
LC-HRMS Profiling of Paralytic Shellfish Toxins in Mytilus galloprovincialis after a Gymnodinium catenatum Bloom


Abstract
:1. Introduction
2. Results
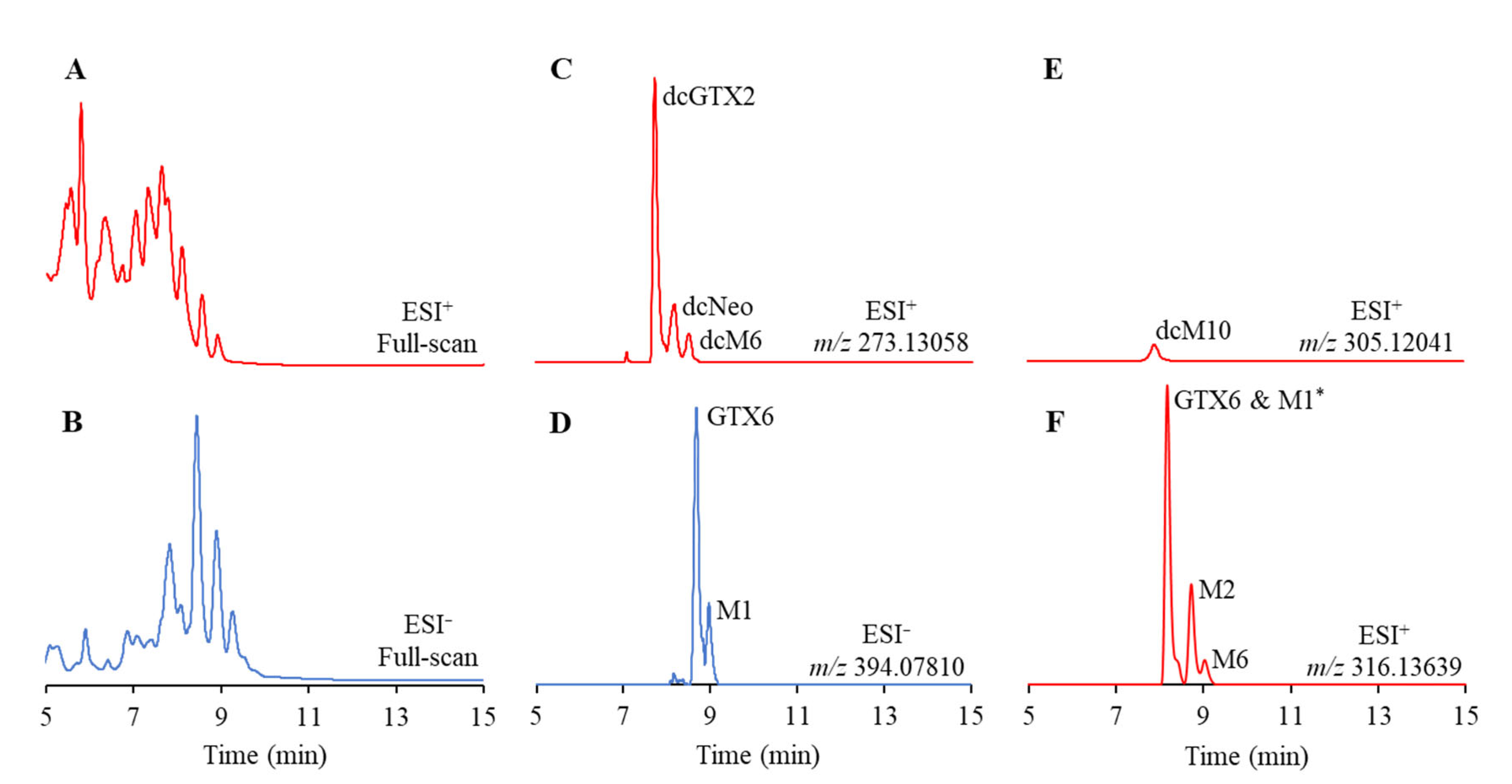
2.1. Accurate Mass-Extracted Ion Chromatogram (AM-XIC)-Based Quantitation
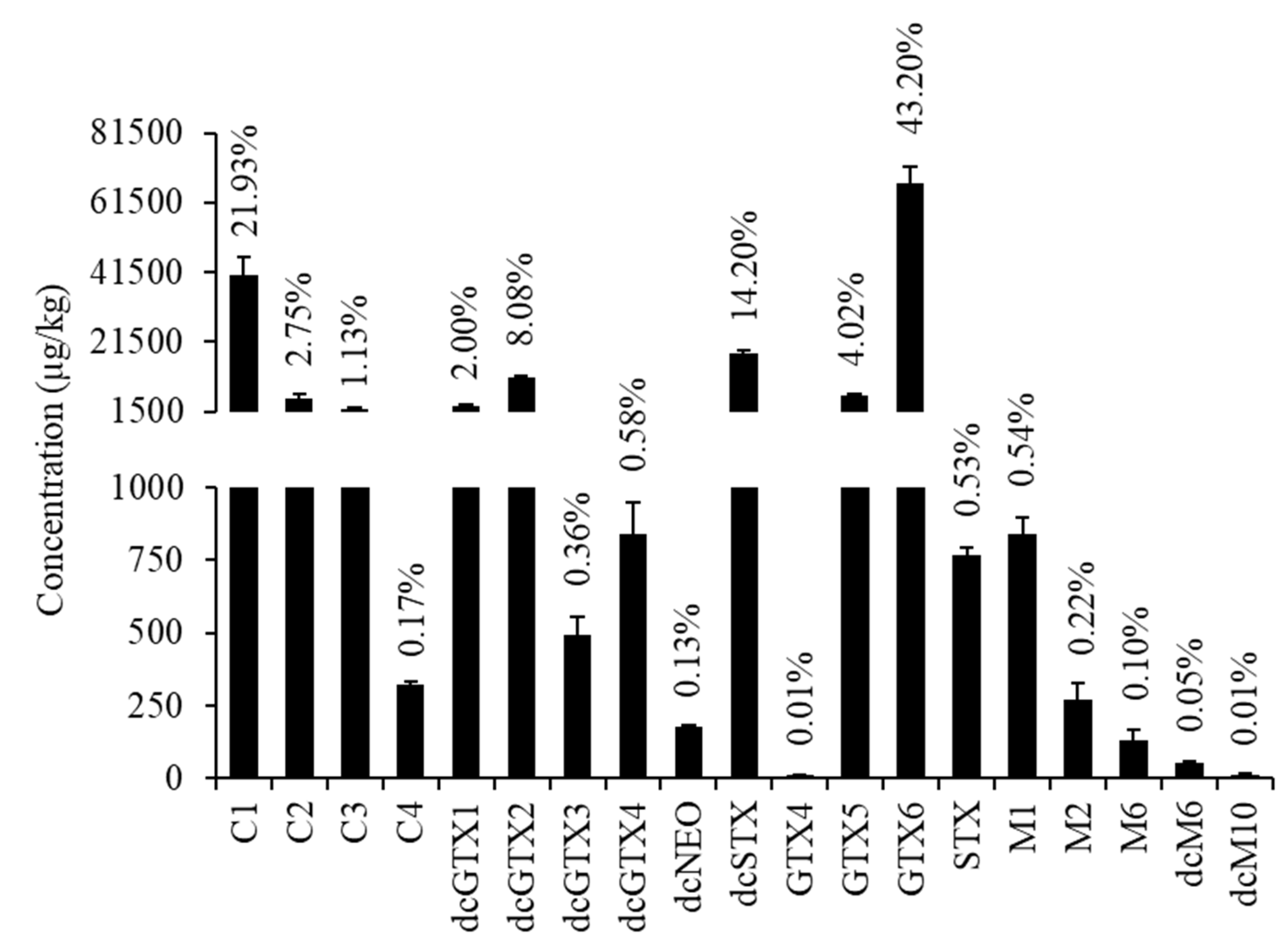
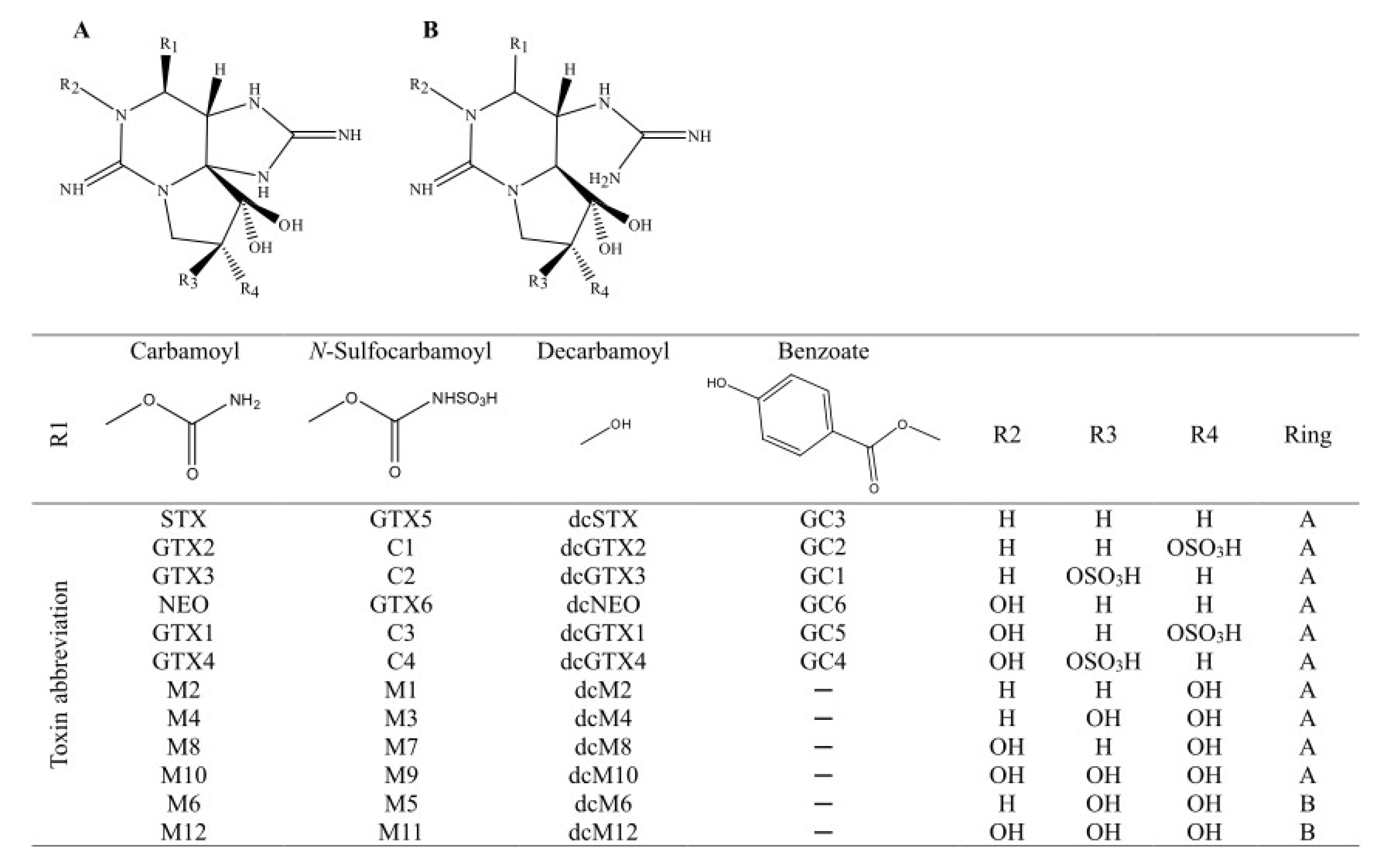
2.2. PSTs Profile
2.3. PST Toxic Equivalent Quantity
2.4. Matrix Effect and Limits of Detection and Quantification
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.3. LC-HRMS Analysis
4.4. PSTs’ Quantitation and Profile Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shumway, S.E. Phycotoxin-related shellfish poisoning: Bivalve molluscs are not the only vectors. Rev. Fish. Sci. 1995, 3, 1–31. [Google Scholar] [CrossRef]
- Bricelj, V.M.; Shumway, S.E. Paralytic Shellfish Toxins in Bivalve Molluscs: Occurrence, Transfer Kinetics, and Biotransformation. Rev. Fish. Sci. 1998, 6, 315–383. [Google Scholar] [CrossRef]
- Deeds, J.R.; Landsberg, J.H.; Etheridge, S.M.; Pitcher, G.C.; Longan, S.W. Non-Traditional Vectors for Paralytic Shellfish Poisoning. Mar. Drugs 2008, 6, 308–348. [Google Scholar] [CrossRef] [PubMed]
- FAO. Food and Nutrition Paper 80; Food and Agriculture Organization of the United Nations: Rome, Italy, 2004. [Google Scholar]
- Codex STAN292-2008; Standard for Live and Raw Bivalve Molluscs. Codex Alimentarius Commission: Rome, Italy, 2008; p. 292.
- Wiese, M.; D’agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y. Chemistry and mechanism of action. In Food Science and Tecnology; Marcel Dekker: New York, NY, USA, 2000; pp. 151–172. [Google Scholar]
- World Health Organization. Toxicity Equivalence Factors for Marine Biotoxins Associated with Bivalve Molluscs; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Selwood, A.I.; Waugh, C.; Harwood, D.T.; Rhodes, L.L.; Reeve, J.; Sim, J.; Munday, R. Acute Toxicities of the Saxitoxin Congeners Gonyautoxin 5, Gonyautoxin 6, Decarbamoyl Gonyautoxin 2 & 3, Decarbamoyl Neosaxitoxin, C-1&2 and C-3& 4 to Mice by Various Routes of Administration. Toxins 2017, 9, 73. [Google Scholar]
- Dell’Aversano, C.; Walter, J.A.; Burton, I.W.; Stirling, D.J.; Fattorusso, E.; Quilliam, M.A. Isolation and structure elucidation of new and unusual saxitoxin analogues from mussels. J. Nat. Prod. 2008, 71, 1518–1523. [Google Scholar] [CrossRef]
- Vale, P. New saxitoxin analogues in the marine environment: Developments in toxin chemistry, detection and biotransformation during the 2000s. Phytochem. Rev. 2010, 9, 525–535. [Google Scholar] [CrossRef]
- Li, A.; Ma, J.; Cao, J.; Wang, Q.; Yu, R.; Thomas, K.; Quilliam, M. Analysis of paralytic shellfish toxins and their metabolites in shellfish from the North Yellow Sea of China. Food Addit. Contam. A 2012, 29, 1455–1464. [Google Scholar] [CrossRef]
- Ding, L.; Qiu, J.; Li, A. Proposed biotransformation pathways for new metabolites of paralytic shellfish toxins based on field and experimental mussel samples. J. Agric. Food Chem. 2017, 65, 5494–5502. [Google Scholar] [CrossRef]
- Quilliam, M.; Li, A.; Lewis, N.; McCarron, P.; Thomas, K.; Walter, J. Biotransformation and chemical degradation of paralytic shellfish toxins in mussels. In Proceedings of the 17th International Conference on Harmful Algae, Florianópolis, Brazil, 9–14 October 2016; pp. 9–14. [Google Scholar]
- Qiu, J.; Meng, F.; Ding, L.; Che, Y.; McCarron, P.; Beach, D.G.; Li, A. Dynamics of paralytic shellfish toxins and their metabolites during timecourse exposure of scallops Chlamys farreri and mussels Mytilus galloprovincialis to Alexandrium pacificum. Aquat. Toxicol. 2018, 200, 233–240. [Google Scholar] [CrossRef]
- Qiu, J.; Rafuse, C.; Lewis, N.I.; Li, A.; Meng, F.; Beach, D.G.; McCarron, P. Screening of cyclic imine and paralytic shellfish toxins in isolates of the genus Alexandrium (Dinophyceae) from Atlantic Canada. Harmful Algae 2018, 77, 108–118. [Google Scholar] [CrossRef] [PubMed]
- EFSA, Scientific opinion of the panel on contaminants in the food chain on a request from the European Commission on Marine biotoxins in shellfish -Saxitoxin group. EFSA J. 2009, 7, 1–76.
- Negri, A.P.; Bolch, C.J.; Geier, S.; Green, D.H.; Park, T.-G.; Blackburn, S.I. Widespread presence of hydrophobic paralytic shellfish toxins in Gymnodinium catenatum. Harmful Algae 2007, 6, 774–780. [Google Scholar] [CrossRef]
- Llewellyn, L.; Negri, A.; Quilliam, M. High affinity for the rat brain sodium channel of newly discovered hydroxybenzoate saxitoxin analogues from the dinoflagellate Gymnodinium catenatum. Toxicon 2004, 43, 101–104. [Google Scholar] [CrossRef] [PubMed]
- AOAC Official Method 2005.06 Quantitative Determination of Paralytic Shellfish Poisoning Toxins in Shellfish Using Pre-chromatographic Oxidation and Liquid Chromatography with Fluorescence Detection; AOAC International: Gaithersburg, MD, USA, 2005.
- Vale, P. Fate of benzoate paralytic shellfish poisoning toxins from Gymnodinium catenatum in shellfish and fish detected by pre-column oxidation and liquid chromatography with fluorescence detection. J. Chromatogr. A 2008, 1190, 191–197. [Google Scholar] [CrossRef]
- Qiu, J.; Wright, E.J.; Thomas, K.; Li, A.; McCarron, P.; Beach, D.G. Semiquantitation of paralytic shellfish toxins by hydrophilic interaction liquid chromatography-mass spectrometry using relative molar response factors. Toxins 2020, 12, 398. [Google Scholar] [CrossRef]
- Turner, A.D.; McNabb, P.S.; Harwood, D.T.; Selwood, A.I.; Boundy, M.J. Single-laboratory validation of a multitoxin ultra-performance LC-hydrophilic interaction LC-MS/MS method for quantitation of paralytic shellfish toxins in bivalve shellfish. J. AOAC Int. 2015, 98, 609–621. [Google Scholar] [CrossRef]
- Boundy, M.J.; Selwood, A.I.; Harwood, D.T.; McNabb, P.S.; Turner, A.D. Development of a sensitive and selective liquid chromatography–mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 2015, 1387, 1–12. [Google Scholar] [CrossRef]
- Etheridge, S.M. Paralytic shellfish poisoning: Seafood safety and human health perspectives. Toxicon 2010, 56, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, I.; Alfonso, A.; González-Jartín, J.M.; Vieytes, M.R.; Botana, L.M. A single run UPLC-MS/MS method for detection of all EU-regulated marine toxins. Talanta 2018, 189, 622–628. [Google Scholar] [CrossRef]
- Costa, C.Q.V.; Afonso, I.I.; Lage, S.; Costa, P.R.; Canário, A.V.M.; Da Silva, J.P. Quantitation Overcoming Matrix Effects of Lipophilic Toxins in Mytilus galloprovincialis by Liquid Chromatography-Full Scan High Resolution Mass Spectrometry Analysis (LC-HR-MS). Mar. Drugs 2022, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Zendong, Z.; McCarron, P.; Herrenknecht, C.; Sibat, M.; Amzil, Z.; Cole, R.B.; Hess, P. High resolution mass spectrometry for quantitative analysis and untargeted screening of algal toxins in mussels and passive samplers. J. Chromatogr. A 2015, 1416, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Révész, Á.; Hevér, H.; Steckel, A.; Schlosser, G.; Szabó, D.; Vékey, K.; Drahos, L. Collision energies: Optimization strategies for bottom-up proteomics. Mass Spectrom. Rev. 2021, e21763. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.R.; Braga, A.C.; Turner, A.D. Accumulation and elimination dynamics of the hydroxybenzoate saxitoxin analogues in mussels Mytilus galloprovincialis exposed to the toxic marine dinoflagellate Gymnodinium catenatum. Toxins 2018, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, J.J.; Iwaoka, W.T.; Liston, J. Enzymatic transformation of PSP toxins in the littleneck clam (Protothacastaminea). Biochem. Biophys. Res. Commun. 1983, 114, 465–472. [Google Scholar] [CrossRef]
- Oshima, Y. Chemical and enzymatic transformation of paralytic shellfish toxins in marine organisms. In Harmful Marine Algal Blooms; Lassus, P., Arzul, G., Gentien, P., Marcaillou, C., Eds.; Lavoisier Publishine Intercept Ltd.: Paris, France, 1995; pp. 475–480. [Google Scholar]
- Rogatsky, E.; Stein, D. Evaluation of matrix effect and chromatography efficiency: New parameters for validation of method development. J. Am. Soc. Mass Spectrom. 2005, 16, 1757–1759. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.R.; Moita, T.; Rodrigues, S.M. Estimating the contribution of N-sulfocarbamoyl paralytic shellfish toxin analogs GTX6 and C3+ 4 to the toxicity of mussels (Mytilus galloprovincialis) over a bloom of Gymnodinium catenatum. Harmful Algae 2014, 31, 35–40. [Google Scholar] [CrossRef]
- Carvalho, I.L.; Pelerito, A.; Ribeiro, I.; Cordeiro, R.; Núncio, M.S.; Vale, P. Paralytic shellfish poisoning due to ingestion of contaminated mussels: A 2018 case report in Caparica (Portugal). Toxicon X 2019, 4, 100017. [Google Scholar] [CrossRef]
- IPMA. Resultados das Determinações de Biotoxinas Marinhas. 2019. Available online: https://www.ipma.pt/pt/bivalves/biotox/ (accessed on 1 September 2022). (In Portuguese).
- IPMA. Resultados das Determinações de Fitoplâncton Nocivo. 2019. Available online: https://www.ipma.pt/pt/bivalves/fito/ (accessed on 1 September 2022). (In Portuguese).
- Costa, P.R.; Robertson, A.; Quilliam, M.A. Toxin profile of Gymnodinium catenatum (Dinophyceae) from the Portuguese coast, as determined by liquid chromatography tandem mass spectrometry. Mar. Drugs 2015, 13, 2046–2062. [Google Scholar] [CrossRef] [Green Version]
- Che, Y.; Ding, L.; Qiu, J.; Ji, Y.; Li, A. Conversion and stability of new metabolites of paralytic shellfish toxins under different temperature and pH conditions. J. Agric. Food Chem. 2020, 68, 1427–1435. [Google Scholar] [CrossRef]
- Artigas, M.L.; Vale, P.J.V.; Gomes, S.S.; Botelho, M.J.; Rodrigues, S.M.; Amorim, A. Profiles of paralytic shellfish poisoning toxins in shellfish from Portugal explained by carbamoylase activity. J. Chromatogr. A 2007, 1160, 99–105. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| PST Analogue | Positive Ion | Calculated Exact Mass | Negative Ion | Calculated Exact Mass | |
|---|---|---|---|---|---|
| Carbamoyl | STX | [M + H]+ | 300.14148 | [M + HCOO]− | 344.1324 |
| GTX2 | [M + H – SO3]+ | 316.13639 | [M – H]− | 394.07810 | |
| GTX3 | [M + H]+ | 396.09321 | |||
| NEO | [M + H]+ | 316.13639 | [M + HCOO]− | 360.12732 | |
| GTX1 | [M + H – SO3]+ | 332.13131 | [M – H]− | 410.07302 | |
| GTX4 | [M + H]+ | 412.08812 | |||
| M2 | [M + H]+ | 316.13639 | [M + HCOO]− | 360.12732 | |
| M4/M8 | [M + H]+ | 332.13131 | [M + HCOO]− | 376.12223 | |
| M10 | [M + H]+ | 348.12622 | [M + HCOO]− | 392.11715 | |
| M6 | [M + H – H2O]+ | 316.13639 | [M + HCOO – H2O]− | 360.12732 | |
| M12 | [M + H] | 350.14187 | [M – H]− | 348.12677 | |
| N–Sulfocarbamoyl | GTX5 | [M + H]+ | 380.09829 | [M – H]− | 378.08319 |
| C1/C2 | [M + H]+ | 476.05002 | [M – H]− | 474.03492 | |
| [M + H – SO3]+ | 396.09321 | ||||
| GTX6 | [M + H]+ | 396.09321 | [M – H]− | 394.07810 | |
| C3/C4 | [M + H]+ | 492.04494 | [M – H]− | 490.02983 | |
| [M + H – SO3]+ | 412.08812 | ||||
| M1 | [M + H]+ | 396.09321 | [M – H]− | 394.07810 | |
| M3/M7 | [M + H]+ | 412.08812 | [M – H]− | 410.07302 | |
| M9 | [M + H]+ | 428.08304 | [M – H]− | 426.34005 | |
| M5 | [M + H – H2O]+ | 396.09321 | [M – H – H2O]− | 394.07810 | |
| M11 | [M + H]+ | 430.09869 | [M–H]− | 428.08358 | |
| Decarbamoyl | dcSTX | [M + H]+ | 257.13566 | [M + HCOO]− | 301.12659 |
| dcGTX2 | [M + H – SO3]+ | 273.13058 | [M – H]− | 351.07229 | |
| dcGTX3 | [M + H]+ | 353.08739 | |||
| dcNEO | [M + H]+ | 273.13058 | [M + HCOO]− | 317.12151 | |
| dcGTX1 | [M + H – SO3]+ | 289.12549 | [M – H]− | 367.06720 | |
| dcGTX4 | [M + H]+ | 369.08231 | |||
| dcM2 | [M + H]+ | 273.13058 | [M + HCOO]− | 317.12151 | |
| dcM4/dcM8 | [M + H]+ | 289.12549 | [M + HCOO]− | 333.11642 | |
| dcM10 | [M + H]+ | 305.12041 | [M + HCOO]− | 349.11134 | |
| dcM6 | [M + H – H2O]+ | 273.13058 | [M + HCOO – H2O]− | 317.12151 | |
| dcM12 | [M + H]+ | 307.13606 | [M + HCOO – H2O]− | 279.14114 | |
| Benzoate | GC1/GC2 | [M + H]+ | 473.10852 | ||
| GC3 | [M + H]+ | 377.15679 | |||
| GC4/GC5 | [M + H]+ | 489.10344 | |||
| GC6 | [M + H]+ | 393.15171 |
| PST Analogue | LOD (µg/kg) | LOQ (µg/kg) | ME ± RSD (%) |
|---|---|---|---|
| C1 | 0.07 | 0.22 | 82 ± 1.0 |
| C2 | 0.02 | 0.06 | 61 ± 4.4 |
| C3 | 0.17 | 0.56 | 53 ± 1.8 |
| C4 | 0.29 | 0.95 | 43 ± 1.8 |
| dcGTX2 | 0.14 | 0.48 | 135 ± 14.8 |
| dcGTX3 | 0.23 | 0.76 | 101 ± 3.1 |
| dcNeo | 0.08 | 0.28 | 73 ± 3.2 |
| dcSTX | 0.07 | 0.22 | 69 ± 8.6 |
| GTX1 | 0.28 | 0.93 | 120 ± 16.3 |
| GTX2 | 0.46 | 1.53 | 106 ± 9.2 |
| GTX3 | 0.35 | 1.17 | 89 ± 5.3 |
| GTX4 | 0.07 | 0.23 | 105 ± 9.2 |
| GTX5 | 0.44 | 1.48 | 79 ± 1.8 |
| GTX6 | 0.13 | 0.43 | 48 ± 6.3 |
| NEO | 0.02 | 0.08 | 79 ± 9.3 |
| STX | 0.01 | 0.04 | 69 ± 6.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lage, S.; Costa, P.R.; Canário, A.V.M.; Da Silva, J.P. LC-HRMS Profiling of Paralytic Shellfish Toxins in Mytilus galloprovincialis after a Gymnodinium catenatum Bloom. Mar. Drugs 2022, 20, 680. https://doi.org/10.3390/md20110680
Lage S, Costa PR, Canário AVM, Da Silva JP. LC-HRMS Profiling of Paralytic Shellfish Toxins in Mytilus galloprovincialis after a Gymnodinium catenatum Bloom. Marine Drugs. 2022; 20(11):680. https://doi.org/10.3390/md20110680
Chicago/Turabian StyleLage, Sandra, Pedro Reis Costa, Adelino V. M. Canário, and José P. Da Silva. 2022. "LC-HRMS Profiling of Paralytic Shellfish Toxins in Mytilus galloprovincialis after a Gymnodinium catenatum Bloom" Marine Drugs 20, no. 11: 680. https://doi.org/10.3390/md20110680