
Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation


Abstract
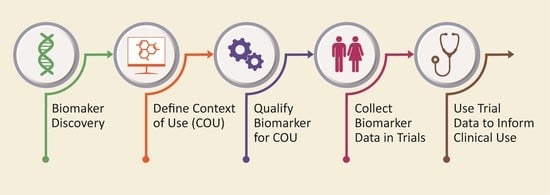
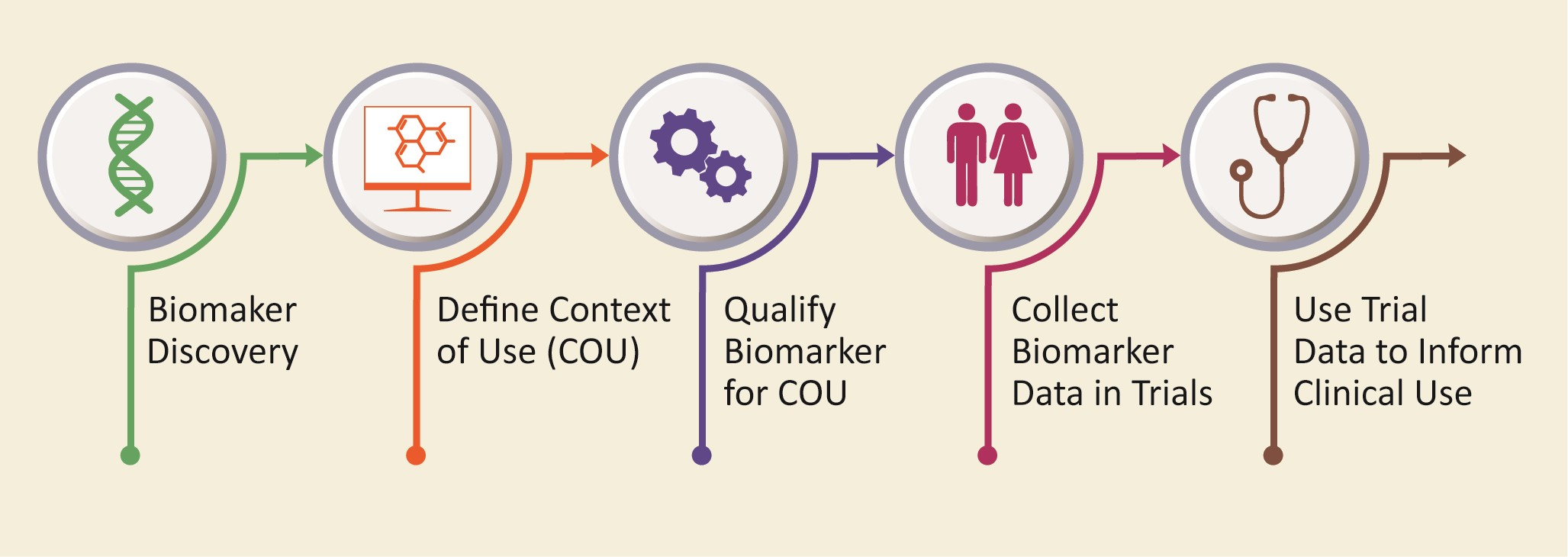
:
1. Introduction
2. Materials and Methods
3. Biomarker Definition and Classification
3.1. Biomarker Definition
3.2. Biomarker Classification
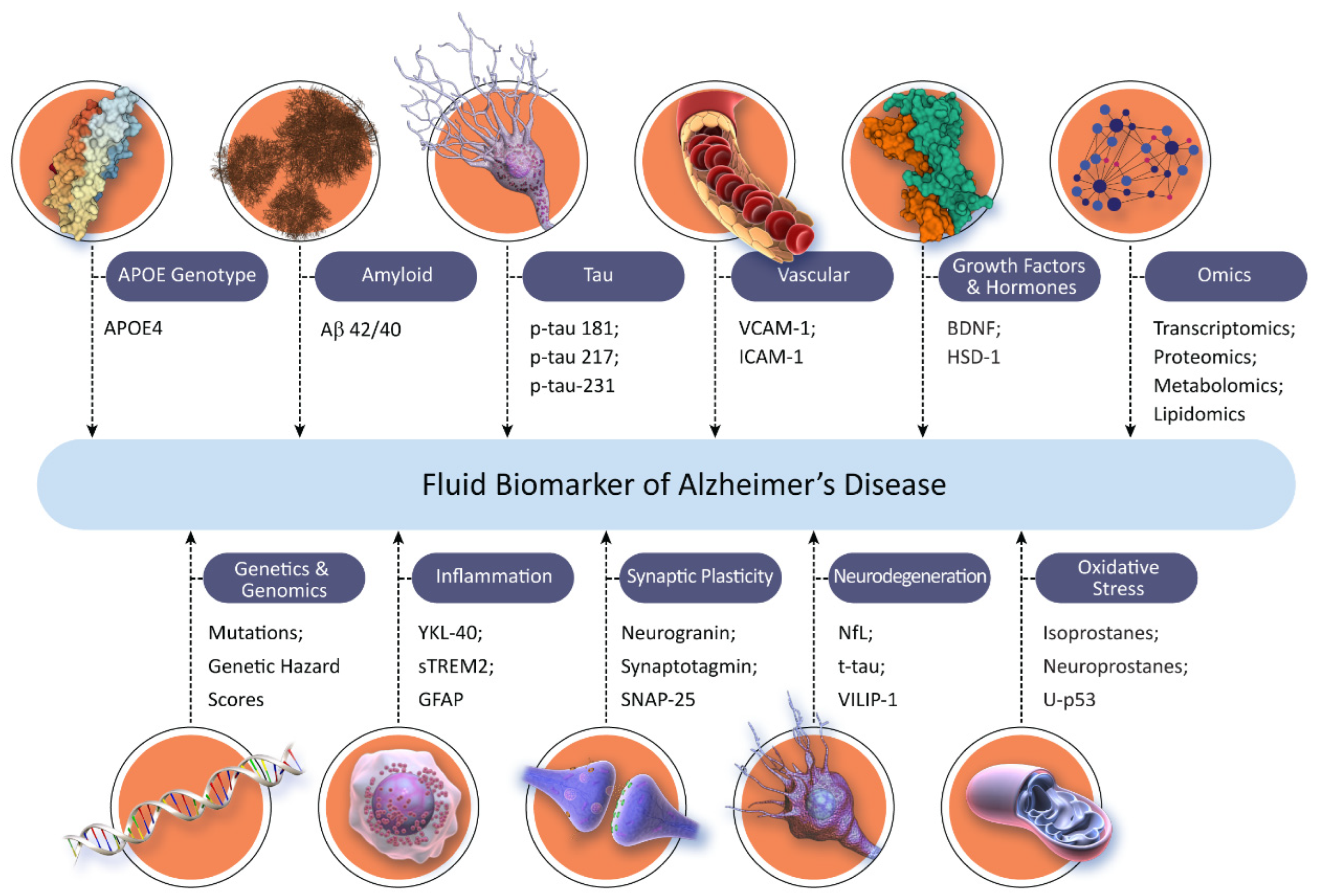
3.3. Biomarkers in Alzheimer’s Disease
3.4. Risk/Susceptibility Biomarkers
3.5. Diagnostic Biomarkers
3.6. Monitoring Biomarkers
3.7. Pharmacodynamic/Response Biomarkers
3.8. Predictive Biomarkers
3.9. Prognostic Biomarkers
3.10. Safety Biomarkers
4. Biomarker Qualification
5. Biomarkers for Use in Clinical Care
5.1. Companion Diagnostic
5.2. In Vitro Diagnostic Devices (IVDs)
5.3. 510(k) Pathway
5.4. Laboratory Developed Test (LDT)
5.5. Research Use Only (RUO) Test
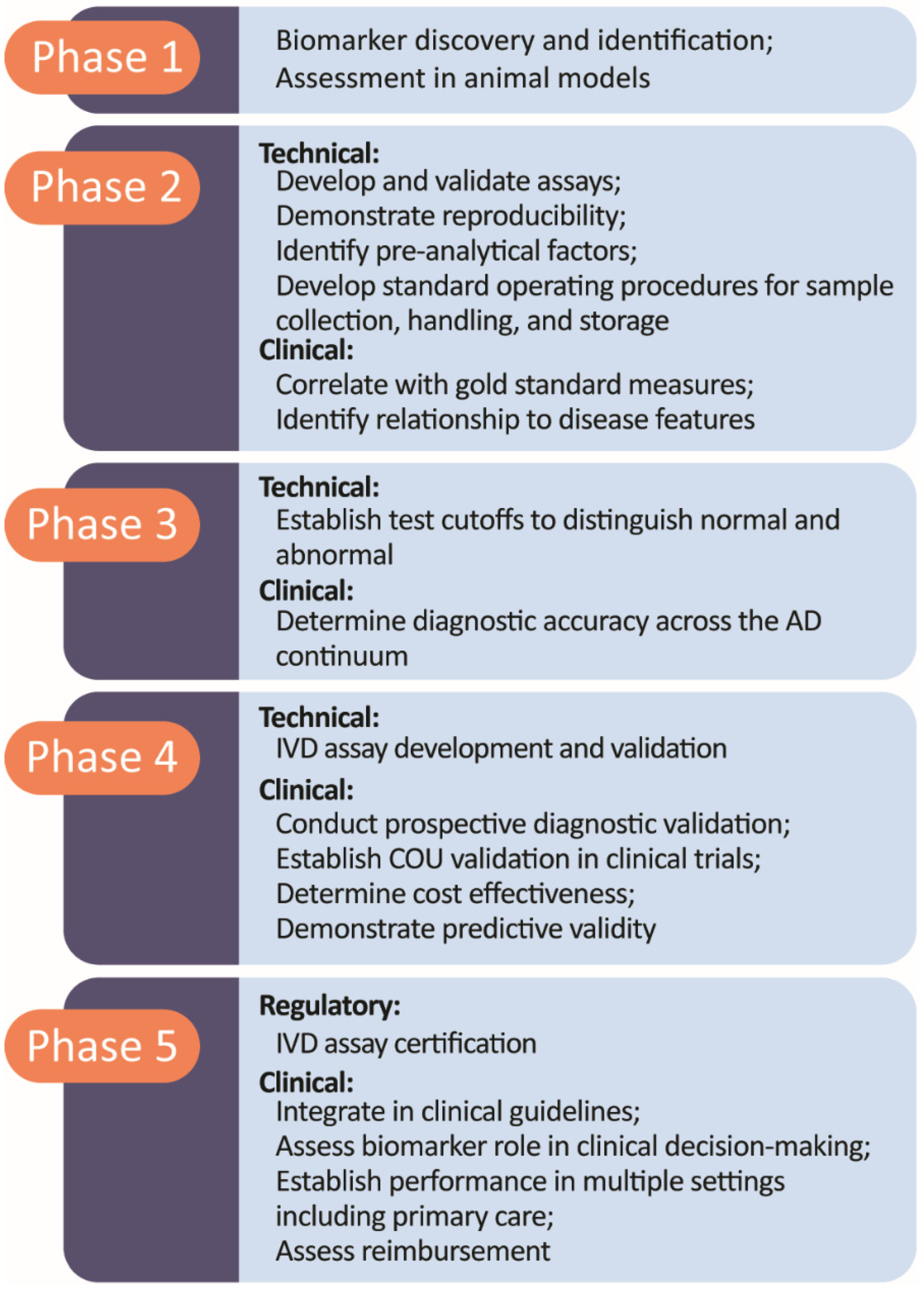
6. Five-Phase Roadmap for Biomarker Development
7. Biomarker Collaborations and Cohorts
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s Disease Facts and Figures; Alzheimer’s Association: Chicago, IL, USA, 2022. [Google Scholar]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. The role of biomarkers in Alzheimer’s disease drug development. Adv. Exp. Med. Biol. 2019, 1118, 29–61. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Biomarker Qualification: Evidentiary Framework. Guidance for Industry and FDA Staff. DRAFT GUIDANCE. U.S. Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER), 2018. Available online: https://www.fda.gov/media/119271/download (accessed on 6 January 2022).
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource [Internet]. Silver Spring (MD): Food and Drug Administration (US); 2016-. Co-Published by National Institutes of Health (US), Bethesda (MD), 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 6 January 2022).
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Valkimadi, P.E.; Pagano, G.; Cousins, O.; Dervenoulas, G.; Politis, M.; Alzheimer’s Disease Neuroimaging Initiative. Applications of amyloid, tau, and neuroinflammation PET imaging to Alzheimer’s disease and mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 5424–5442. [Google Scholar] [CrossRef] [Green Version]
- Matthews, D.C.; Ritter, A.; Thomas, R.G.; Andrews, R.D.; Lukic, A.S.; Revta, C.; Kinney, J.W.; Tousi, B.; Leverenz, J.B.; Fillit, H.; et al. Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer’s dementia. Alzheimers Dement. 2021, 7, e12106. [Google Scholar] [CrossRef]
- Reiman, E.M.; Alzheimer’s Disease Biomarkers Working Group for the Alliance for Aging Research. Fluorodeoxyglucose positron emission tomography: Emerging roles in the evaluation of putative Alzheimer’s disease-modifying treatments. Neurobiol. Aging 2011, 32 (Suppl. S1), S44–S47. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, D.S.; Ashton, N.J.; Blennow, K.; Zetterberg, H.; Simren, J.; Lantero-Rodriguez, J.; Karikari, T.K.; Hiniker, A.; Rissman, R.A.; Salmon, D.P.; et al. Plasma biomarkers for Alzheimer’s Disease in relation to neuropathology and cognitive change. Acta Neuropathol. 2022, 143, 487–503. [Google Scholar] [CrossRef]
- Syrjanen, J.A.; Campbell, M.R.; Algeciras-Schimnich, A.; Vemuri, P.; Graff-Radford, J.; Machulda, M.M.; Bu, G.; Knopman, D.S.; Jack, C.R., Jr.; Petersen, R.C.; et al. Associations of amyloid and neurodegeneration plasma biomarkers with comorbidities. Alzheimers Dement. 2022, 18, 1128–1140. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Petersen, M.; Hall, J.; Johnson, L.A. Medical comorbidities and ethnicity impact plasma Alzheimer’s disease biomarkers: Important considerations for clinical trials and practice. Alzheimers Dement. 2022. [Google Scholar] [CrossRef]
- Grothe, M.J.; Moscoso, A.; Ashton, N.J.; Karikari, T.K.; Lantero-Rodriguez, J.; Snellman, A.; Zetterberg, H.; Blennow, K.; Scholl, M.; Alzheimer’s Disease Neuroimaging Initiative. Associations of fully automated CSF and novel plasma biomarkers with Alzheimer isease neuropathology at autopsy. Neurology 2021, 97, e1229–e1242. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Grinberg, L.T.; Boxer, A.; Ossenkoppele, R.; Jonsson, M.; Seeley, W.; Ehrenberg, A.; Spina, S.; Janelidze, S.; Rojas-Martinex, J.; et al. Cerebrospinal fluid biomarkers in autopsy-confirmed Alzheimer disease and frontotemporal lobar degeneration. Neurology 2022, 98, e1137–e1150. [Google Scholar] [CrossRef]
- Hu, Y.; Kirmess, K.M.; Meyer, M.R.; Rabinovici, G.D.; Gatsonis, C.; Siegel, B.A.; Whitmer, R.A.; Apgar, C.; Hanna, L.; Kanekiyo, M.; et al. Assessment of a plasma amyloid probability score to estimate amyloid positron emission tomography findings among adults with cognitive impairment. JAMA Netw. Open 2022, 5, e228392. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.; Gordon, B.A.; Hornbeck, R.; Norton, J.B.; Levitch, D.; Louden, A.; Ziegemeier, E.; Laforce, R., Jr.; Chhatwal, J.; Day, G.S.; et al. Discovery and validation of autosomal dominant Alzheimer’s disease mutations. Alzheimers Res. Ther. 2018, 10, 67. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Qin, W.; Li, W.; Wang, Q.; Gong, M.; Li, T.; Shi, Y.; Song, Y.; Li, Y.; Li, F.; Jia, J. Race-related association between APOE genotype and Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimers Dis. 2021, 83, 897–906. [Google Scholar] [CrossRef]
- Dickson, S.P.; Hendrix, S.B.; Brown, B.L.; Ridge, P.G.; Nicodemus-Johnson, J.; Hardy, M.L.; McKeany, A.M.; Booth, S.B.; Fortna, R.R.; Kauwe, J.S.K.; et al. GenoRisk: A polygenic risk score for Alzheimer’s disease. Alzheimers Dement 2021, 7, e12211. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Abdalla, N. Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimers Dement. 2018, 14, 981–988. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef]
- Sevigny, J.; Suhy, J.; Chiao, P.; Chen, T.; Klein, G.; Purcell, D.; Oh, J.; Verma, A.; Sampat, M.; Barakos, J. Amyloid PET screening for enrichment of early-stage Alzheimer disease clinical trials: Experience in a phase 1b clinical trial. Alzheimer Dis. Assoc. Disord. 2016, 30, 1–7. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019, 76, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate use recommendations. J. Prev. Alzheimers Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Cummings, J.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Apostolova, L.G.; Hendrix, S.; Sabbagh, M.; Selkoe, D.; Weiner, M.; Salloway, S.; et al. Aducanumab: Appropriate use recommendations update. J. Prev. Alzheimer’s Dis. 2022, 9, 221–230. [Google Scholar] [CrossRef]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of aducanumab for Alzheimer isease-the FDA’s perspective. JAMA Intern. Med. 2021, 181, 1276–1278. [Google Scholar] [CrossRef]
- Bateman, R.J.; Siemers, E.R.; Mawuenyega, K.G.; Wen, G.; Browning, K.R.; Sigurdson, W.C.; Yarasheski, K.E.; Friedrich, S.W.; Demattos, R.B.; May, P.C.; et al. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann. Neurol. 2009, 66, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Portelius, E.; Zetterberg, H.; Dean, R.A.; Marcil, A.; Bourgeois, P.; Nutu, M.; Andreasson, U.; Siemers, E.; Mawuenyega, K.G.; Sigurdson, W.C.; et al. Amyloid-beta (1-15/16) as a marker for gamma-secretase inhibition in Alzheimer’s disease. J. Alzheimers Dis. 2012, 31, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Leuzy, A.; Janelidze, S.; Mattsson-Carlgren, N.; Palmqvist, S.; Jacobs, D.; Cicognola, C.; Stomrud, E.; Vanmechelen, E.; Dage, J.L.; Hansson, O. Comparing the clinical utility and diagnostic performance of CSF P-Tau181, P-Tau217, and P-Tau231 assays. Neurology 2021, 97, e1681–e1694. [Google Scholar] [CrossRef]
- Marks, J.D.; Syrjanen, J.A.; Graff-Radford, J.; Petersen, R.C.; Machulda, M.M.; Campbell, M.R.; Algeciras-Schimnich, A.; Lowe, V.; Knopman, D.S.; Jack, C.R., Jr.; et al. Comparison of plasma neurofilament light and total tau as neurodegeneration markers: Associations with cognitive and neuroimaging outcomes. Alzheimers Res. Ther. 2021, 13, 199. [Google Scholar] [CrossRef]
- Tarawneh, R.; D’Angelo, G.; Macy, E.; Xiong, C.; Carter, D.; Cairns, N.J.; Fagan, A.M.; Head, D.; Mintun, M.A.; Ladenson, J.H.; et al. Visinin-like protein-1: Diagnostic and prognostic biomarker in Alzheimer disease. Ann. Neurol. 2011, 70, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, W.; Middeldorp, J.; Kooijman, L.; Sluijs, J.A.; Kooi, E.J.; Moeton, M.; Freriks, M.; Mizee, M.R.; Hol, E.M. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 492–510. [Google Scholar] [CrossRef] [Green Version]
- Benedet, A.L.; Mila-Aloma, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J.; et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021, 78, 1471–1483. [Google Scholar] [CrossRef]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [Green Version]
- Rauchmann, B.S.; Schneider-Axmann, T.; Alexopoulos, P.; Perneczky, R.; Alzheimer’s Disease Neuroimaging Initiative. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol. Aging 2019, 74, 182–190. [Google Scholar] [CrossRef] [Green Version]
- McGrowder, D.A.; Miller, F.; Vaz, K.; Nwokocha, C.; Wilson-Clarke, C.; Anderson-Cross, M.; Brown, J.; Anderson-Jackson, L.; Williams, L.; Latore, L.; et al. Cerebrospinal fluid biomarkers of Alzheimer’s disease: Current evidence and future perspectives. Brain Sci. 2021, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Ji, B.; Kong, Y.; Qin, L.; Ren, W.; Guan, Y.; Ni, R. PET imaging of neuroinflammation in Alzheimer’s disease. Front. Immunol. 2021, 12, 739130. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Spires-Jones, T.; Zetterberg, H.; Blennow, K.; Caggiano, A.; DeKosky, S.T.; Fillit, H.; Harrison, J.E.; Schneider, L.S.; Scheltens, P.; et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Camporesi, E.; Nilsson, J.; Brinkmalm, A.; Becker, B.; Ashton, N.J.; Blennow, K.; Zetterberg, H. Fluid biomarkers for synaptic dysfunction and loss. Biomark Insights 2020, 15, 1177271920950319. [Google Scholar] [CrossRef]
- Kong, Y.; Zhang, S.; Huang, L.; Zhang, C.; Xie, F.; Zhang, Z.; Huang, Q.; Jiang, D.; Li, J.; Zhou, W.; et al. Positron emission computed tomography imaging of synaptic vesicle glycoprotein 2A in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 731114. [Google Scholar] [CrossRef]
- Drake, J.D.; Chambers, A.B.; Ott, B.R.; Daiello, L.A.; Alzheimer’s Disease Neuroimaging Initiative. Peripheral markers of vascular endothelial dysfunction show independent but additive relationships with brain-based biomarkers in association with functional impairment in Alzheimer’s disease. J. Alzheimers Dis. 2021, 80, 1553–1565. [Google Scholar] [CrossRef]
- Musaeus, C.S.; Gleerup, H.S.; Hogh, P.; Waldemar, G.; Hasselbalch, S.G.; Simonsen, A.H. Cerebrospinal fluid/plasma albumin ratio as a biomarker for blood-brain barrier impairment across neurodegenerative dementias. J. Alzheimers Dis. 2020, 75, 429–436. [Google Scholar] [CrossRef]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C. Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): A systematic review and meta-analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Raikes, A.C.; Hernandez, G.D.; Matthews, D.C.; Lukic, A.S.; Law, M.; Shi, Y.; Schneider, L.S.; Brinton, R.D. Exploratory imaging outcomes of a phase 1b/2a clinical trial of allopregnanolone as a regenerative therapeutic for Alzheimer’s disease: Structural effects and functional connectivity outcomes. Alzheimers Dement. 2022, 8, e12258. [Google Scholar] [CrossRef]
- Marek, G.J.; Katz, D.A.; Meier, A.; Greco, N.t.; Zhang, W.; Liu, W.; Lenz, R.A. Efficacy and safety evaluation of HSD-1 inhibitor ABT-384 in Alzheimer’s disease. Alzheimers Dement. 2014, 10, S364–S373. [Google Scholar] [CrossRef]
- Sultzer, D.L.; Melrose, R.J.; Riskin-Jones, H.; Narvaez, T.A.; Veliz, J.; Ando, T.K.; Juarez, K.O.; Harwood, D.G.; Brody, A.L.; Mandelkern, M.A. Cholinergic receptor binding in Alzheimer disease and healthy aging: Assessment in vivo with positron emission tomography imaging. Am. J. Geriatr. Psychiatry 2017, 25, 342–353. [Google Scholar] [CrossRef]
- Volkow, N.D.; Ding, Y.S.; Fowler, J.S.; Gatley, S.J. Imaging brain cholinergic activity with positron emission tomography: Its role in the evaluation of cholinergic treatments in Alzheimer’s dementia. Biol. Psychiatry 2001, 49, 211–220. [Google Scholar] [CrossRef]
- Kanel, P.; Bedard, M.A.; Aghourian, M.; Rosa-Neto, P.; Soucy, J.P.; Albin, R.L.; Bohnen, N.I. Molecular imaging of the cholinergic system in Alzheimer and Lewy body dementias: Expanding views. Curr. Neurol. Neurosci. Rep. 2021, 21, 52. [Google Scholar] [CrossRef]
- Smith, G.S.; Workman, C.I.; Protas, H.; Su, Y.; Savonenko, A.; Kuwabara, H.; Gould, N.F.; Kraut, M.; Joo, J.H.; Nandi, A.; et al. Positron emission tomography imaging of serotonin degeneration and beta-amyloid deposition in late-life depression evaluated with multi-modal partial least squares. Transl. Psychiatry 2021, 11, 473. [Google Scholar] [CrossRef]
- Cummings, J.L.; Henchcliffe, C.; Schaier, S.; Simuni, T.; Waxman, A.; Kemp, P. The role of dopaminergic imaging in patients with symptoms of dopaminergic system neurodegeneration. Brain 2011, 134, 3146–3166. [Google Scholar] [CrossRef] [Green Version]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef]
- Piccirella, S.; Van Neste, L.; Fowler, C.; Masters, C.L.; Fripp, J.; Doecke, J.D.; Xiong, C.; Uberti, D.; Kinnon, P. A Conformational variant of p53 (U-p53AZ) as blood-based biomarker for the prediction of the onset of symptomatic Alzheimer’s disease. J. Prev. Alzheimer’s Dis. 2022, 9, 469–479. [Google Scholar] [CrossRef]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational altered p53 as an early marker of oxidative stress in Alzheimer’s disease. PLoS ONE 2012, 7, e29789. [Google Scholar] [CrossRef]
- Zarrouk, A.; Debbabi, M.; Bezine, M.; Karym, E.M.; Badreddine, A.; Rouaud, O.; Moreau, T.; Cherkaoui-Malki, M.; El Ayeb, M.; Nasser, B.; et al. Lipid biomarkers in Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 303–312. [Google Scholar] [CrossRef]
- Wong, M.W.; Braidy, N.; Poljak, A.; Sachdev, P.S. The application of lipidomics to biomarker research and pathomechanisms in Alzheimer’s disease. Curr. Opin. Psychiatry 2017, 30, 136–144. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Moller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Braendgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Ryan, J. Micro RNA as a potential blood-based epigenetic biomarker for Alzheimer’s disease. Clin. Biochem. 2018, 58, 5–14. [Google Scholar] [CrossRef]
- Briels, C.T.; Stam, C.J.; Scheltens, P.; Gouw, A.A. The predictive value of normal EEGs in dementia due to Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2021, 8, 1038–1048. [Google Scholar] [CrossRef]
- Holiga, S.; Abdulkadir, A.; Kloppel, S.; Dukart, J. Functional magnetic resonance imaging in Alzheimer’ disease drug development. Methods Mol. Biol. 2018, 1750, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Prvulovic, D.; Teipel, S.J.; Bokde, A.L. Recent developments of functional magnetic resonance imaging research for drug development in Alzheimer’s disease. Prog. Neurobiol. 2011, 95, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nuaimi, A.H.; Bluma, M.; Al-Juboori, S.S.; Eke, C.S.; Jammeh, E.; Sun, L.; Ifeachor, E. Robust EEG based biomarkers to detect Alzheimer’s disease. Brain Sci. 2021, 11, 1026. [Google Scholar] [CrossRef] [PubMed]
- Badhwar, A.; McFall, G.P.; Sapkota, S.; Black, S.E.; Chertkow, H.; Duchesne, S.; Masellis, M.; Li, L.; Dixon, R.A.; Bellec, P. A multiomics approach to heterogeneity in Alzheimer’s disease: Focused review and roadmap. Brain 2020, 143, 1315–1331. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 2020, 105, 975–991.e7. [Google Scholar] [CrossRef]
- Hampel, H.; Nistico, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef]
- Cotta Ramusino, M.; Perini, G.; Altomare, D.; Barbarino, P.; Weidner, W.; Salvini Porro, G.; Barkhof, F.; Rabinovici, G.D.; van der Flier, W.M.; Frisoni, G.B.; et al. Outcomes of clinical utility in amyloid-PET studies: State of art and future perspectives. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2157–2168. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Pontecorvo, M.J.; Devous, M.D., Sr.; Lu, M.; Arora, A.K.; Truocchio, S.P.; Aldea, P.; Flitter, M.; Locascio, T.; Devine, M.; et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020, 77, 829–839. [Google Scholar] [CrossRef]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef]
- Samper-Gonzalez, J.; Burgos, N.; Bottani, S.; Fontanella, S.; Lu, P.; Marcoux, A.; Routier, A.; Guillon, J.; Bacci, M.; Wen, J.; et al. Reproducible evaluation of classification methods in Alzheimer’s disease: Framework and application to MRI and PET data. Neuroimage 2018, 183, 504–521. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Barker, P.B. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol. Biol. 2011, 711, 203–226. [Google Scholar] [CrossRef] [Green Version]
- Fossati, S.; Ramos Cejudo, J.; Debure, L.; Pirraglia, E.; Sone, J.Y.; Li, Y.; Chen, J.; Butler, T.; Zetterberg, H.; Blennow, K.; et al. Plasma tau complements CSF tau and P-tau in the diagnosis of Alzheimer’s disease. Alzheimers Dement. 2019, 11, 483–492. [Google Scholar] [CrossRef]
- Mielke, M.M.; Syrjanen, J.A.; Blennow, K.; Zetterberg, H.; Vemuri, P.; Skoog, I.; Machulda, M.M.; Kremers, W.K.; Knopman, D.S.; Jack, C., Jr.; et al. Plasma and CSF neurofilament light: Relation to longitudinal neuroimaging and cognitive measures. Neurology 2019, 93, e252–e260. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; Lee, J.M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 2012, 78, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Cummings, J.; Blennow, K.; Gao, P.; Jack, C.R., Jr.; Vergallo, A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat. Rev. Neurol. 2021, 17, 580–589. [Google Scholar] [CrossRef]
- Cummings, J.; Fox, N. Defining disease modifying therapy for Alzheimer’s disease. J. Prev. Alzheimers Dis. 2017, 4, 109–115. [Google Scholar] [CrossRef]
- Food and Drug Administration. In Vitro Companion Diagnostic Devices: Guidance for Industry and Food and Drug Administration Staff. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research Center for Drug Evaluation and Research, 2014. Available online: https://www.fda.gov/files/medical%20devices/published/In-Vitro-Companion-Diagnostic-Devices---Guidance-for-Industry-and-Food-and-Drug-Administration-Staff%29.pdf (accessed on 6 January 2022).
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-related imaging abnormalities in 2 Phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Zetterberg, H.; Skillback, T.; Mattsson, N.; Trojanowski, J.Q.; Portelius, E.; Shaw, L.M.; Weiner, M.W.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer isease progression. JAMA Neurol. 2016, 73, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ebenau, J.L.; Pelkmans, W.; Verberk, I.M.W.; Verfaillie, S.C.J.; van den Bosch, K.A.; van Leeuwenstijn, M.; Collij, L.E.; Scheltens, P.; Prins, N.D.; Barkhof, F.; et al. Association of CSF, plasma, and imaging markers of neurodegeneration with clinical progression in people with subjective cognitive ecline. Neurology 2022, 98, e1315–e1326. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Pontecorvo, M.J.; Devous, M.D., Sr.; Arora, A.K.; Galante, N.; McGeehan, A.; Devadanam, C.; Salloway, S.P.; Doraiswamy, P.M.; Curtis, C.; et al. Aggregated tau measured by visual interpretation of flortaucipir positron emission tomography and the associated risk of clinical progression of mild cognitive impairment and Alzheimer disease: Results from 2 phase III clinical trials. JAMA Neurol. 2021, 78, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Qualification Process for Drug Development Tools. Guidance for Industry and FDA Staff. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER), 2020. Available online: https://www.fda.gov/media/133511/download (accessed on 6 January 2022).
- Kraus, V.B. Biomarkers as drug development tools: Discovery, validation, qualification and use. Nat. Rev. Rheumatol. 2018, 14, 354–362. [Google Scholar] [CrossRef]
- Food and Drug Administration. Recommended Content and Format of Non-Clinical Bench Performance Testing Information in Premarket Submissions. Guidance for Industry and Food and Drug Administration Staff. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research, 2019. Available online: https://www.fda.gov/media/113230/download (accessed on 6 January 2022).
- Jorgensen, J.T. Companion and complementary iagnostics: Clinical and regulatory perspectives. Trends Cancer 2016, 2, 706–712. [Google Scholar] [CrossRef]
- Jorgensen, J.T. Companion and complementary diagnostics: An important treatment decision tool in precision medicine. Expert. Rev. Mol. Diagn. 2020, 20, 557–559. [Google Scholar] [CrossRef]
- Pew Charitable Trusts. What Are In Vitro Diagnostic Tests and How Are They Regulated. A Brief from the Pew Charitable Trusts. May 2019. Available online: https://www.pewtrusts.org/-/media/assets/2019/05/what-are-in-vitro-diagnostic-tests-and-how-are-they-regulated.pdf (accessed on 6 January 2022).
- Food and Drug Administration. Acceptance of Clinical Data to Support Medical Device Applications and Submissions. Frequently Asked Questions. Guidance for Industry and Food and Drug Administration Staff. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research Office of Good Clinical Practice, 2018. Available online: https://www.hhs.gov/guidance/document/acceptance-clinical-data-support-medical-device-applications-and-submissions-frequently (accessed on 6 January 2022).
- Leitao, M.J.; Silva-Spinola, A.; Santana, I.; Olmedo, V.; Nadal, A.; Le Bastard, N.; Baldeiras, I. Clinical validation of the Lumipulse G cerebrospinal fluid assays for routine diagnosis of Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 91. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]. Guidance for Industry and Food and Drug Administration Staff. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research, 2011. Available online: https://www.fda.gov/media/82395/download (accessed on 6 January 2022).
- Food and Drug Administration. Draft Guidance for Industry, Food and 2 Drug Administration Staff, and Clinical 3 Laboratories: Framework for Regulatory Oversight of 7 Laboratory Developed Tests (LDTs). U.S. Department of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, Office of In Vitro Diagnostics and Radiological Health, Center for Biologics Evaluation and Research, 2014. Available online: https://www.fda.gov/media/89841/download (accessed on 6 January 2022).
- Food and Drug Administration. Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only. Guidance for Industry and Food and Drug Administration Staff. U.S. Department of Health and Human Services Food; Drug Administration Center for Devices and Radiological Health Office of In Vitro Diagnostic Device Evaluation and Safety; Center for Biologics Evaluation and Research, 2013. Available online: https://www.fda.gov/media/87374/download (accessed on 6 January 2022).
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; Del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Boccardi, M.; Barkhof, F.; Blennow, K.; Cappa, S.; Chiotis, K.; Demonet, J.F.; Garibotto, V.; Giannakopoulos, P.; Gietl, A.; et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017, 16, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Boccardi, M.; Dodich, A.; Albanese, E.; Gayet-Ageron, A.; Festari, C.; Ashton, N.J.; Bischof, G.N.; Chiotis, K.; Leuzy, A.; Wolters, E.E.; et al. The strategic biomarker roadmap for the validation of Alzheimer’s diagnostic biomarkers: Methodological update. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2070–2085. [Google Scholar] [CrossRef]
- Garibotto, V.; Herholz, K.; Boccardi, M.; Picco, A.; Varrone, A.; Nordberg, A.; Nobili, F.; Ratib, O.; Geneva Task Force for the Roadmap of Alzheimer’s, B. Clinical validity of brain fluorodeoxyglucose positron emission tomography as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 183–195. [Google Scholar] [CrossRef]
- Ashton, N.J.; Leuzy, A.; Karikari, T.K.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2140–2156. [Google Scholar] [CrossRef]
- Bischof, G.N.; Dodich, A.; Boccardi, M.; van Eimeren, T.; Festari, C.; Barthel, H.; Hansson, O.; Nordberg, A.; Ossenkoppele, R.; Sabri, O.; et al. Clinical validity of second-generation tau PET tracers as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2110–2120. [Google Scholar] [CrossRef]
- Wolters, E.E.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Hansson, O.; Nordberg, A.; Frisoni, G.B.; Garibotto, V.; Ossenkoppele, R. Clinical validity of increased cortical uptake of [(18)F] flortaucipir on PET as a biomarker for Alzheimer’s disease in the context of a structured 5-phase biomarker development framework. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2097–2109. [Google Scholar] [CrossRef]
- Chiotis, K.; Saint-Aubert, L.; Boccardi, M.; Gietl, A.; Picco, A.; Varrone, A.; Garibotto, V.; Herholz, K.; Nobili, F.; Nordberg, A.; et al. Clinical validity of increased cortical uptake of amyloid ligands on PET as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Ten Kate, M.; Barkhof, F.; Boccardi, M.; Visser, P.J.; Jack, C.R., Jr.; Lovblad, K.O.; Frisoni, G.B.; Scheltens, P.; Geneva Task Force for the Roadmap of Alzheimer’s, B. Clinical validity of medial temporal atrophy as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 167–182.e1. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.; Rainey-Smith, S.R.; Bird, S.; Bomke, J.; Bourgeat, P.; Brown, B.M.; Burnham, S.C.; Bush, A.I.; Chadunow, C.; Collins, S.; et al. Fifteen years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study: Progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer’s disease. J. Alzheimers Dis. Rep. 2021, 5, 443–468. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Morris, J.C.; et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019, 15, 106–152. [Google Scholar] [CrossRef]
- Van Maurik, I.S.; Rhodius-Meester, H.F.M.; Teunissen, C.E.; Scheltens, P.; Barkhof, F.; Palmqvist, S.; Hansson, O.; van der Flier, W.M.; Berkhof, J. Biomarker testing in MCI patients-deciding who to test. Alzheimers Res. Ther. 2021, 13, 14. [Google Scholar] [CrossRef]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Abeta42/Abeta40 and Abeta42/Abeta38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Measurement |
|---|---|
| Risk/susceptibility | Indicates the potential for developing a disease or medical condition in an individual who does not currently have a clinically apparent disease or medical condition |
| Diagnosis | Detects or confirms the presence of a disease or condition or identifies an individual with a subtype of the disease |
| Monitoring | Measured serially to assess the status of a disease or medical condition for evidence of exposure to a medical product or environmental agent or to detect an effect of a medical product or biological agent |
| Pharmacodynamic/response | Changes in response to exposure to a medical product or an environmental agent |
| Predictive | The presence or change in the biomarker predicts an individual or group of individuals more likely to experience a favorable or unfavorable effect from the exposure to a medical product or environmental agent |
| Prognostic | Identifies the likelihood of a clinical event, disease recurrence, or disease progression in patients with a disease or medical condition |
| Safety | Measured before or after an exposure to a medical intervention or environmental agent to indicate the likelihood, presence, or extent of a toxicity as an adverse event |
| CADRO Category | Fluid Biomarkers | Imaging, Digital, and Device-Based Biomarkers |
|---|---|---|
| Amyloid beta | Inhibition of production of CSF Aβ by beta and gamma secretase inhibitors; increase in Aβ 1–15/16 by gamma secretase inhibitors | Amyloid PET |
| Tau | CSF and plasma p-tau 181, p-tau 217, and p-tau 231 | Tau PET |
| APOE, lipids, lipoprotein receptors | Lipid peroxidation, isoprostanes, and lipidomics | None identified |
| Neurotransmitter receptors | None identified | Nicotinic cholinergic receptor PET, muscarinic receptor PET, dopamine transporter SPECT and PET, acetylcholine (VCHAT) and serotonin vesicular transporter PET |
| Neurogenesis | None identified | MRI measures of hippocampus; fractional and quantitative anisotropy |
| Inflammation | CSF and plasma GFAP, CSF YKL40, sTREM2, and MCP-1 | TSPO PET and evolving ligands |
| Oxidative stress | Lipid peroxidation, isoprostanes, neuroprostanes, and u-P53 | None identified |
| Proteostasis/proteinopathies | CSF Aβ and proteomics | None identified |
| Metabolism and bioenergetics | Metabolomics | FDG PET |
| Vasculature | Plasma VCAM-1 and ICAM-1; CSF/plasma albumin ratio to assess blood–brain barrier | MRI |
| Growth factors and hormones | Brain-derived neurotrophic factor (BDNF), HSD-1, and trial-specific hormones | MRI measures of hippocampal volume |
| Synaptic plasticity/neuroprotection | Neurogranin, synaptotagmin, and SNAP-25 | SV2A PET |
| Cell death | Total tau, neurofilament light, VILIP-1, and GAP-43 | Structural MRI (including hippocampal volume), FDG PET, and MR spectroscopy (NAA) |
| Gut-brain axis | Changes in blood amino acids and inflammatory cells | Changes in the microbe composition of the microbiome |
| Circadian rhythm | None identified | Polysomnography and actigraphy |
| Epigenetic regulators | MicroRNA | None identified |
| Amyloid (A) | Tau (T) | Neurodegeneration (N) | |
|---|---|---|---|
| Imaging | Amyloid PET | Tau PET | FDG PET; MRI; spectroscopy |
| CSF | Aβ 42/40 | p-tau (181, 217) | Total tau; NfL; VILIP-1 |
| Plasma | Aβ 42/40 | p-tau (181, 217) | Total tau; NfL |
| Pathway | Characteristic |
|---|---|
| Companion diagnostic | Required for appropriate use of a specific agent |
| In vitro diagnostic device (IVD) | Review by the FDA varies according to level of risk associated with the biomarker |
| 510(k) pathway | Shown to be substantially equivalent to an approved IVD with performance characteristics at least as good as the approved IVD |
| Laboratory Developed Test (LDT) | Performed in a single laboratory; relatively limited FDA review |
| Research Use Only (RUO) | Cannot be used in diagnosis; may be used to gather additional information on the biomarker |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cummings, J.; Kinney, J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina 2022, 58, 952. https://doi.org/10.3390/medicina58070952
Cummings J, Kinney J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina. 2022; 58(7):952. https://doi.org/10.3390/medicina58070952
Chicago/Turabian StyleCummings, Jeffrey, and Jefferson Kinney. 2022. "Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation" Medicina 58, no. 7: 952. https://doi.org/10.3390/medicina58070952