
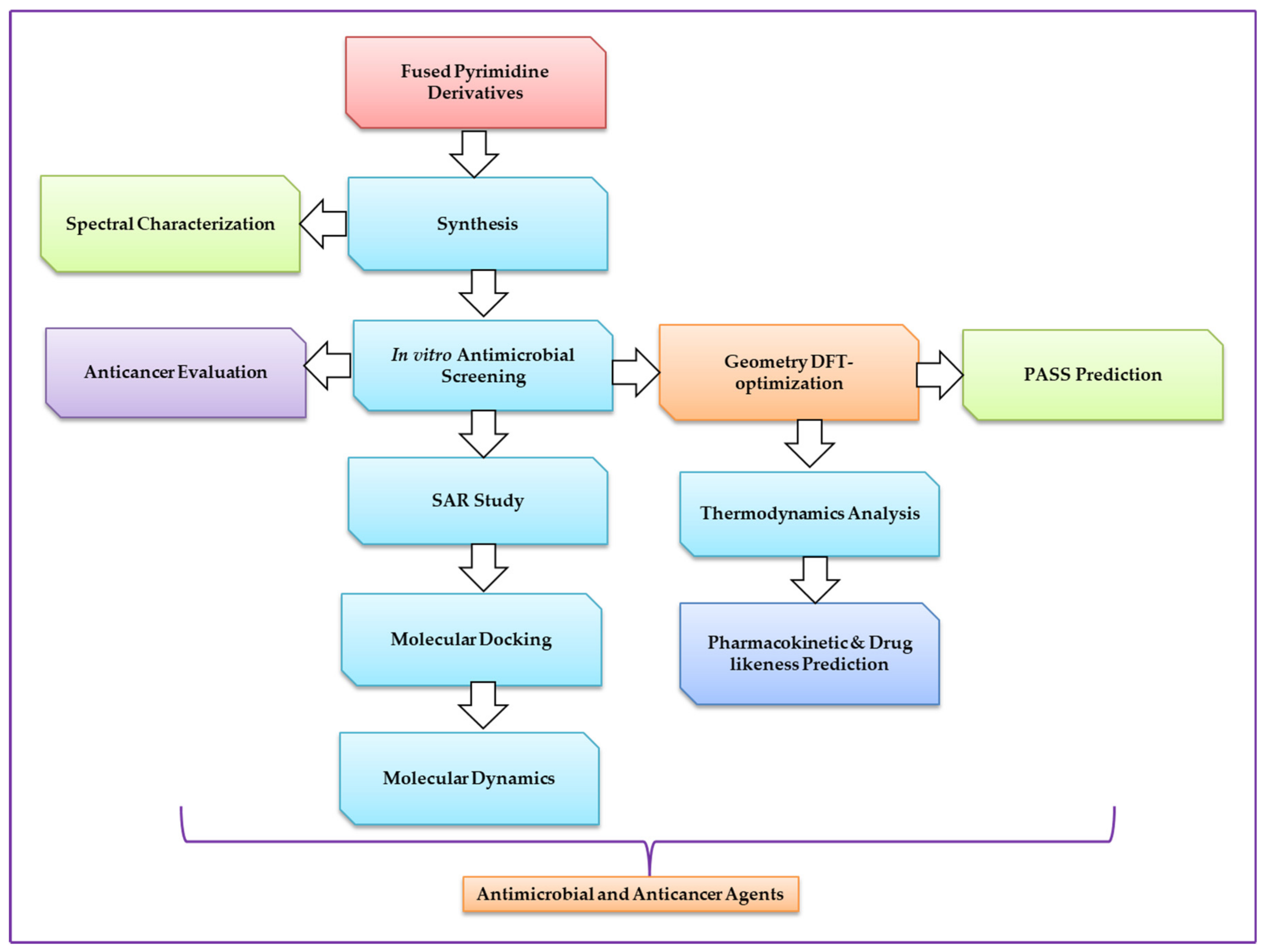
Uridine Derivatives: Synthesis, Biological Evaluation, and In Silico Studies as Antimicrobial and Anticancer Agents
 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
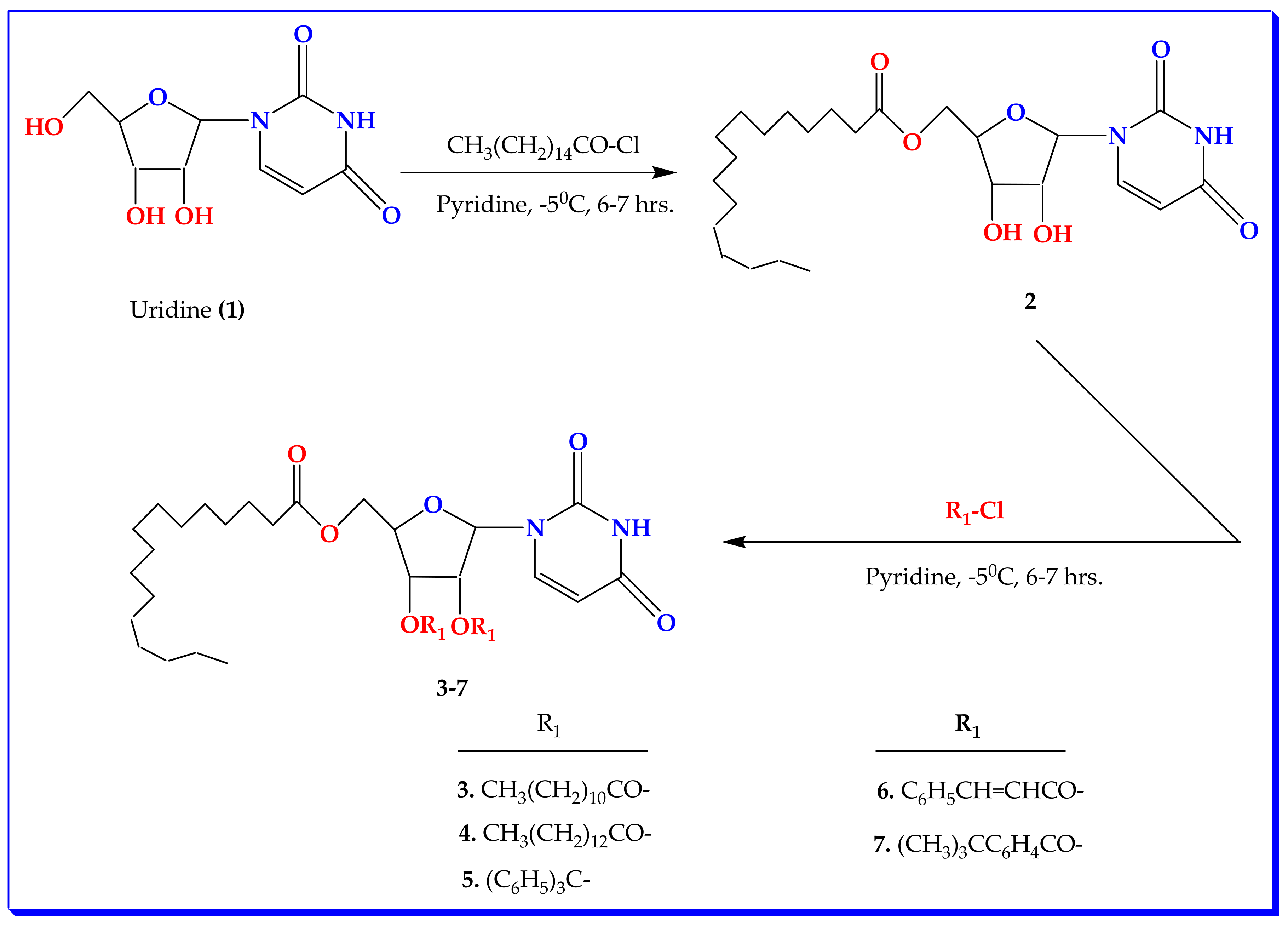
2.1. Chemistry and Characterization
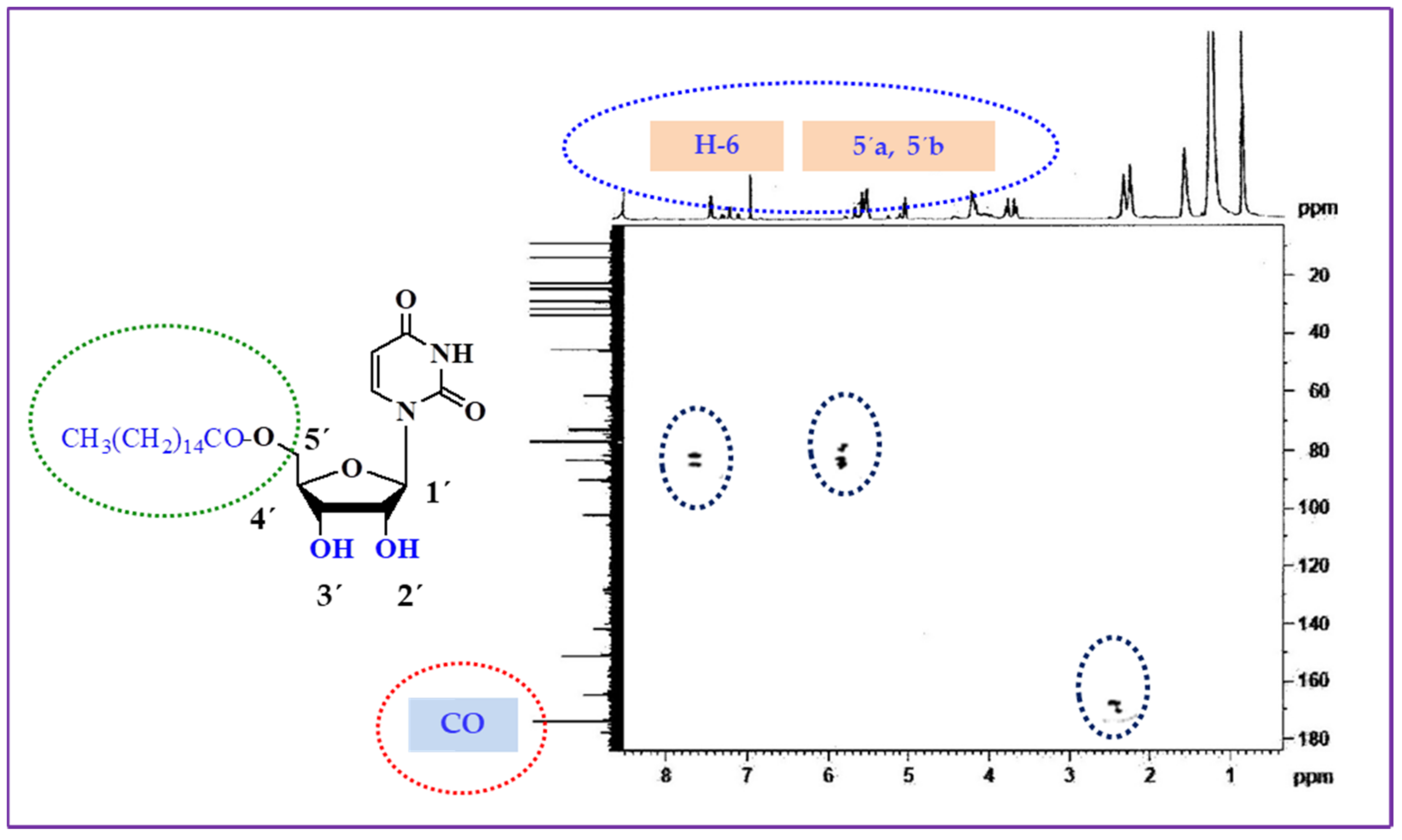
2.2. Two-Dimensional NMR Analysis
2.3. Antibacterial Activity Analyses
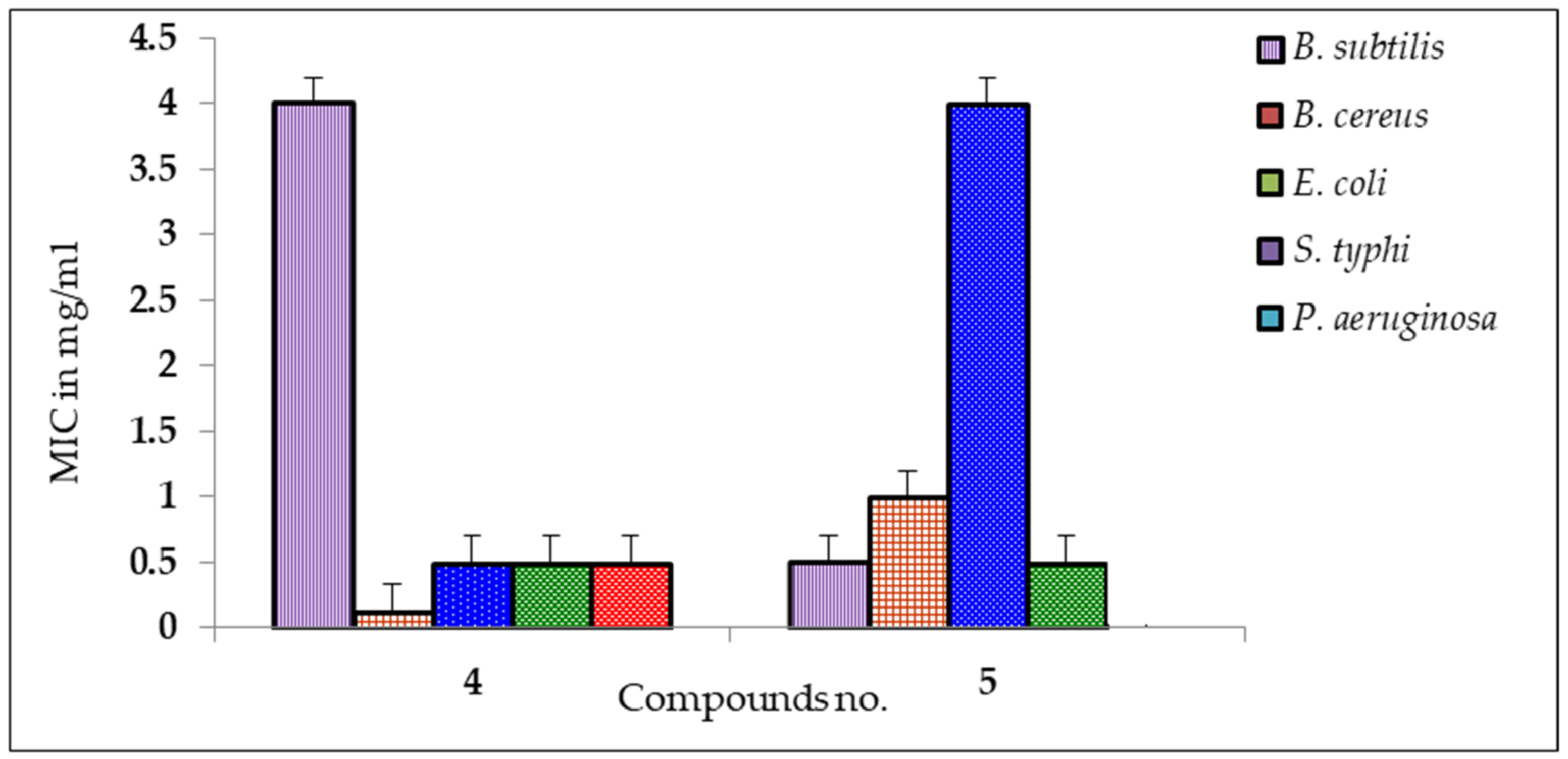
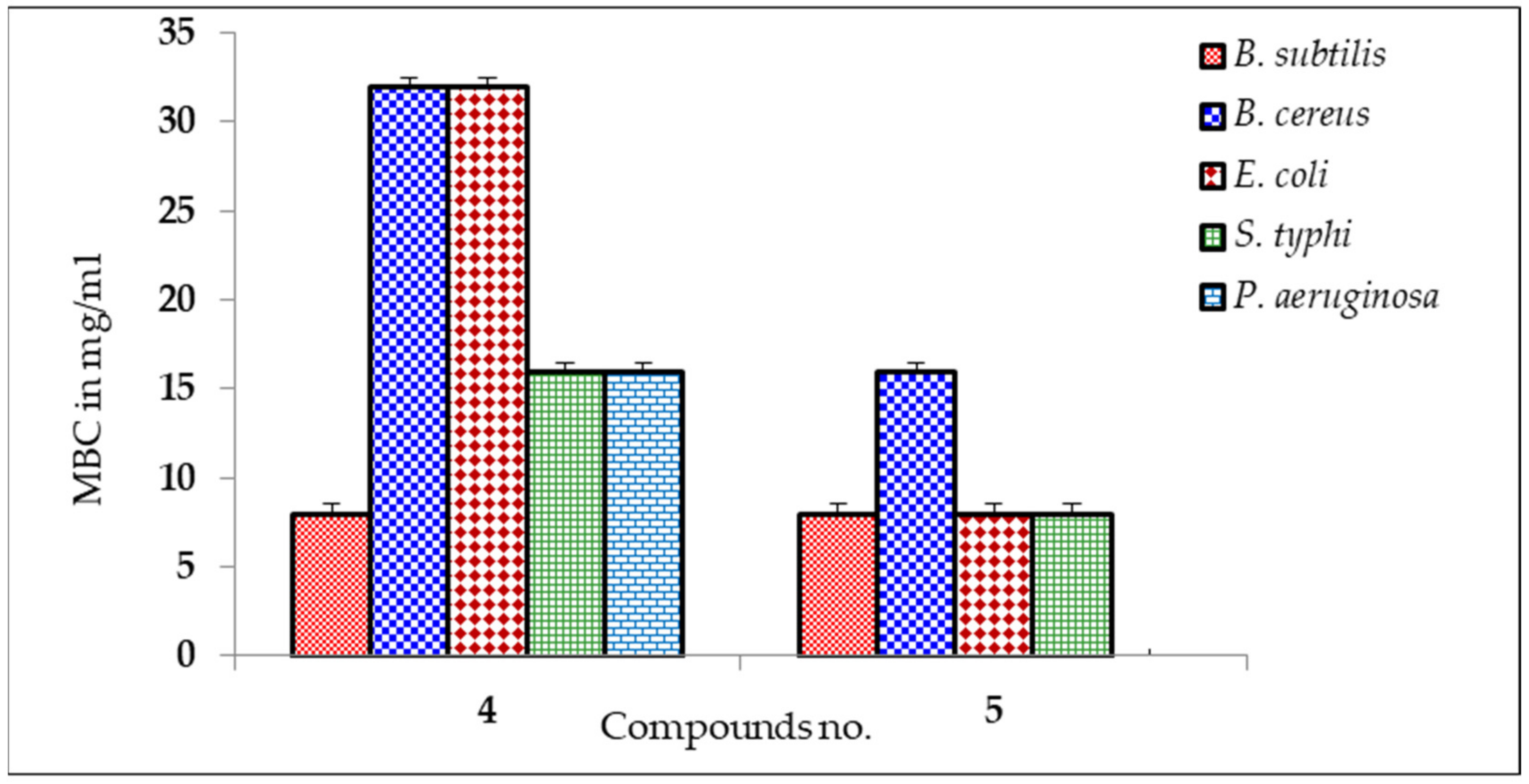
2.4. MIC and MBC Screening
2.5. Antifungal Susceptibility
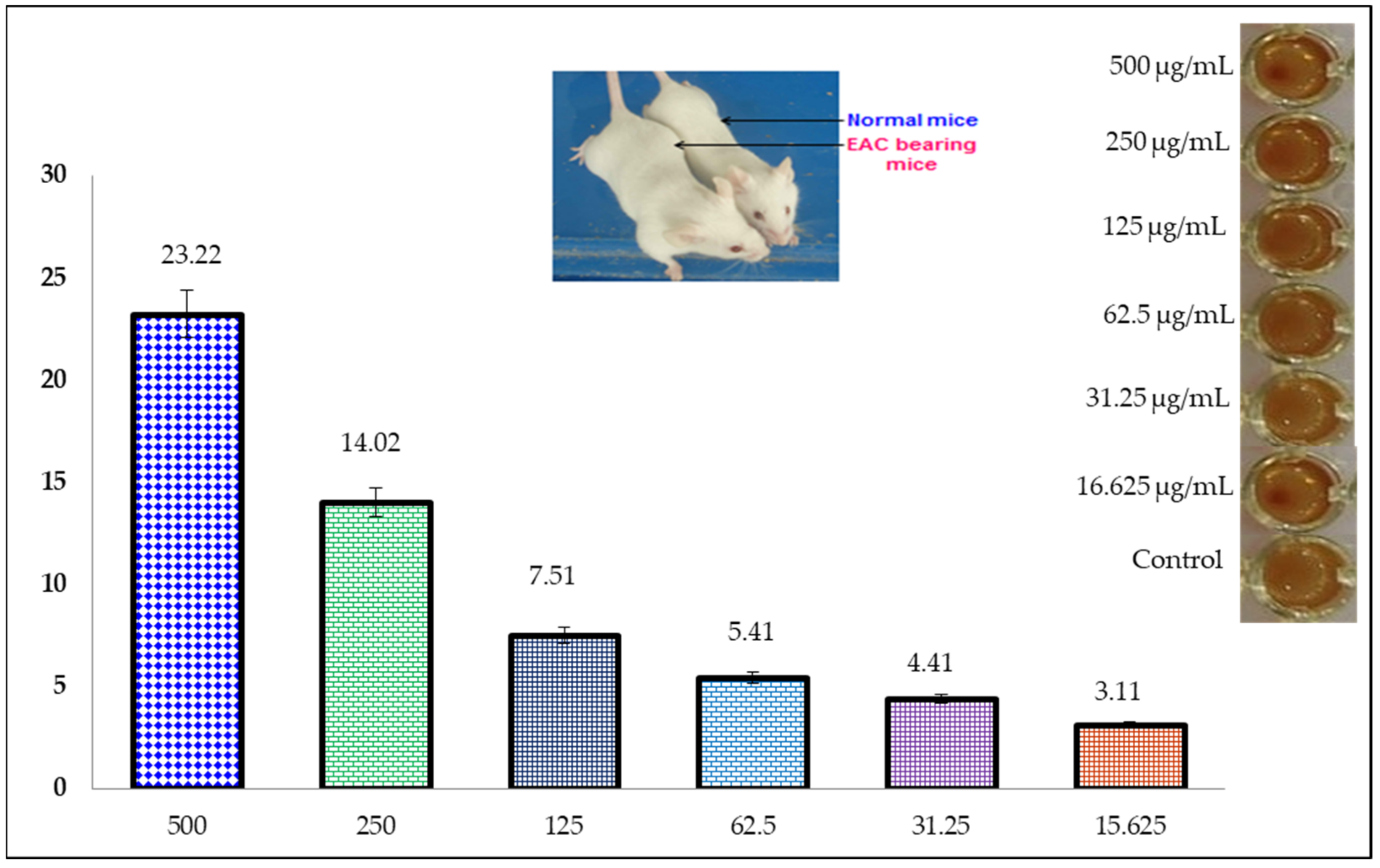
2.6. Anticancer Activity (MTT Assay)
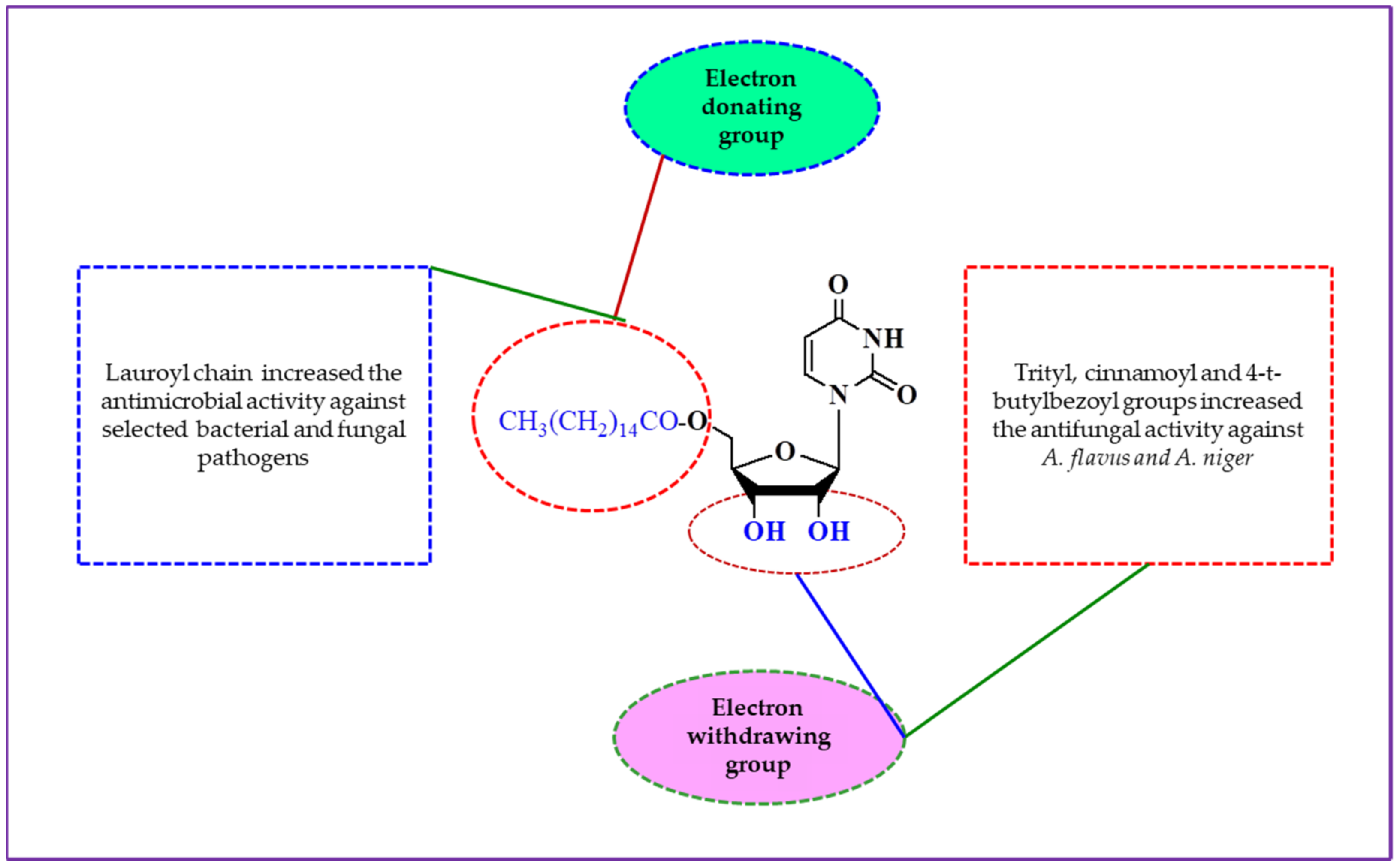
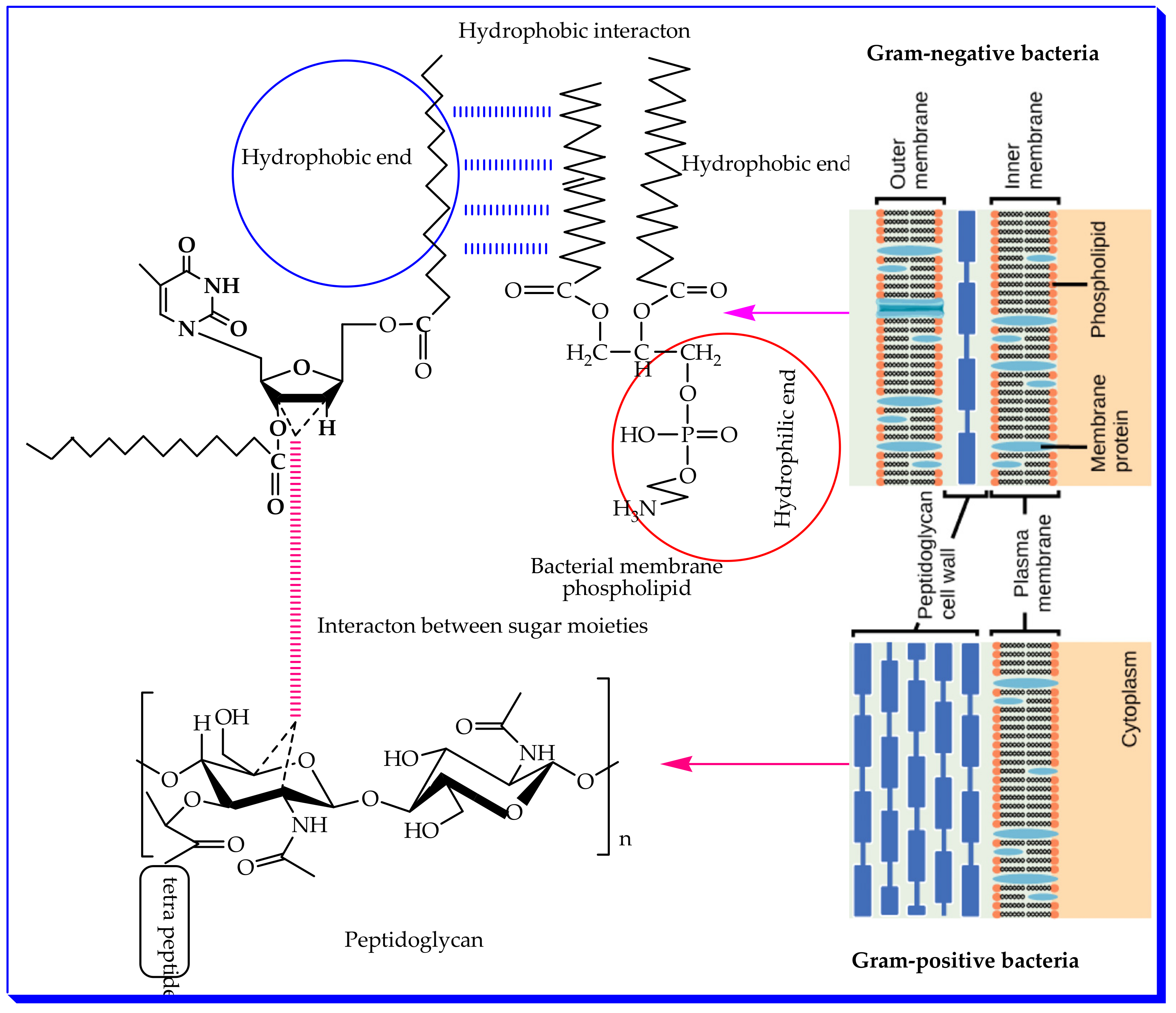
2.7. Structure–Activity Relationship (SAR)
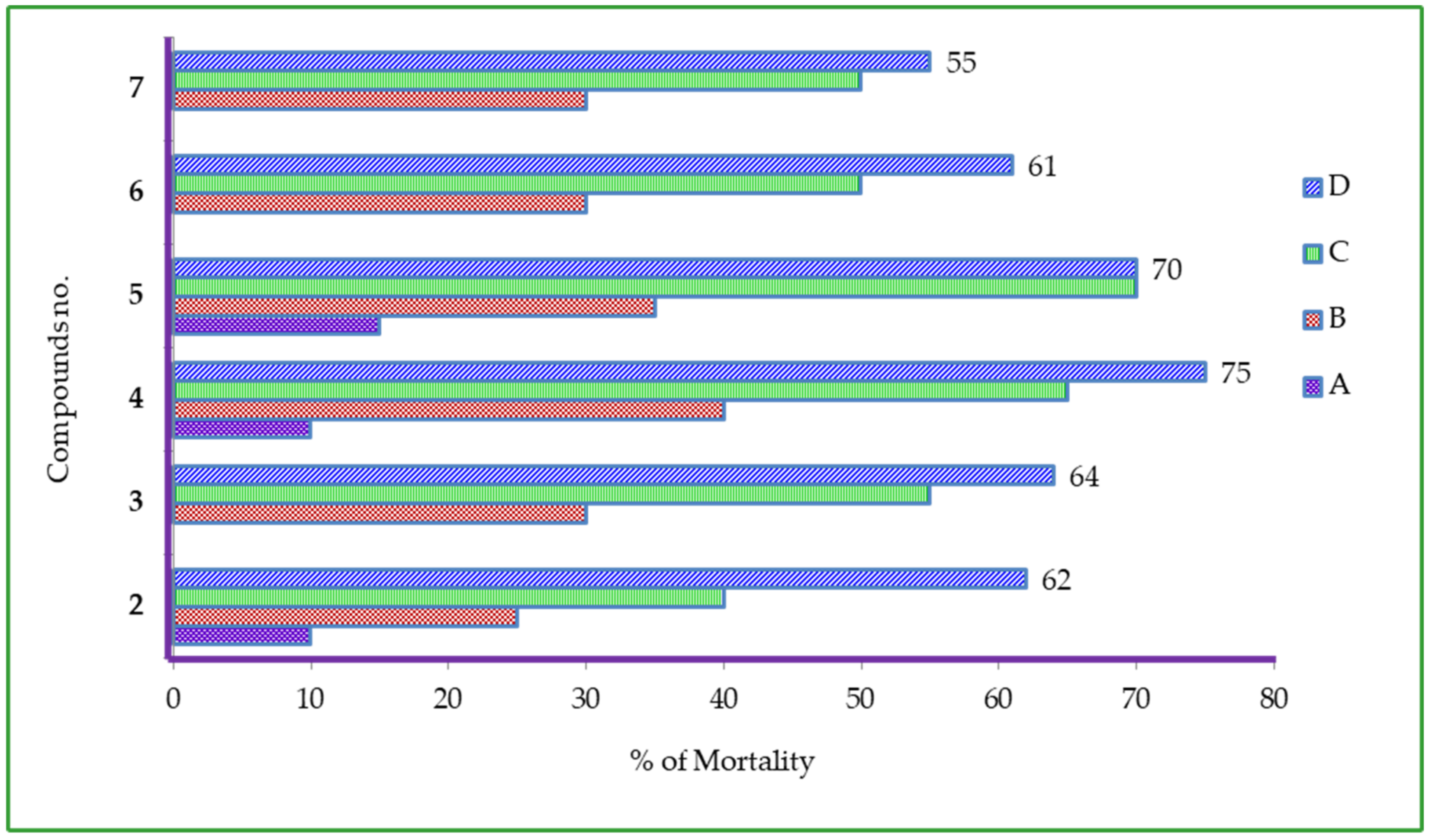
2.8. Cytotoxic Activity of Uridine Derivatives
2.9. Predicted Biological Properties (PASS)
2.10. Thermodynamic Study
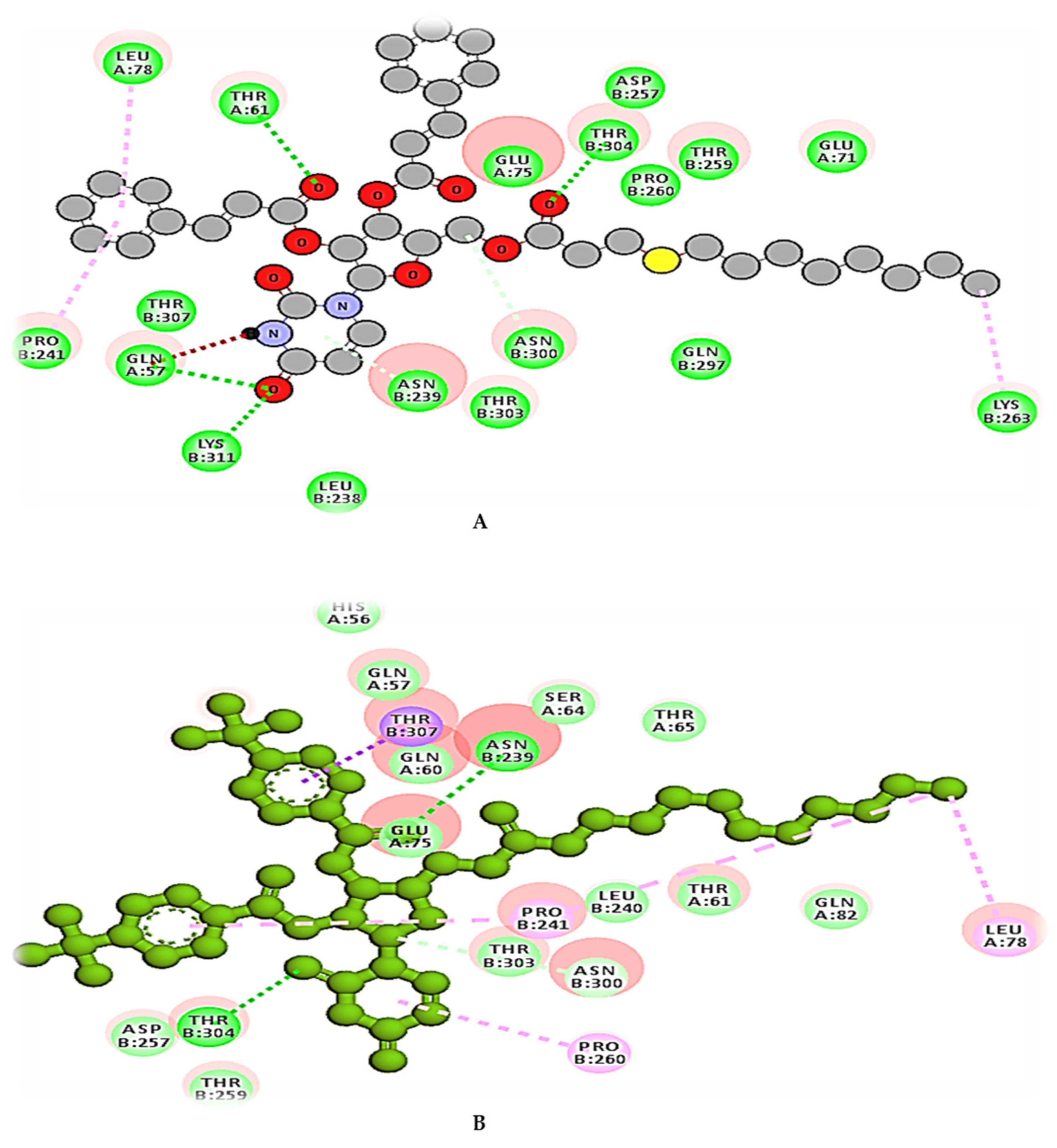
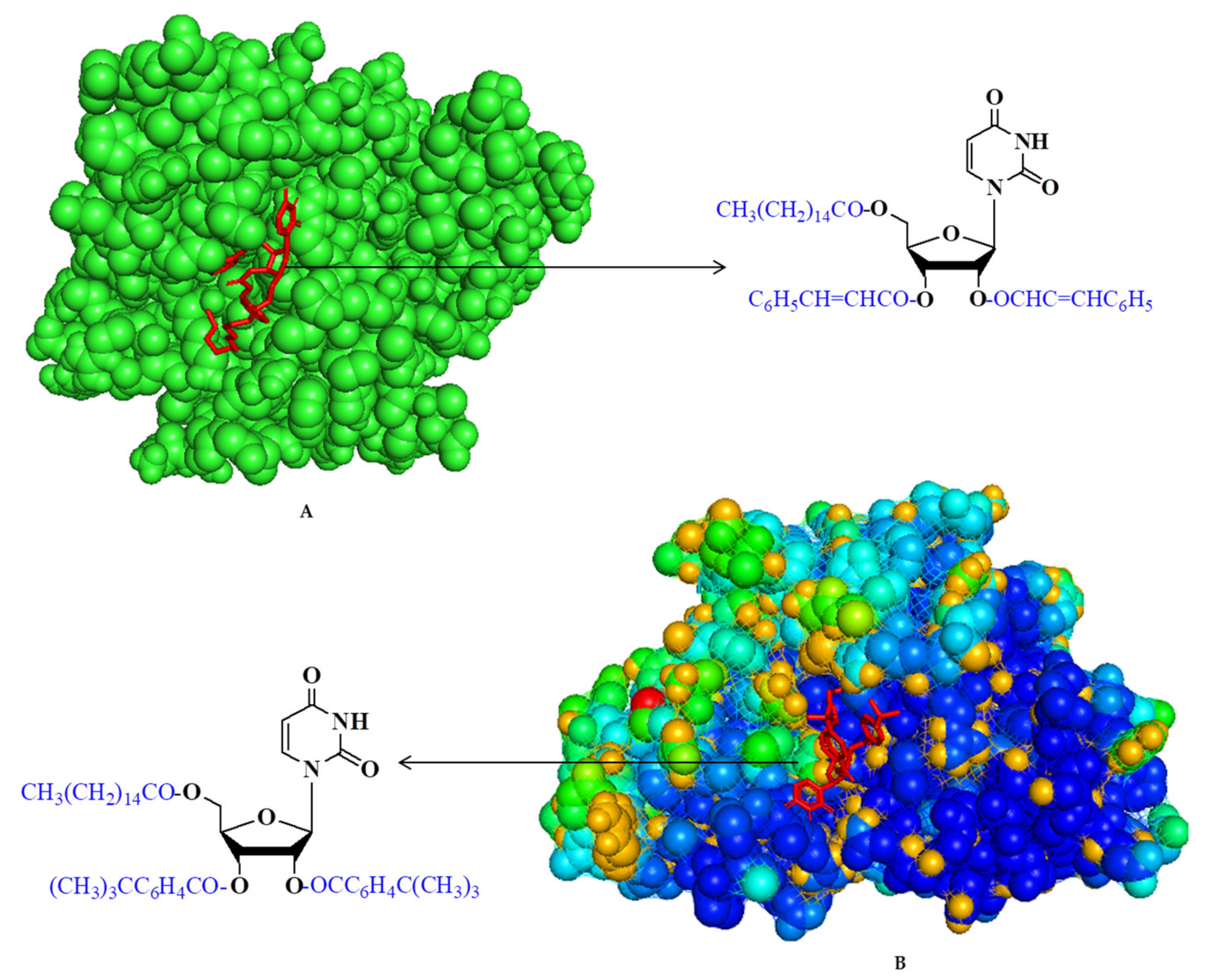
2.11. Molecular Docking Simulation
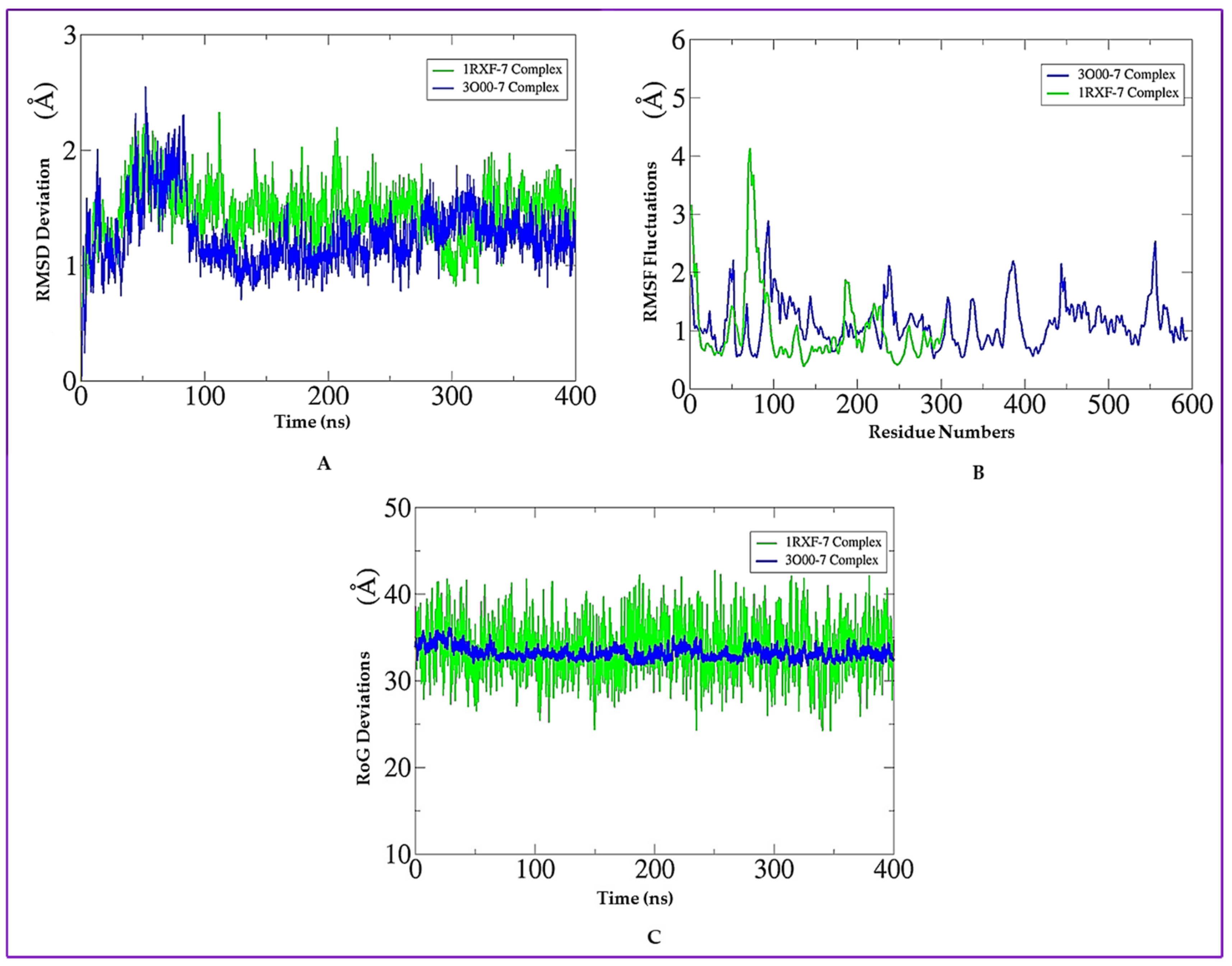
2.12. Molecular Dynamics (MD) Simulation
2.13. Binding Free Energy Calculation
2.14. Pharmacokinetic Profile and Drug-Likeness Exploration
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Uridine Derivatives
5′-Oxo-palmitoyluridine (2)
3.3. General Procedure for the Synthesis of 2′,3′-Di-oxo-acyl Uridine Derivatives (3–7)
3.3.1. 2′,3′-Di-oxo-lauroyl-5′-oxo-palmitoyluridine (3)
3.3.2. 2′,3′-Di-oxo-myristoyl-5′-oxo-palmitoyluridine (4)
3.3.3. 5′-oxo-Palmitoyl-2′,3′-di-oxo-trityluridine (5)
3.3.4. 2′,3′-Di-oxo-cinnamoyl-5′-oxo-palmitoyluridine (6)
3.3.5. 2′,3′-Di-oxo-(4-t-butylbenzoyl)-5′-oxo-palmitoyluridine (7)
3.4. In Vitro Antimicrobial Experiment
3.4.1. Antibacterial Susceptibility
3.4.2. Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
3.4.3. Screening of Mycelial Growth
3.5. Experimental Animals and Ethical Clearance
3.6. Antiproliferative Activity against EAC Cells by MTT Assay
3.7. Growth Inhibition Assay of Ehrlich ascites Carcinoma (EAC) Cells
3.8. Structure–Activity Relationship (SAR) Study
3.9. Cytotoxic Activity Evaluation
3.10. PASS Enumeration and Bioactivity
3.11. Geometry DFT Optimization
3.12. Protein Selection and Molecular Docking
3.13. MD Simulations
3.14. Binding Free Energy Calculation
3.15. Pharmacokinetic and Drug-Likeness Prediction
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koszytkowska-Stawinska, M.; Buchowicz, W. Multicomponent Reactions in Nucleoside Chemistry. Beilstein J. Org. Chem. 2014, 10, 1706–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordheim, L.P.; Ben Larbi, S.; Fendrich, O.; Ducrot, C.; Bergeron, E.; Dumontet, C. Gemcitabine is Active against Clinical Multiresistant Staphylococcus aureus Strains and is Synergistic with Gentamicin. Int. J. Antimicrob. Agents 2012, 39, 444–447. [Google Scholar] [CrossRef]
- Meanwell, M.; Silverman, S.M.; Lehmann, J.; Adluri, B.; Wang, Y.; Cohen, R.; Campeau, L.-C.; Britton, R. A Short de Novo Synthesis of Nucleoside Analogs. Science 2020, 369, 725–730. [Google Scholar] [CrossRef]
- Shagir, A.C.; Bhuiyan, M.M.R.; Ozeki, Y.; Kawsar, S.M.A. Simple and Rapid Synthesis of Some Nucleoside Derivatives: Structural and Spectral Characterization. Curr. Chem. Lett. 2016, 5, 83–92. [Google Scholar]
- Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G.J. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogs. Molecules 2020, 25, 2050. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, M.Z.H.; Chowdhury, T.S.; Misbah, M.M.H.; Ferdous, J.; Dey, S.; Kawsar, S.M.A. Synthesis of New Series of Pyrimidine Nucleoside Derivatives Bearing the Acyl Moieties as Potential Antimicrobial Agents. Pharmacia 2021, 68, 23–34. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the Development of Nucleoside and Nucleotide Analogs for Cancer and Viral Diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Lamont, I.L. Nucleoside Analogues as Antibacterial Agents. Front. Microbiol. 2019, 10, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenchenko, A.A.; Drenichev, M.S.; Il’icheva, I.A. Antiviral and Antimicrobial Nucleoside Derivatives: Structural Features and Mechanisms of Action. Mol. Biol. 2021, 55, 786–812. [Google Scholar] [CrossRef] [PubMed]
- Dobolyi, A.; Juhasz, G.; Kovacs, Z.; Kardos, J. Uridine Function in the Central Nervous System. Curr. Top. Med. Chem. 2011, 11, 1058–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamuy, R.; Berman, B. Topical Antiviral Agents for Herpes Simplex Virus Infections. Drugs Today 1998, 34, 1013–1025. [Google Scholar] [CrossRef]
- Rabasseda, X. Brivudine: A Herpes Virostatic with Rapid Antiviral Activity and Once-Daily Dosing. Drugs Today 2003, 39, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Yuen, G.J.; Weller, S.; Pakes, G.E. A Review of the Pharmacokinetics of Abacavir. Clin. Pharmacokinet. 2008, 47, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Maowa, J.; Hosen, M.A.; Alam, A.; Rana, K.M.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Pharmacokinetics and Molecular Docking Studies of Uridine Derivatives as SARS-COV-2 Mpro Inhibitors. Phys. Chem. Res. 2021, 9, 385–412. [Google Scholar]
- Koszalka, G.W.; Daluge, S.M.; Boyd, F.L. Advances in Nucleoside and Nucleotide Antiviral Therapies. Ann. Rep. Med. Chem. 1998, 33, 163–171. [Google Scholar]
- Chan, K.Y.; Kinghorm, A.B.; Hollenstein, M.; Tanner, J.A. Chemical Modifications for a Next Generation of Nucleic Acid Aptamers. ChemBioChem 2022, 23, e202200006. [Google Scholar] [CrossRef]
- Mikhailopulo, I.A.; Miroshnikov, A.I. New Trends in Nucleoside Biotechnology. Acta Nat. 2010, 2, 36–58. [Google Scholar] [CrossRef] [Green Version]
- Alam, A.; Hosen, M.A.; Hosen, A.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Synthesis, Characterization, and Molecular Docking against a Receptor Protein FimH of Escherichia coli (4XO8) of Thymidine Derivatives. J. Mex. Chem. Soc. 2021, 65, 256–276. [Google Scholar] [CrossRef]
- Arbour, C.A.; Imperiali, B. Uridine Natural Products: Challenging TargetS AND Inspiration for Novel Small Molecule Inhibitors. Bioorg. Med. Chem. 2020, 28, 115661. [Google Scholar] [CrossRef]
- Thatipamula, R.K.; Narsimha, S.; Battula, K.; Chary, V.R.; Mamidala, E.; Reddy, N.V. Synthesis, Anticancer and Antibacterial Evaluation of Novel (Isopropylidene) Uridine[1,2,3]triazole Hybrids. J. Saudi Chem. Soc. 2017, 21, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Rahman, M.S.; Chowdhury, M.E. Antimicrobial Screening Studies of Some Derivatives of Methyl α-D-Glucopyranoside. Pak. J. Sci. Ind. Res. 2009, 52, 138–142. [Google Scholar]
- Brab, A.W.; Leavy, T.M.; Robins, L.I.; Guan, Z.; Six, D.A.; Zhou, P.; Bertozzi, C.R.; Raetz, C.R.H. Uridine-based Inhibitors as New Leads for Antibiotics Targeting E. coli LpxC. Biochemistry 2009, 48, 3068–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirajul, M.I.; Arifuzzaman, M.; Monjur, M.R.; Atiar, M.R.; Kawsar, S.M.A. Novel Methyl 4,6-O-Benzylidene-α-D-Glucopyranoside Derivatives: Synthesis, Structural Characterization and Evaluation of Antibacterial Activities. Hacettepe J. Biol. Chem. 2019, 47, 153–164. [Google Scholar]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Kabir, A.K.M.S.; Kawsar, S.M.A.; Bhuiyan, M.M.R.; Hossain, S.; Rahman, M.S. Biological Evaluation of Some Uridine Derivatives. Chittagong Univ. J. Sci. 2003, 27, 105–111. [Google Scholar]
- Farhana, Y.; Amin, M.R.; Hosen, A.; Kawsar, S.M.A. Bromobenzoylation of Methyl α-D-Mannopyranoside: Synthesis and Spectral Characterization. J. Sib. Fed. Univ. Chem. 2021, 14, 171–183. [Google Scholar]
- Kawsar, S.M.A.; Islam, M.M.; Chowdhury, S.A.; Hasan, T.; Hossain, M.K.; Manchur, M.A.; Ozeki, Y. Design and Newly Synthesis of Some 1,2-O-Isopropylidene-α-D-Glucofuranose Derivatives: Characterization and Antibacterial Screening Studies. Hacettepe J. Biol. Chem. 2013, 41, 195–206. [Google Scholar]
- Saleh, S.S.; Salihi, S.S.A.; Mohammed, I.A. Biological Activity Study for Some Heterocyclic Compounds and their Impact on the Gram Positive and Negative Bacteria. Energy Procedia 2019, 157, 296–306. [Google Scholar] [CrossRef]
- Li, W.R.; Xie, X.B.; Shi, Q.S.; Zeng, H.Y.; Ou-Yang, Y.S.; Chen, Y.B. Antibacterial Activity and Mechanism of Silver Nanoparticles on Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 85, 1115–1122. [Google Scholar] [CrossRef]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; De Clercq, E.; Pannecouque, C.; Balzarini, J. Synthesis, Antimycobacterial, Antiviral, Antimicrobial Activities, and QSAR Studies of Isonicotinic acid-1-(substituted phenyl)-ethylidene/cycloheptylidene Hydrazides. Med. Chem. Res. 2012, 21, 1935–1952. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Huq, E.; Nahar, N. Cytotoxicity Assessment of the Aerial Parts of Macrotyloma uniflorum Linn. Int. J. Pharmacol. 2008, 4, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Kawsar, S.M.A.; Matsumoto, R.; Fujii, Y.; Matsuoka, H.; Masuda, N.; Iwahara, C.; Yasumitsu, H.; Kanaly, R.A.; Sugawara, S.; Hosono, M.; et al. Cytotoxicity and Glycan-Binding Profile of α-D-Galactose-Binding Lectin from the Eggs of a Japanese Sea Hare (Aplysia kurodai). Protein J. 2011, 30, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-Glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Molecular Electrostatic Potentials and Chemical Reactivity. Rev. Comput. Chem. 1991, 2, 273–312. [Google Scholar]
- Saha, S.; Banerjee, S.; Ganguly, S. Molecular docking studies of some novel hydroxamic acid derivatives. Int. J. ChemTech Res. 2010, 2, 932–936. [Google Scholar]
- Kawsar, S.M.A.; Hosen, M.A.; Fujii, Y.; Ozeki, Y. Thermochemical, DFT, Molecular Docking and Pharmacokinetic Studies of Methyl β-D-Galactopyranoside Esters. J. Comput. Chem. Mol. Model. 2020, 4, 452–462. [Google Scholar] [CrossRef]
- Farhana, Y.; Amin, M.R.; Hosen, M.A.; Bulbul, M.Z.H.; Dey, S.; Kawsar, S.M.A. Monosaccharide Derivatives: Synthesis, Antimicrobial, PASS, Antiviral, and Molecular Docking studies Against SARS-COV-2 Mpro Inhibitors. J. Cellul. Chem. Technol. 2021, 55, 477–499. [Google Scholar]
- Judge, V.; Narasimhan, B.; Ahuja, M.; Sriram, D.; Yogeeswari, P.; Clercq, E.D.; Pannecouque, C.; Balzarini, J. Synthesis, Antimycobacterial, Antiviral, Antimicrobial Activity and QSAR Studies of N(2)-acyl Isonicotinic Acid Hydrazide Derivatives. Med. Chem. 2013, 9, 53–76. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-Dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Binding Mode Analysis, Dynamic Simulation and Binding Free Energy Calculations of the MurF ligase from Acinetobacter Baumannii. J. Mol. Graph. Model. 2017, 77, 72–85. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Hosen, M.A. An Optimization and Pharmacokinetic Studies of Some Thymidine Derivatives. Turk. Comput. Theor. Chem. 2020, 4, 59–66. [Google Scholar] [CrossRef]
- Kim, E.; Kang, W. Pharmacokinetics of Uridine Following Ocular, Oral and Intravenous Administration in Rabbits. Biomol. Ther. 2013, 21, 170–172. [Google Scholar] [CrossRef] [Green Version]
- Clinical Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 9th ed.; CLSI Documents M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Grover, R.K.; Moore, J.D. In-Vitro Efficacy of Certain Essential Oils and Plant Extracts Against Three Major Pathogens of Jatropha curcas L. Phytopathology 1962, 52, 876–879. [Google Scholar]
- Hasan, I.; Asaduzzaman, A.K.M.; Swarna, R.R.; Fujii, Y.; Ozeki, Y.; Uddin, M.B.; Kabir, S.R. MytiLec-1 Shows Glycan-Dependent Toxicity against Brine Shrimp Artemia and Induces Apoptotic Death of Ehrlich Ascites Carcinoma Cells In Vivo. Mar. Drugs 2019, 17, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, W.A. The Effects of Aliphatic Alcohols on the Biophysical and Biochemical Correlates of Membrane Function. Adv. Exp. Med. Biol. 1975, 56, 195. [Google Scholar]
- Kim, Y.M.; Farrah, S.; Baney, R.H. Structure–Antimicrobial Activity Relationship for Silanols, a New Class of Disinfectants, Compared with Alcohols and Phenols. Int. J. Antimicrob. Agents 2007, 29, 217. [Google Scholar] [CrossRef]
- McLaughlin, J.L. Crown-Gall Tumors in Potato Discs and Brine Shrimp Lethality: Two Simple Bioassays for Higher Plant Screening and Fractionation. In Methods in Plant Biochemistry: Assays for Bioactivity; Hostettmann, K., Ed.; Academic Press: Cambridge, MA, USA, 1991; Volume 6, pp. 1–32. [Google Scholar]
- Kumaresan, S.; Senthilkumar, V.; Stephen, A.; Balakumar, B.S. GC–MS Analysis and PASS-Assisted Prediction of Biological Activity Spectra of Extract of Phomopsis sp. Isolated from Andrographis paniculata. World J. Pharm. Res. 2015, 4, 1035–1053. [Google Scholar]
- Seeberger, P.H.; Werz, D.B. Synthesis and Medical Applications of Oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, M.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Delano, W.L. The PyMOL Molecular Graphics System; De-Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H.; Hong, C.C. (Eds.) Chemical Biology: Methods and Protocols; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- Kawsar, S.M.A.; Kumer, A. Computational Investigation of Methyl α-D-Glucopyranoside Derivatives as Inhibitor against Bacteria, Fungi and COVID-19 (SARS-2). J. Chil. Chem. Soc. 2021, 66, 5206–5214. [Google Scholar] [CrossRef]
- Accelrys. Version ADS 4.0; Accelrys: San Diego, CA, USA, 2017. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- He, X.; Liu, S.; Lee, T.-S.; Ji, B.; Man, V.H.; York, D.M.; Wang, J. Fast, Accurate, and Reliable Protocols for Routine Calculations of Protein–Ligand Binding Affinities in Drug Design Projects Using AMBER GPU-TI with ff14SB/GAFF. ACS Omega 2020, 5, 4611–4619. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (ppm) | (J Hz) | (HSQC) δC (ppm) | HMBC |
|---|---|---|---|---|
| Ar-NH | 9.00 | (1H, s, -NH) | 127.09 | H: 6 |
| H-6 | 7.28 | (1H, m) | 89.11 | Ar-CO |
| H-1′ | 6.85 | (d, J = 5.5) | 141.41 | H: 6 |
| OH-2′ | 6.78 | (1H, s) | 103.97 | H: 3′ |
| H-5′a | 6.01 | (1H, dd, J = 2.2,and 12.0 Hz) | 79.21 | H: 4′ |
| H-5′b | 5.46 | (1H, dd, J = 2.3, and 12.0 Hz) | 81.01 | H: 4′ |
| OH-3′ | 5.05 | (1H, s) | 77.54 | H: 4′, |
| H-5 | 4.77 | (1H, d, J = 8.2 Hz) | 84.73 | H: 5′a, 5′b |
| C-2′ | 4.46 | (1H, d, J = 5.6 Hz) | 87.07 | H: 3′,5′a, |
| C-3′ | 4.38 | (1H, dd, J = 7.4, and 5.5 Hz) | 83.47 | H: 4′ |
| CH3(CH2)13CH2CO- | - | - | 169.36 | H: 5′a, 5′b |
| Diameter of Inhibition Zones (in mm) | |||||
|---|---|---|---|---|---|
| Entry | B. subtilis (G + ve) | S. aureus (G + ve) | E. coli (G − ve) | S. typhi (G − ve) | P. aeruginosa (G − ve) |
| 1 | NI | NI | NI | NI | NI |
| 2 | NI | NI | NI | NI | NI |
| 3 | NI | NI | NI | * 11.0 ± 0.0 | NI |
| 4 | * 17 ± 0.20 | * 15 ± 0.50 | * 17 ± 0.00 | * 18 ± 0.75 | 7 ± 0.25 |
| 5 | * 13 ± 0.30 | * 16 ± 0.20 | * 10 ± 0.75 | * 12 ± 0.25 | NI |
| 6 | NI | NI | NI | NI | NI |
| 7 | NI | NI | NI | 9 ± 0.00 | NI |
| Azithromycin | ** 18 ± 0.10 | ** 17 ± 0.50 | ** 17 ± 0.50 | ** 18 ± 0.90 | ** 15 ± 0.10 |
| Entry | Millimeters of Fungal Mycelial Growth Inhibition in DMSO (20 μg/μL). | |
|---|---|---|
| Aspergillus niger | Aspergillus flavus | |
| 1 | NI | NI |
| 2 | NI | NI |
| 3 | NI | NI |
| 4 | NI | 31 ± 0.15 |
| 5 | * 64 ± 0.44 | * 66 ± 0.39 |
| 6 | * 72 ± 0.95 | 53 ± 0.33 |
| 7 | * 66 ± 0.41 | * 63 ± 0.10 |
| Nystatin | 59 ± 0.00 ** | 61 ± 0.00 ** |
| Biological Activity | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Antiviral | Antibacterial | Antifungal | Anticarcinogenic | ||||
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| 1 | 0.611 | 0.369 | 0.117 | 0.030 | 0.412 | 0.078 | 0.646 | 0.037 |
| 2 | 0.785 | 0.235 | 0.563 | 0.044 | 0.229 | 0.028 | 0.434 | 0.010 |
| 3 | 0.687 | 0.201 | 0.669 | 0.031 | 0.478 | 0.011 | 0.552 | 0.063 |
| 4 | 0.714 | 0.153 | 0.562 | 0.024 | 0.572 | 0.021 | 0.720 | 0.014 |
| 5 | 0.696 | 0.521 | 0.618 | 0.050 | 0.500 | 0.091 | 0.535 | 0.025 |
| 6 | 0.806 | 0.117 | 0.850 | 0.009 | 0.730 | 0.054 | 0.826 | 0.006 |
| 7 | 0.862 | 0.224 | 0.733 | 0.067 | 0.751 | 0.019 | 0.798 | 0.003 |
| Entry | MF | MW | E | H | G | p |
|---|---|---|---|---|---|---|
| 1 | C9H12N2O6 | 244.20 | −862.035 | −862.036 | −772.103 | 4.255 |
| 2 | C25H42O7N2 | 483.62 | −1512.432 | −1512.432 | −1512.512 | 4.823 |
| 3 | C49H86O9N2 | 848.23 | −2105.423 | −2105.424 | −2105.532 | 3.942 |
| 4 | C53H94O9N2 | 922.34 | −3248.546 | −3248.546 | −3248.697 | 3.255 |
| 5 | C63H70O7N2 | 967.26 | −1735.579 | −1735.580 | −1735.669 | 4.918 |
| 6 | C43H54O9N2 | 742.91 | −3926.037 | −3927.036 | −3925.138 | 5.217 |
| 7 | C47H66O9N2 | 803.05 | −2237.369 | −2237.375 | −2237.405 | 3.254 |
| Entry | 1RXF (kcal/mol) | 3000 (kcal/mol) |
|---|---|---|
| 1 | −6.5 | −6.3 |
| 2 | −5.7 | −6.9 |
| 3 | −4.5 | −5.3 |
| 4 | −6.4 | −6.9 |
| 5 | −5.4 | −5.3 |
| 6 | −6.0 | −6.6 |
| 7 | −6.8 | −7.8 |
| E. coli 1RXF | ||||
|---|---|---|---|---|
| Entry | Bond Category | Residues in Contact | Interaction Type | Distance (Å) |
| 1 | H | Asp185 | CH | 2.3027 |
| H | Ser260 | C | 3.4238 | |
| Hydrophobic | Ile192 | PA | 5.0799 | |
| Hydrophobic | Leu204 | PA | 4.7740 | |
| Hydrophobic | Val245 | PA | 4.6041 | |
| Hydrophobic | Val262 | PA | 4.9465 | |
| 2 | H | Trp297 | CH | 2.2888 |
| H | Arg306 | CH | 2.9698 | |
| H | Thr308 | CH | 2.8950 | |
| H | Gly282 | C | 3.7125 | |
| H | Asp284 | PDH | 2.9446 | |
| Hydrophobic | Arg242 | A | 4.2592 | |
| 3 | H | Gly236 | CH | 2.8534 |
| Hydrophobic | Lys239 | A | 4.6475 | |
| Hydrophobic | Ala240 | A | 4.6703 | |
| Hydrophobic | Tyr184 | PA | 4.8027 | |
| Hydrophobic | Phe283 | PA | 5.3891 | |
| Hydrophobic | Trp297 | PA | 4.5201 | |
| Hydrophobic | Trp297 | PA | 4.9643 | |
| Hydrophobic | Cys281 | PA | 5.4869 | |
| 4 | H | Tyr184 | CH | 2.4128 |
| H | Trp297 | CH | 1.9265 | |
| H | Thr308 | CH | 2.3072 | |
| H | Asp284 | CH | 3.0389 | |
| H | Tyr184 | C | 3.4162 | |
| Electrostatic | Arg306 | PCa | 4.4532 | |
| Hydrophobic | Phe283 | PS | 3.5573 | |
| 5 | H | Arg160 | CH | 2.2907 |
| H | Arg160 | CH | 2.6698 | |
| H | Arg160 | CH | 2.2443 | |
| H | Arg162 | CH | 2.0553 | |
| H | Arg162 | CH | 2.4242 | |
| H | Asn304 | CH | 2.2870 | |
| Hydrophobic | Ile305 | A | 4.2841 | |
| Hydrophobic | Val245 | A | 4.2461 | |
| Hydrophobic | Arg74 | A | 3.9921 | |
| Hydrophobic | Phe164 | PA | 4.8948 | |
| 6 | H | Arg306 | CH | 2.3238 |
| H | Arg306 | CH | 2.9836 | |
| H | Thr308 | C | 3.0353 | |
| H | Tyr184 | C | 3.6460 | |
| Hydrophobic | Tyr184 | PPS | 3.9134 | |
| Hydrophobic | Phe283 | PPS | 4.0173 | |
| Hydrophobic | Lys239 | A | 4.4264 | |
| Hydrophobic | Arg242 | PA | 5.2713 | |
| Hydrophobic | Ala228 | PA | 4.9831 | |
| 7 | H | Arg160 | CH | 2.3553 |
| H | Arg160 | CH | 2.0783 | |
| H | Asn301 | CH | 2.1739 | |
| H | Asn304 | CH | 1.9583 | |
| H | Ile305 | CH | 1.9083 | |
| Hydrophobic | Ile192 | A | 4.7525 | |
| Hydrophobic | Leu204 | A | 3.7404 | |
| Hydrophobic | Val245 | A | 4.9226 | |
| S. typhi 3000 | ||||
|---|---|---|---|---|
| Entry | Bond Category | Residues in Contact | Interaction Type | Distance (Å) |
| 1 | H | Arg41, Ser107 | CH | 2.1563 |
| H | Gly42, Ser107 | CH | 2.0607 | |
| H | Thr43, Ser110 | CH | 2.1484 | |
| H | Thr43, Ser110 | CH | 1.7969 | |
| H | Gln50, Glu237 | PA | 1.8046 | |
| Hydrophobic | Gly42, Ala108 | PA | 3.3389 | |
| 2 | H | Gly42, Ala108 | C | 3.3389 |
| H | Ser47 | C | 3.4860 | |
| H | Ser93 | C | 3.6352 | |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Ala89 | PS | 3.4540 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| 3 | H | Asp257 | CH | 2.7677 |
| H | Gly42, Ala108 | C | 3.3389 | |
| H | Asn300 | C | 3.6065 | |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| Hydrophobic | Pro112, Ile46 | A | 4.4930 | |
| Hydrophobic | Pro66 | A | 5.2447 | |
| Hydrophobic | Pro260 | PA | 4.7411 | |
| 4 | H | Gln50, Glu237 | CH | 1.8046 |
| H | Asn314 | CH | 2.4977 | |
| H | Ser49 | CH | 2.6312 | |
| H | Gly42, Ala108 | C | 3.3389 | |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| Hydrophobic | Pro112, Ile46 | A | 4.4930 | |
| 5 | H | Thr43, Ser110 | CH | 1.7969 |
| H | Gln50, Glu237 | CH | 1.8046 | |
| H | Lys92 | CH | 2.9600 | |
| H | Gly42, Ala108 | C | 3.3389 | |
| H | Ser47 | C | 3.4875 | |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Ser93 | PS | 3.5662 | |
| Hydrophobic | Ala89 | A | 4.5921 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| Hydrophobic | Pro112, Ile46 | A | 4.4930 | |
| 6 | H | Gly42, Ala108 | C | 3.3389 |
| H | Asn300 | C | 3.4289 | |
| H | Asn239 | PDH | 2.5559 | |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| Hydrophobic | Pro112, Ile46 | A | 4.4930 | |
| Hydrophobic | Lys263 | A | 4.1809 | |
| Hydrophobic | Leu78 | PA | 5.3726 | |
| Hydrophobic | Pro241 | PA | 5.1424 | |
| 7 | H | Asn300 | C | 3.4437 |
| Hydrophobic | Ile46, Phe117 | PS | 3.9645 | |
| Hydrophobic | Thr307 | PS | 3.5375 | |
| Hydrophobic | Ala111, Ile46 | A | 5.4224 | |
| Hydrophobic | Pro112, Ile46 | A | 4.4930 | |
| Hydrophobic | Leu78 | A | 5.2374 | |
| Hydrophobic | Pro241 | A | 4.1761 | |
| Hydrophobic | Pro241 | PA | 4.8967 | |
| Energy Parameters | 1RXF-7 Complex | 3000-7 Complex |
|---|---|---|
| MM-GBSA | ||
| VDWAALS | −28.65 | −42.17 |
| EEL | −15.44 | −10.27 |
| Delta G solv | 19.39 | 26.13 |
| Delta total | −24.7 | −26.31 |
| MM-PBSA | ||
| VDWAALS | −28.65 | −42.17 |
| EEL | −15.44 | −10.27 |
| Delta G solv | 18.37 | 21.26 |
| Delta total | −25.72 | −31.18 |
| Entry | Water Solubility (log mol/L) | Caco-2 Permeability | Intestinal Absorption | Skin Permeability |
|---|---|---|---|---|
| 1 | −1.008 | −0.050 | 78.332 | −3.13 |
| 2 | −2.649 | 0.371 | 81.147 | −1.08 |
| 3 | −2.401 | 1.014 | 84.657 | −1.06 |
| 4 | −2.375 | 0.710 | 85.980 | −2.33 |
| 5 | −3.679 | 0.417 | 91.364 | −2.14 |
| 6 | −3.601 | 0.268 | 89.004 | −1.91 |
| 7 | −4.987 | 0.854 | 93.229 | −1.00 |
| Entry | Distribution | Execration | |||
|---|---|---|---|---|---|
| VDss | BBB Permeability | CNS Permeability | Total Clearance | Renal OCT2 Substrate | |
| 1 | 0.017 | −0.783 | −1.90 | 0.319 | No |
| 2 | −0.229 | −0.041 | −2.23 | 0.279 | No |
| 3 | −0.531 | −0.345 | −2.71 | 0.851 | No |
| 4 | 0.182 | −1.931 | −3.06 | 0.966 | No |
| 5 | −0.316 | −0.007 | −3.21 | 1.073 | No |
| 6 | −0.563 | −1.778 | −3.65 | 1.741 | No |
| 7 | −0.598 | −1.510 | −3.37 | 1.434 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munia, N.S.; Alanazi, M.M.; El Bakri, Y.; Alanazi, A.S.; Mukhrish, Y.E.; Hasan, I.; Kawsar, S.M.A. Uridine Derivatives: Synthesis, Biological Evaluation, and In Silico Studies as Antimicrobial and Anticancer Agents. Medicina 2023, 59, 1107. https://doi.org/10.3390/medicina59061107
Munia NS, Alanazi MM, El Bakri Y, Alanazi AS, Mukhrish YE, Hasan I, Kawsar SMA. Uridine Derivatives: Synthesis, Biological Evaluation, and In Silico Studies as Antimicrobial and Anticancer Agents. Medicina. 2023; 59(6):1107. https://doi.org/10.3390/medicina59061107
Chicago/Turabian StyleMunia, Nasrin S., Mohammed M. Alanazi, Youness El Bakri, Ashwag S. Alanazi, Yousef E. Mukhrish, Imtiaj Hasan, and Sarkar M. A. Kawsar. 2023. "Uridine Derivatives: Synthesis, Biological Evaluation, and In Silico Studies as Antimicrobial and Anticancer Agents" Medicina 59, no. 6: 1107. https://doi.org/10.3390/medicina59061107