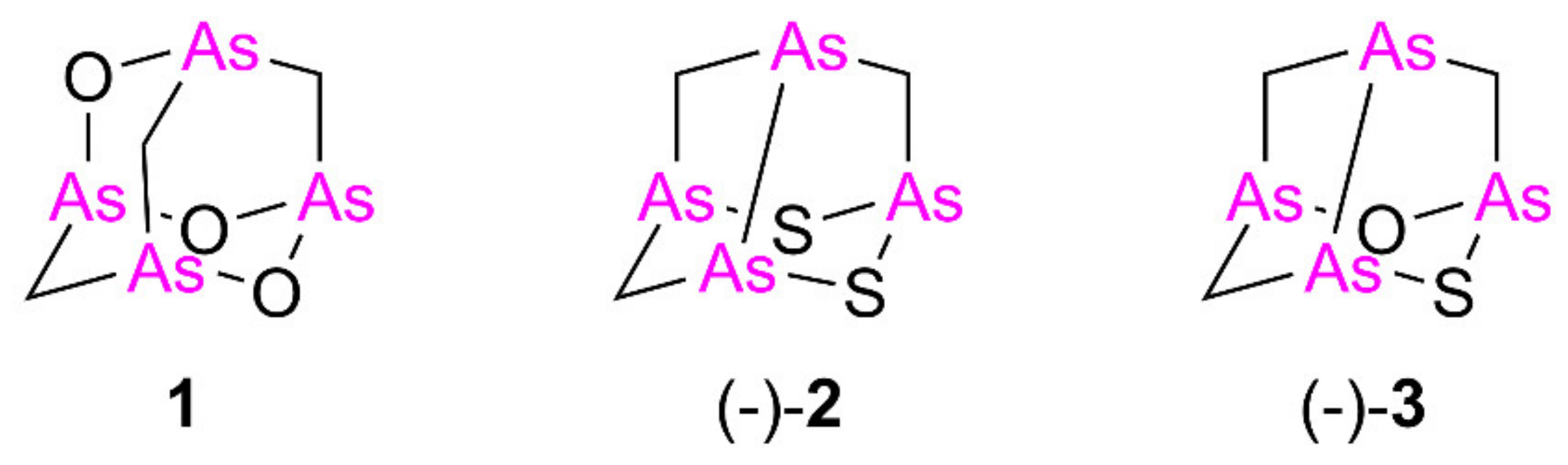
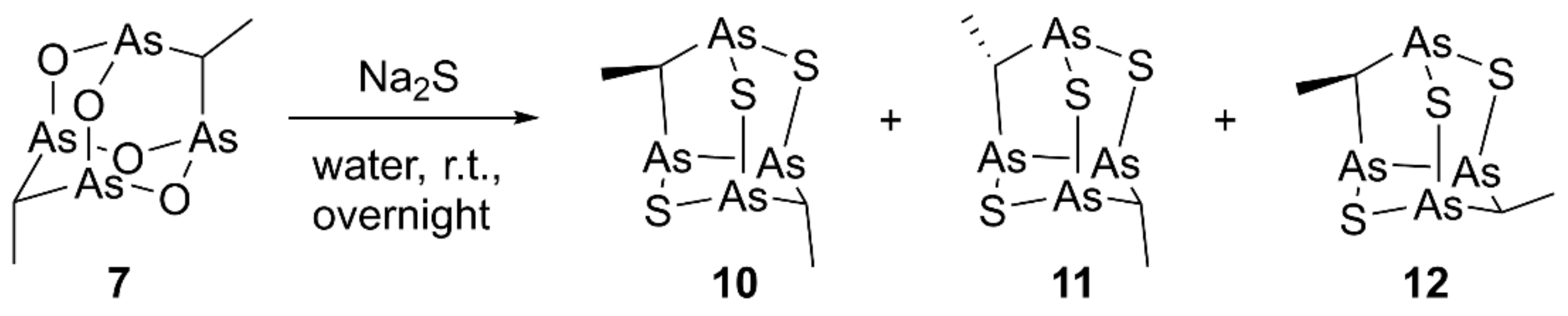
2.1.2. Synthesis of Arsenic–Sulfur Compounds 10–12
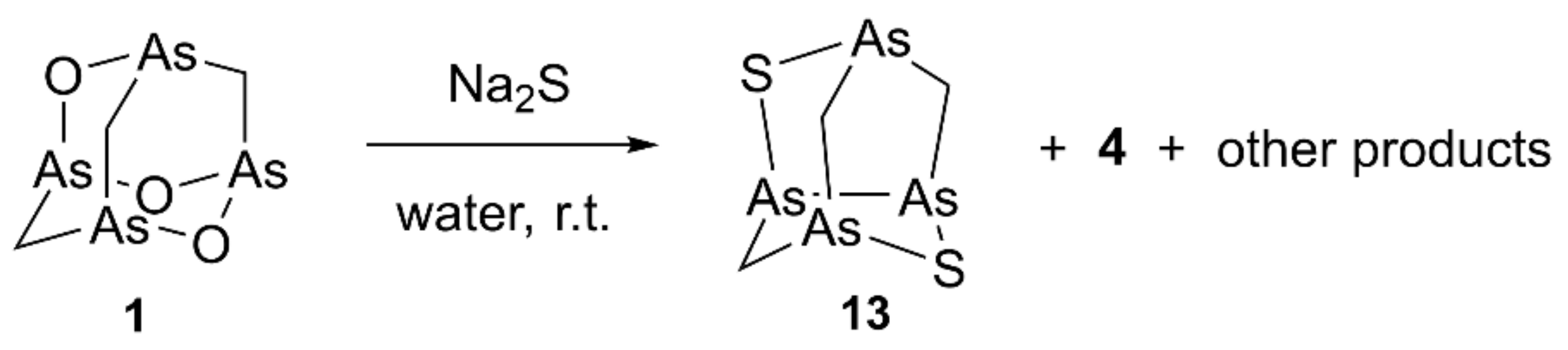
2.1.3. Identification of Natural Arsenicin D and Biogenetic Consideration
2.2. Biological Evaluation
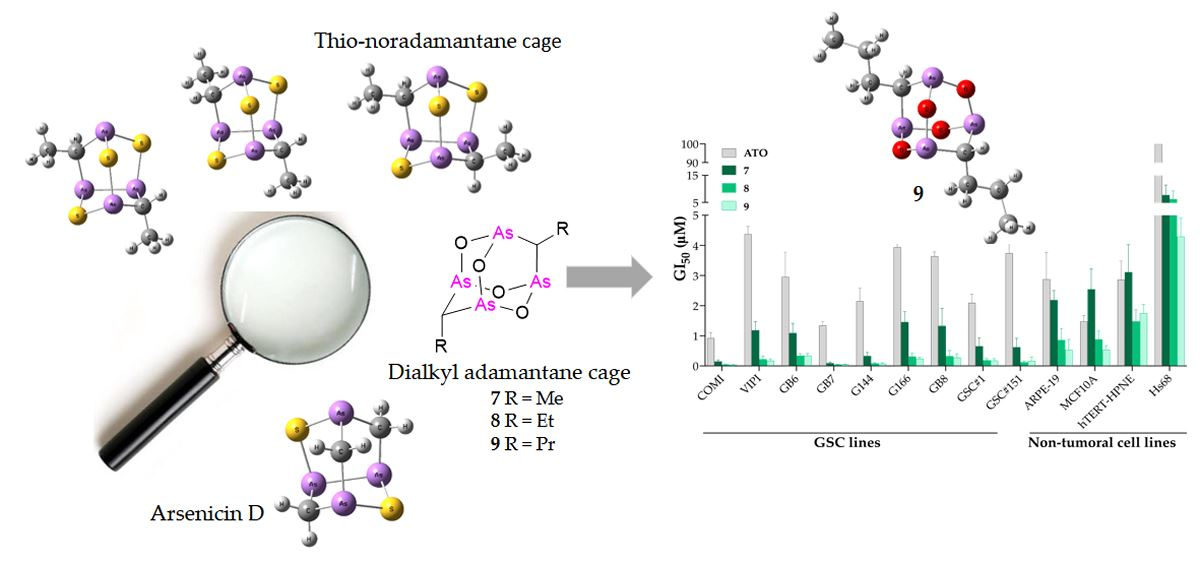
2.2.1. Alkyl Polyarsenicals Potently and Selectively Inhibit the Growth of GSCs
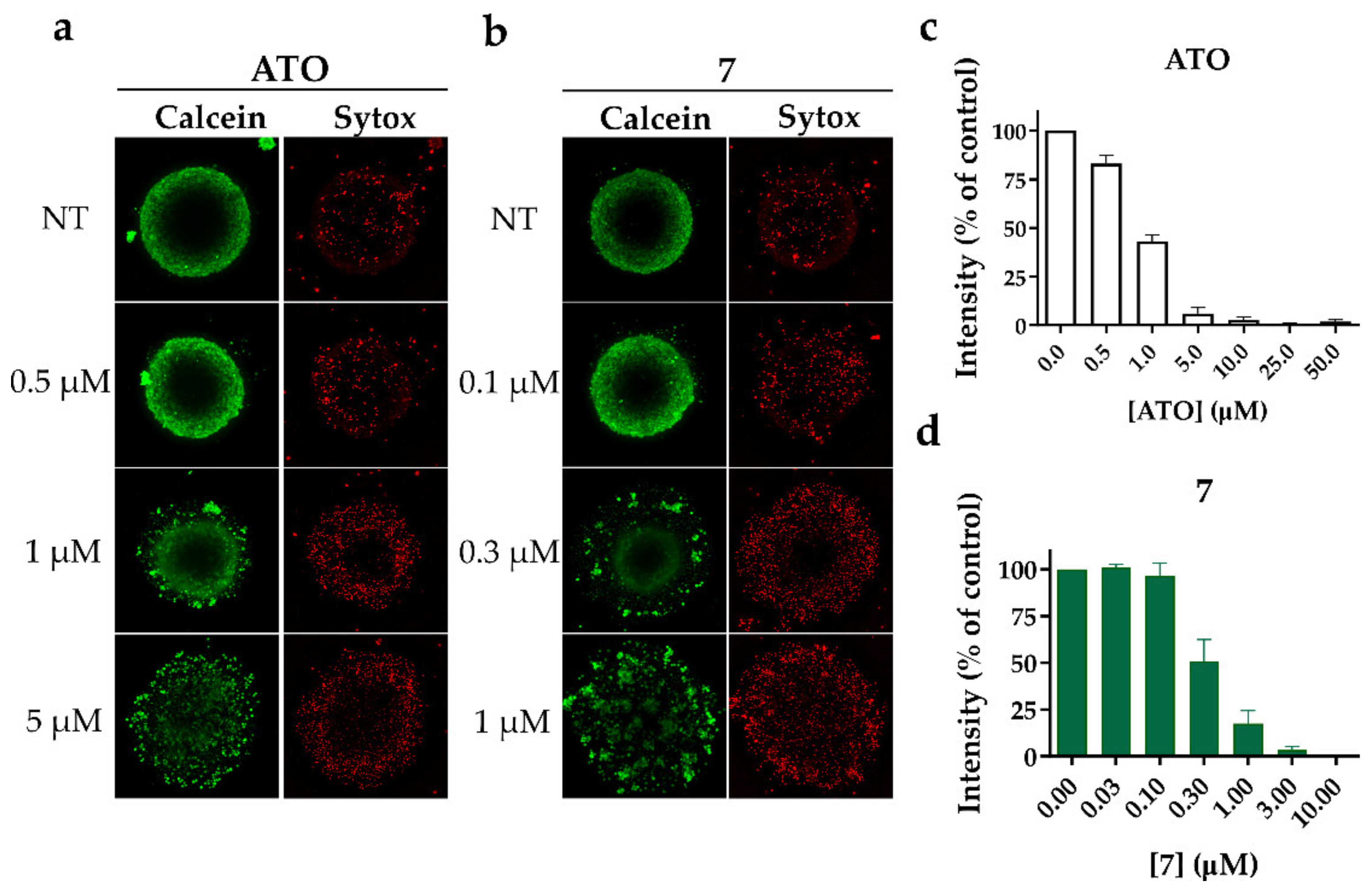
2.2.2. Alkyl Polyarsenicals Display Cytotoxic Effects also on GSC 3D Models
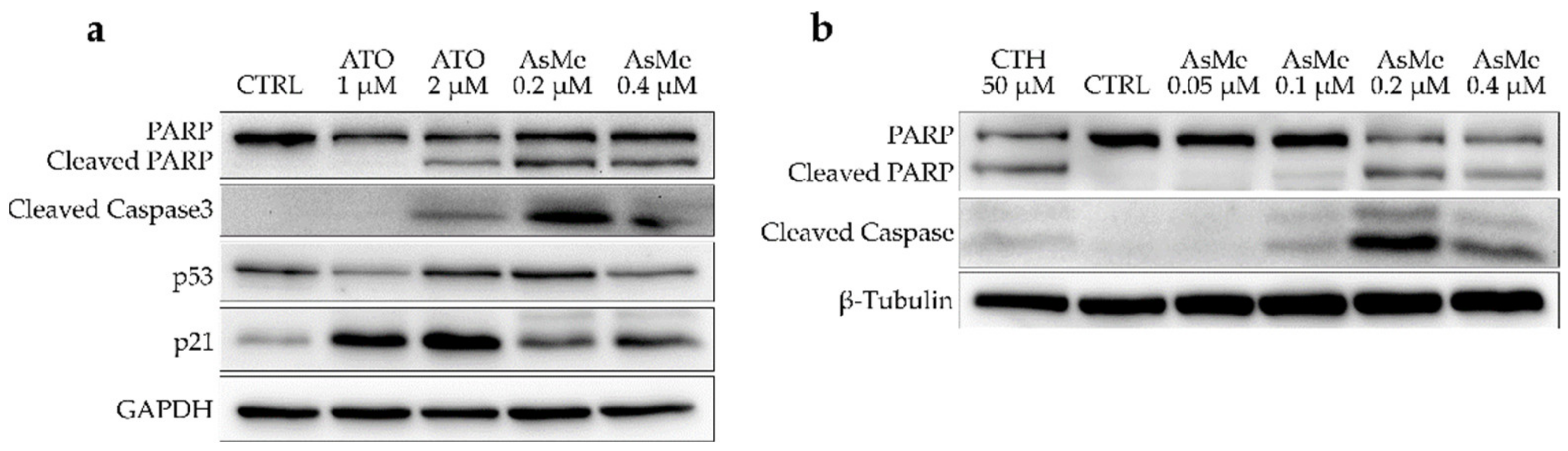
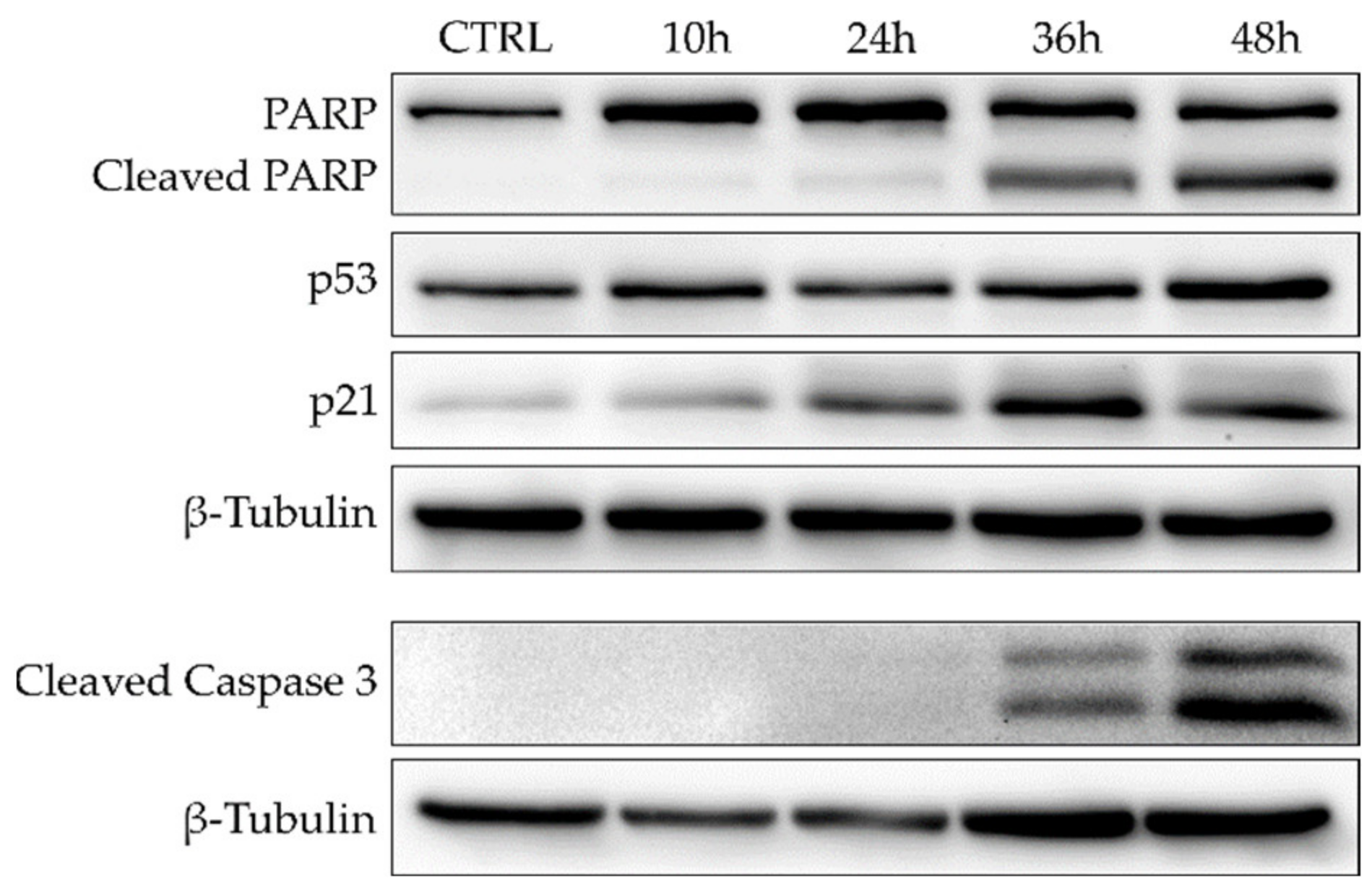
2.2.3. Alkyl Polyarsenicals Induce Apoptosis in GSCs
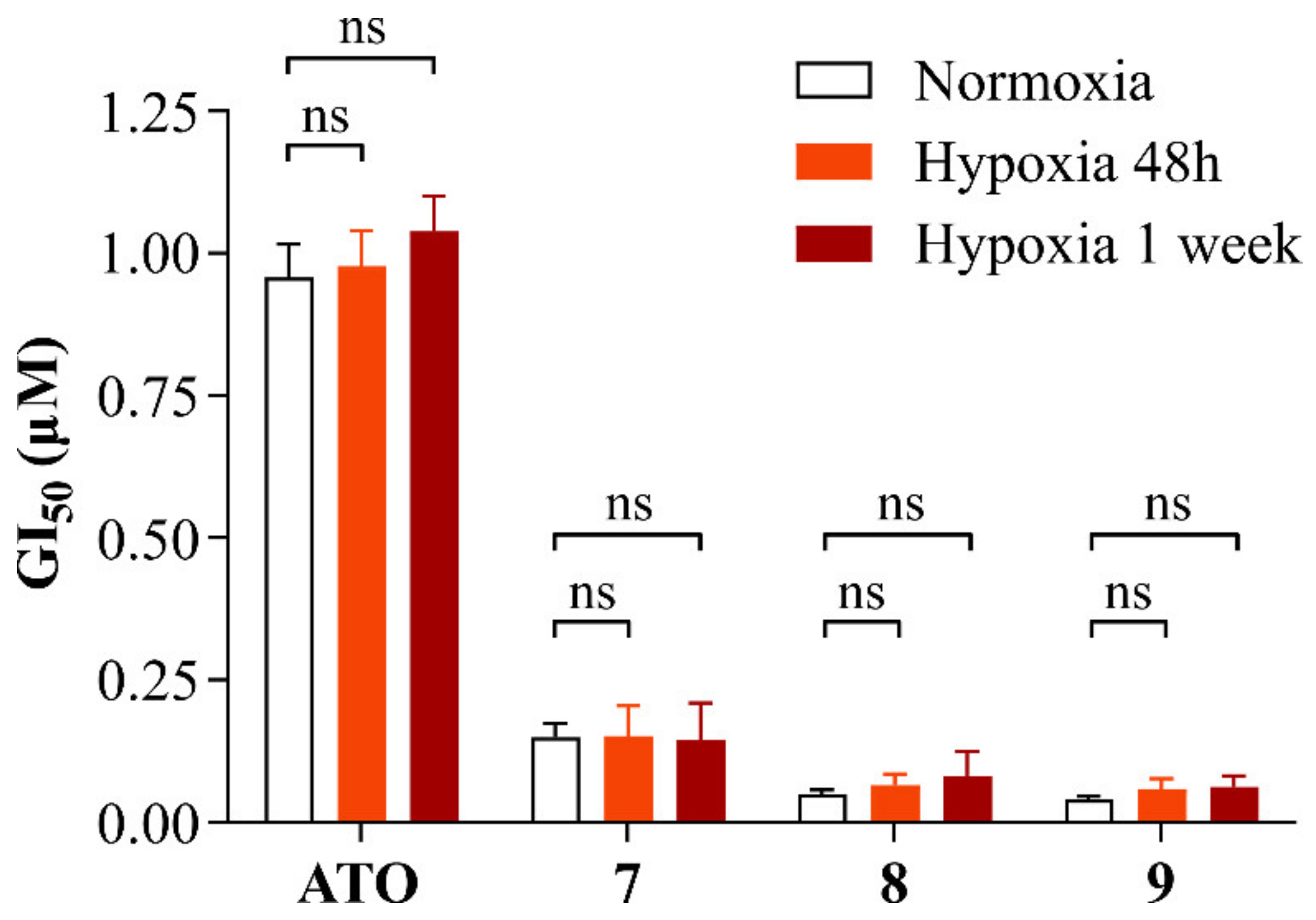
2.2.4. Alkyl Polyarsenicals Maintain their Potency under Hypoxic Conditions
2.3. Physicochemical Properties and ADME Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Procedures
3.1.2. Synthesis of Alkyl Analogs 7–9
9,10-Dimethyl-2,4,6,8-tetraoxa-1,3,5,7-tetraarsatricyclo[3.3.1.13,7]decane (7)
9,10-Diethyl-2,4,6,8-tetraoxa-1,3,5,7-tetraarsatricyclo[3.3.1.13,7]decane (8)
9,10-Dipropyl-2,4,6,8-tetraoxa-1,3,5,7-tetraarsatricyclo[3.3.1.13,7]decane (9)
3.1.3. Synthesis of Sulfur Analogs 10–12
3.1.4. Identification of Arsenicin D
Arsenicin D (2,6-dithia-1,3,5,7-tetraarsatricyclo[3.3.1.03,7]nonane, 13)
3.2. Computational Details
3.3. Biological Evaluation
3.3.1. Cell Culture
3.3.2. Preparation of Solutions of the Compounds to Be Tested
3.3.3. Treatments Performed Using Echo 650 and Data Analysis
3.3.4. Treatment of Gliomaspheres
3.3.5. Western Blot
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mancini, I.; Guella, G.; Frostin, M.; Hnawia, E.; Laurent, D.; Debitus, C.; Pietra, F. On the First Polyarsenic Organic Compound from Nature: Arsenicin A from the New Caledonian Marine Sponge Echinochalina bargibanti. Chem. Eur. J. 2006, 12, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Tähtinen, P.; Guella, G.; Saielli, G.; Debitus, C.; Hnawia, E.; Mancini, I. New Sulfur-Containing Polyarsenicals from the New Caledonian Sponge Echinochalina bargibanti. Mar. Drugs 2018, 16, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tähtinen, P.; Saielli, G.; Guella, G.; Mancini, I.; Bagno, A. Computational NMR Spectroscopy of Organoarsenicals and the Natural Polyarsenic Compound Arsenicin A. Chem. Eur. J. 2008, 14, 10445–10452. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Mariotto, G.; Rossi, B.; Viliani, G. Vibrational analysis as a powerful tool in structure elucidation of polyarsenicals: A DFT-based investigation of arsenicin A. Phys. Chem. Chem. Phys. 2009, 11, 2420–2427. [Google Scholar] [CrossRef]
- Mancini, I.; Defant, A. Bioactive poly(arsenic) compounds. In Progress in Molecular and Subcellular Biology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 175–195. [Google Scholar]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in medicine: Past, present and future. Biometals 2022, 1–19. [Google Scholar] [CrossRef]
- Chin, L.; Kumana, C.R.; Kwong, Y.-L.; Gill, H. The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics 2022, 14, 1945. [Google Scholar] [CrossRef]
- Sönksen, M.; Kerl, K.; Bunzen, H. Current Status and Future Prospects of Nanomedicine for Arsenic Trioxide Delivery to Solid Tumors. Med. Res. Rev. 2022, 42, 374–398. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, Z. Arsenic trioxide as a novel anti-glioma drug: A review. Cell. Mol. Biol. Lett. 2020, 25, 44. [Google Scholar] [CrossRef]
- Lu, D.; Coote, M.L.; Ho, J.; Kilah, N.L.; Lin, C.Y.; Salem, G.; Weir, M.L.; Willis, A.C.; Wild, S.B.; Dilda, P.J. Resolution and Improved Synthesis of (±)-Arsenicin A: A Natural Adamantane-Type Tetraarsenical Possessing Strong Anti-Acute Promelocytic Leukemia Cell Line Activity. Organometallics 2012, 31, 1808–1816. [Google Scholar] [CrossRef]
- Lu, D.; Arulmozhiraja, S.; Coote, M.L.; Rae, A.D.; Salem, G.; Willis, A.C.; Wild, S.B.; Benhenda, S.; Breitenbach, V.L.; De Thé, H.; et al. Sulfur Derivatives of the Natural Polyarsenical Arsenicin a: Biologically Active, Organometallic Arsenic-Sulfur Cages Related to the Minerals Realgar and Uzonite. Organometallics 2015, 34, 829–840. [Google Scholar] [CrossRef]
- Mancini, I.; Planchestainer, M.; Defant, A. Synthesis and In-vitro Anticancer Evaluation of Polyarsenicals Related to the Marine Sponge Derived Arsenicin A. Sci. Rep. 2017, 7, 11548. [Google Scholar] [CrossRef]
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the Treatment of Glioblastoma: Past, Present and Emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [Google Scholar] [CrossRef] [PubMed]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Marx, M.B.L.; Pritzkow, H.; Keppler, B.K. Struktur von S-9,10-Dimethyl-1,3,5,7-tetraarsa-2,4,6,8-tetraoxaadamantan und 9,10-Diethyl-1,3,5,7-tetraarsa-2,4,6,8-tetraoxaadamantan. Z. Anorg. Allg. Chem. 1996, 622, 1097–1100. [Google Scholar] [CrossRef]
- Toomsalu, E.; Burk, P. Critical Test of Some Computational Methods for Prediction of NMR 1H and 13C Chemical Shifts. J. Mol. Model. 2015, 21, 244–264. [Google Scholar] [CrossRef]
- Butt, N.Z.; Mohsin, F. Determination of Sulphide in Arabian Seawater using UV-Visible Spectrophotometer. Nucleus 2021, 58, 37–41. [Google Scholar]
- Fan, C.; Liu, G.; Long, Y.; Rosen, B.; Cai, Y. Thiolation in arsenic metabolism: A chemical perspective. Metallomics 2018, 10, 1368–1382. [Google Scholar] [CrossRef]
- Bagno, A. Computing the NMR Spectra of the Sponge Metabolite Arsenicin A: When Simple Becomes Difficult. J. Phys. Org. Chem. 2010, 23, 1016–1021. [Google Scholar] [CrossRef]
- Sighel, D.; Notarangelo, M.; Aibara, S.; Re, A.; Ricci, G.; Guida, M.; Soldano, A.; Adami, V.; Ambrosini, C.; Broso, F.; et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep. 2021, 35, 109024. [Google Scholar] [CrossRef]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of in-vitro bioassay methods: Application in herbal drug research. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: Cambridge, MA, USA, 2021; Chapter 6; Volume 46, pp. 273–307. [Google Scholar]
- Sighel, D.; Battistini, G.; Rosatti, E.F.; Vigna, J.; Pavan, M.; Belli, R.; Peroni, D.; Alessandrini, F.; Longhi, S.; Pancher, M.; et al. Streptogramin A derivatives as mitochondrial translation inhibitors to suppress glioblastoma stem cell growth. Eur. J. Med. Chem. 2023, 246, 114979. [Google Scholar] [CrossRef]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Raso, A.; Mascelli, S.; Nozza, P.; Garrè, M.L.; Pitto, F.; Ravetti, J.L.; et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int. J. Cancer 2015, 136, 1445–1457. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro-Oncology 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranmandura, H.; Xu, S.; Sawata, T.; Hao, W.H.; Liu, H.; Bu, N.; Ogra, Y.; Lou, Y.J.; Suzuki, N. Mitochondria are the main target organelle for trivalent monomethylarsonous acid (MMA(III))-induced cytotoxicity. Chem. Res. Toxicol. 2011, 24, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [Green Version]
- Swiss ADME. Available online: http://www.swissadme.ch/ (accessed on 9 January 2023).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717–42728. [Google Scholar] [CrossRef] [Green Version]
- Carmignani, M.; Volpe, A.R.; Aldea, M.; Soritau, O.; Irimie, A.; Florian, I.S.; Tomuleasa, C.; Baritchii, A.; Petrushev, B.; Crisan, G.; et al. Glioblastoma stem cells: A new target for metformin and arsenic trioxide. J. Biol. Regul. Homeost. Agents 2014, 28, 1–15. [Google Scholar]
- Molsft, L.L.C. Available online:https://www.molsoft.com/ (accessed on 2 March 2023).
- Molinspiration. Available online: https://www.molinspiration.com/ (accessed on 2 March 2023).
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-Functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Handy, N.C.; Cohen, A. Left-Right correlation energy. J. Mol. Phys. 2001, 99, 403–412. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.J.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scalmani, G.; Scuseria, G.E. Can short-range hybrids describe long-range-dependent properties? J. Chem. Phys. 2009, 131, 044108. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ditchfield, R. Molecular Orbital Theory of Magnetic Shielding and Magnetic Susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Baio, G.; Neumaier, C.E.; Vagge, S.; Corvò, R.; Brisigotti, M.P.; Ravetti, J.L.; et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 2014, 135, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative Analysis of DNA Repair in Stem and Nonstem Glioma Cell Cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Carén, H.; Stricker, S.H.; Bulstrode, H.; Gagrica, S.; Johnstone, E.; Bartlett, T.E.; Feber, A.; Wilson, G.; Teschendorff, A.E.; Bertone, P.; et al. Glioblastoma stem cells respond to differentiation cues but fail to undergo commitment and terminal cell-cycle arrest. Stem Cell Rep. 2015, 5, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Conti, L.; Crisafulli, L.; Caldera, V.; Tortoreto, M.; Brilli, E.; Conforti, P.; Zunino, F.; Magrassi, L.; Schiffer, D.; Cattaneo, E. REST Controls Self-Renewal and Tumorigenic Competence of Human Glioblastoma Cells. PLoS ONE 2012, 7, e38486. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandris, Q.G.; Biffoni, M.; Martini, M.; Runci, D.; Buccarelli, M.; Cenci, T.; Signore, M.; Stancato, L.; Olivi, A.; De Maria, R.; et al. The clinical value of patient-derived glioblastoma tumorspheres in predicting treatment response. Neuro-Oncology 2017, 19, 1097–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Minimized Structure | Exptl. | Calcd. | Δ | Assignment |
|---|---|---|---|---|---|
| 10 |  | 48.8 | 57.00 | 8.20 | C(7) |
| 43.0 | 52.29 | 9.29 | C(1) | ||
| 20.0 | 19.28 | −0.52 | C(8) | ||
| 13.3 | 12.41 | −0.89 | C(2) | ||
| 3.97 | 3.57 | −0.40 | H-C(7) | ||
| 2.51 | 2.42 | −0.09 | H-(C1) | ||
| 2.12 | 2.43 | 0.31 | H3C(2) | ||
| 1.11 | 1.04 | −0.07 | H3C(8) | ||
| 11 |  | 48.8 | 56.71 | 7.91 | C(1), C(7) |
| 19.8 | 19.05 | −0.75 | C(2), C(8) | ||
| 3.76 | 3.27 | −0.49 | H-C(1), H-C(7) | ||
| 1.07 | 0.98 | −0.09 | H3C(2), H3C(8) | ||
| 12 |  | 42.5 | 51.95 | 9.45 | C(1), C(7) |
| 12.9 | 12.37 | −0.53 | C(2), C(8) | ||
| 2.61 | 2.56 | −0.05 | H-C(1), H-C(7) | ||
| 2.21 | 2.54 | 0.33 | H3C(2), H3C(8) |
| ATO | 7 | Ratio b ATO/7 | 8 | Ratio b ATO/8 | 9 | Ratio b ATO/9 | ||
|---|---|---|---|---|---|---|---|---|
| GSC lines | COMI | 0.93 ± 0.18 | 0.15 ± 0.05 | 6.2 | 0.05 ± 0.02 | 18.6 | 0.04 ± 0.01 | 23.2 |
| VIPI | 4.37 ± 0.26 | 1.19 ± 0.28 | 3.7 | 0.22 ± 0.11 | 19.9 | 0.18 ± 0.06 | 24.3 | |
| GB6 | 2.96 ± 0.82 | 1.10 ± 0.32 | 2.7 | 0.34 ± 0.07 | 8.7 | 0.34 ± 0.07 | 8.7 | |
| GB7 | 1.35 ± 0.13 | 0.09 ± 0.03 | 15.0 | 0.05 ± 0.01 | 27.0 | 0.05 ± 0.01 | 27.0 | |
| G144 | 2.15 ± 0.43 | 0.33 ± 0.12 | 6.5 | 0.09 ± 0.02 | 23.9 | 0.08 ± 0.02 | 26.9 | |
| G166 | 3.95 ± 0.31 | 1.46 ± 0.34 | 2.7 | 0.31 ± 0.11 | 12.7 | 0.24 ± 0.05 | 16.5 | |
| GB8 | 3.65 ± 0.14 | 1.33 ± 0.58 | 2.7 | 0.33 ± 0.18 | 11.1 | 0.28 ± 0.11 | 13.0 | |
| GSC#1 | 2.10 ± 0.28 | 0.66 ± 0.28 | 3.2 | 0.19 ± 0.06 | 11.1 | 0.18 ± 0.06 | 11.7 | |
| GSC#151 | 4.0 ± 2.0 | 0.62 ± 0.29 | 6.5 | 0.12 ± 0.04 | 33.3 | 0.17 ± 0.13 | 23.5 | |
| Non-tumor cell lines | ARPE-19 | 2.88 ± 0.89 | 2.19 ± 0.32 | 0.86 ± 0.38 | 0.54 ± 0.34 | |||
| MCF10A | 1.49 ± 0.18 | 2.54 ± 0.67 | 0.89 ± 0.27 | 0.53 ± 0.14 | ||||
| hTERT-HPNE | 2.87 ± 0.62 | 3.12 ± 0.91 | 1.48 ± 0.38 | 1.75 ± 0.29 | ||||
| Hs68 | >100 | 7.78 ± 3.64 | 6.2 ± 3.0 | 4.29 ± 0.61 | ||||
| SI c | ARPE-19 | 0.7–3.1 | 1.5–24.3 | 2.5–17.2 | 1.6–13.5 | |||
| MCF10A | 0.3–1.6 | 1.7–28.2 | 2.6–17.8 | 1.6–13.3 | ||||
| hTERT-HPNE | 0.7–3.1 | 2.1–34.7 | 4.4–29.6 | 5.1–43.8 | ||||
| Hs68 | nd | 5.3–86.4 | 18.2–124.0 | 12.6–107.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vigna, J.; Sighel, D.; Rosatti, E.F.; Defant, A.; Pancher, M.; Sidarovich, V.; Quattrone, A.; Mancini, I. Expanding the Chemical Space of Arsenicin A-C Related Polyarsenicals and Evaluation of Some Analogs as Inhibitors of Glioblastoma Stem Cell Growth. Mar. Drugs 2023, 21, 186. https://doi.org/10.3390/md21030186
Vigna J, Sighel D, Rosatti EF, Defant A, Pancher M, Sidarovich V, Quattrone A, Mancini I. Expanding the Chemical Space of Arsenicin A-C Related Polyarsenicals and Evaluation of Some Analogs as Inhibitors of Glioblastoma Stem Cell Growth. Marine Drugs. 2023; 21(3):186. https://doi.org/10.3390/md21030186
Chicago/Turabian StyleVigna, Jacopo, Denise Sighel, Emanuele Filiberto Rosatti, Andrea Defant, Michael Pancher, Viktoryia Sidarovich, Alessandro Quattrone, and Ines Mancini. 2023. "Expanding the Chemical Space of Arsenicin A-C Related Polyarsenicals and Evaluation of Some Analogs as Inhibitors of Glioblastoma Stem Cell Growth" Marine Drugs 21, no. 3: 186. https://doi.org/10.3390/md21030186