
Effect of Autophagy Inhibitors on Radiosensitivity in DNA Repair-Proficient and -Deficient Glioma Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Assessment of Autophagy by Acridine Orange Staining
2.4. Evaluation of Senescence by β-Galactosidase Staining
2.5. Determination of γH2AX Intensity as a Marker of DNA Damage
2.6. Evaluation of Apoptosis
2.7. Cell Cycle Analysis
2.8. Statistical Analysis
3. Results
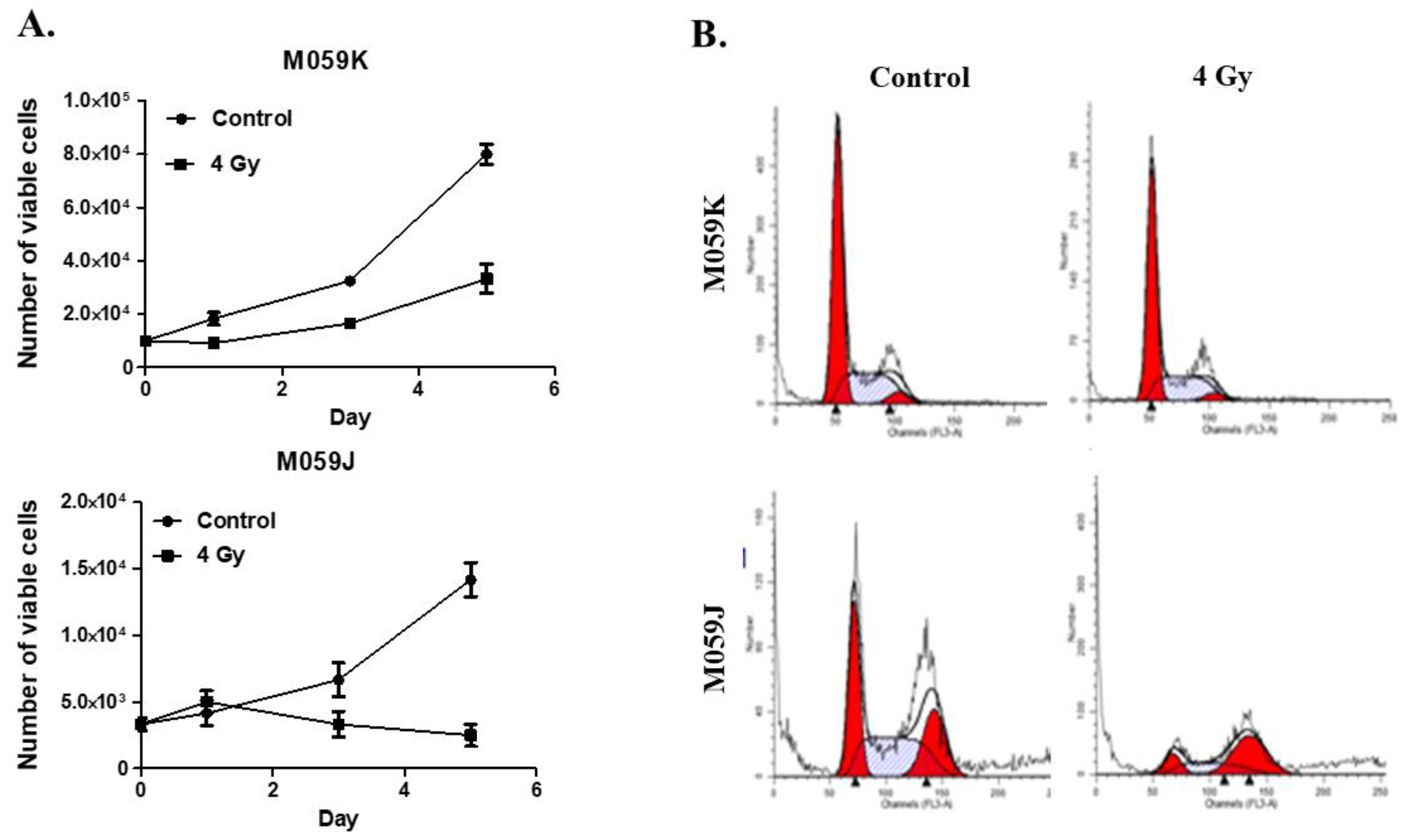
3.1. Differential Response of M059K and M059J Cell Lines to Radiation
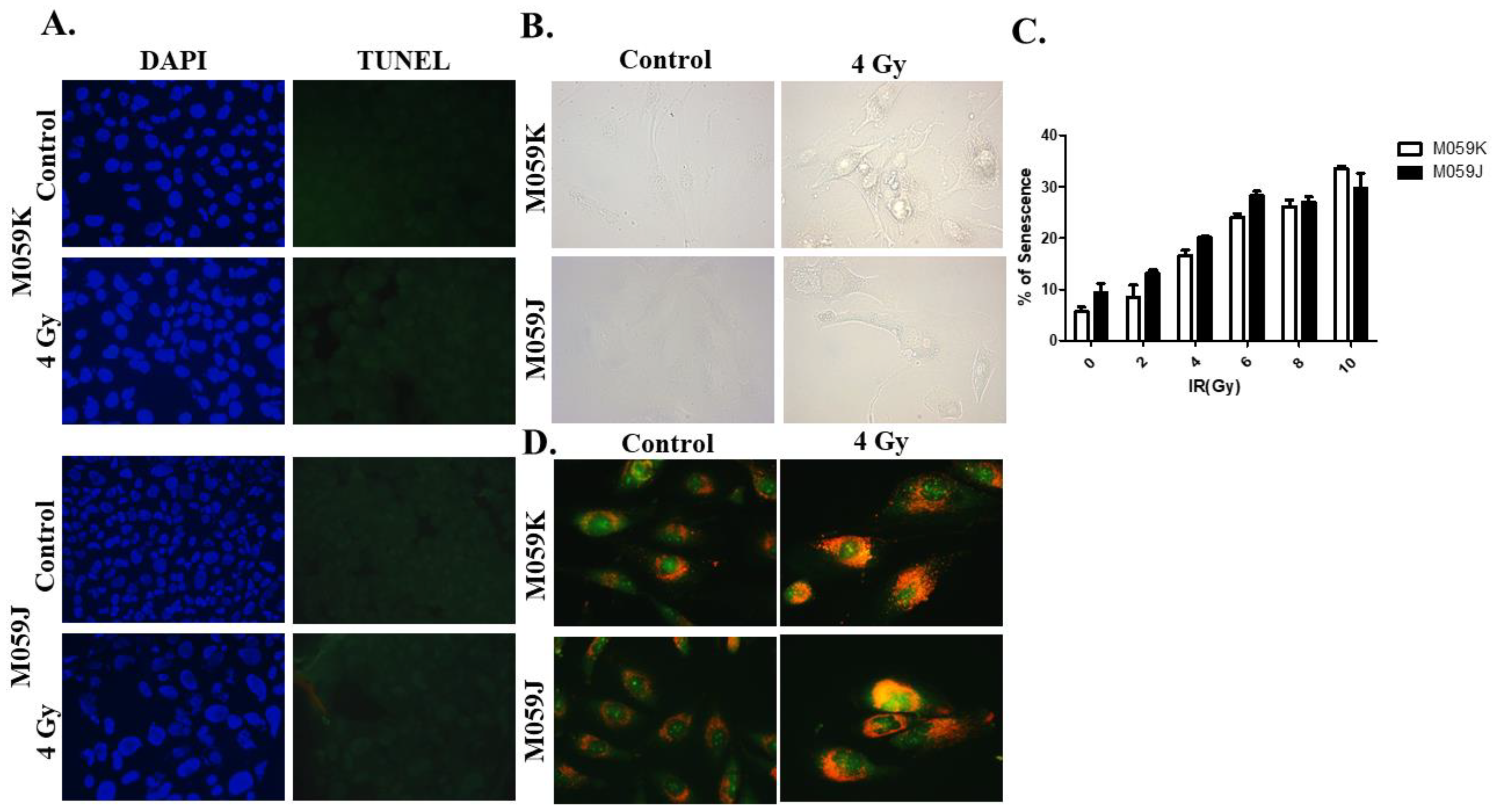
3.2. Induction of Autophagy, but Neither Apoptosis or Senescence, by Ionizing Radiation in DNA-PKcs-Expressing and DNA-PKcs-Not Expressing Cells
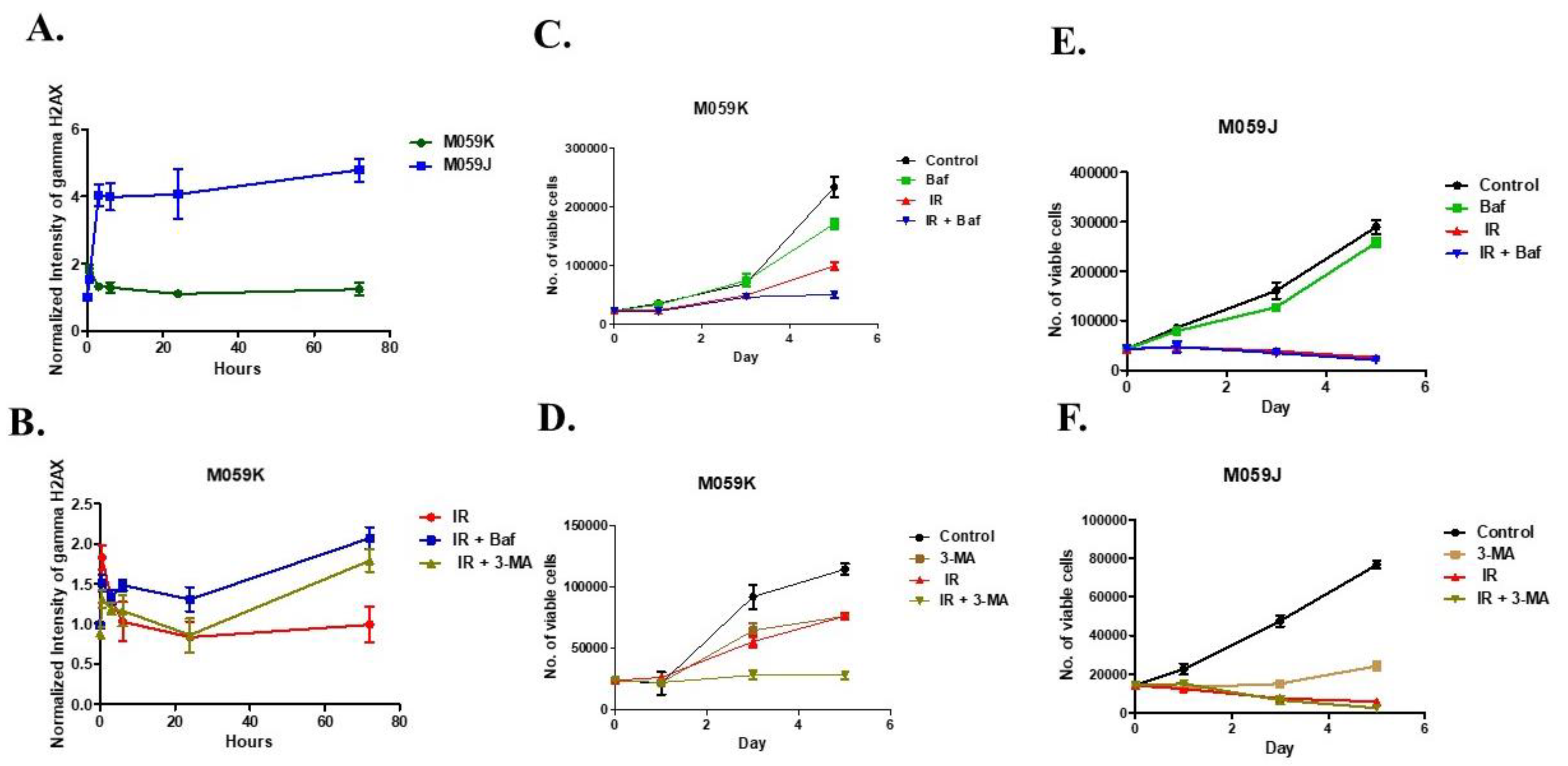
3.3. Effects of Autophagy Inhibitors on Radiosensitization and DNA Repair Capacity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.; McGale, P.; Correa, C.; Taylor, C.; Arriagada, R.; Clarke, M.; Cutter, D.; Davies, C.; Ewertz, M.; Godwin, J.; et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet 2011, 378, 1707–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grégoire, V.; Langendijk, J.A.; Nuyts, S. Advances in radiotherapy for head and neck cancer. J. Clin. Oncol. 2015, 33, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Le Chevalier, T.; Arriagada, R.; Quoix, E.; Ruffle, P.; Martin, M.; Tarayre, M.; Marie-josé, L.T.; Douillard, J.Y.; Laplanche, A. Radiotherapy alone versus combined chemotherapy and radiotherapy in nonresectable non-small-cell lung cancer: First analysis of a randomized trial in 353 patients. J. Natl. Cancer Inst. 1991, 83, 417–423. [Google Scholar] [CrossRef]
- Curran, W.J.; Paulus, R.; Langer, C.J.; Komaki, R.; Lee, J.S.; Hauser, S.; Movsas, B.; Wasserman, T.; Rosenthal, S.A.; Gore, E.; et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: Randomized phase III trial RTOG 9410. J. Natl. Cancer Inst. 2011, 103, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Abe, O.; Abe, R.; Enomoto, K.; Kikuchi, K.; Koyama, H.; Masuda, H.; Nomura, Y.; Sakai, K.; Sugimachi, K.; Tominaga, T.; et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 366, 2087–2106. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular stress responses in radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chircop, M.; Speidel, D. Cellular stress responses in cancer and cancer therapy. Front. Oncol. 2014, 4, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellette, M.M.; Zhou, S.; Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics 2022, 12, 656. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Zhang, X.-R.; Ma, Y.-L.; Zhao, Z.-J.; Zhao, R.; Wang, Y.-Y. Nrf2 promotes esophageal squamous cell carcinoma (ESCC) resistance to radiotherapy through the CaMKIIα-associated activation of autophagy. Cell Biosci. 2020, 10, 90. [Google Scholar] [CrossRef]
- Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic autophagy in cancer therapy. Int. J. Mol. Sci. 2014, 15, 10034–10051. [Google Scholar] [CrossRef]
- Ulasov, I.; Fares, J.; Timashev, P.; Lesniak, M.S. Editing Cytoprotective Autophagy in Glioma: An Unfulfilled Potential for Therapy. Trends Mol. Med. 2019, 26, 252–262. [Google Scholar] [CrossRef]
- Xu, J.; Patel, N.H.; Saleh, T.; Cudjoe, E.K.; Alotaibi, M.; Wu, Y.; Lima, S.; Hawkridge, A.M.; Gewirtz, D.A. Differential Radiation Sensitivity in p53 Wild-Type and p53-Deficient Tumor Cells Associated with Senescence but not Apoptosis or (Nonprotective) Autophagy. Radiat. Res. 2018, 190, 538–557. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.; Guan, J.L. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol. Cancer Res. 2011, 9, 1232–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Yan, T.; Schupp, J.E.; Seo, Y.; Kinsella, T.J. DNA mismatch repair initiates 6-thioguanine-induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 2007, 13, 1315–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gámez, J.A.; Rodríguez-Vargas, J.M.; Quiles-Pérez, R.; Aguilar-Quesada, R.; Martín-Oliva, D.; De Murcia, G.; De Murcia, J.M.; Almendros, A.; Ruiz De Almodóvar, M.; Oliver, F.J. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a potential therapy for malignant glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Hoppe, B.S.; Jensen, R.B.; Kirchgessner, C.U. Complementation of the radiosensitive M059J cell line. Radiat. Res. 2000, 153, 125–130. [Google Scholar] [CrossRef]
- Chan, D.W.; Gately, D.P.; Urban, S.; Galloway, A.M.; Lees-Miller, S.P.; Yen, T.; Allalunis-Turner, J. Lack of correlation between ATM protein expression and tumour cell radiosensitivity. Int. J. Radiat. Biol. 1998, 74, 217–224. [Google Scholar] [CrossRef]
- Gately, D.P.; Hittle, J.C.; Chan, G.K.T.; Yen, T.J. Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol. Biol. Cell 1998, 9, 2361–2374. [Google Scholar] [CrossRef] [Green Version]
- Mitrakas, A.G.; Kalamida, D.; Giatromanolaki, A.; Pouliliou, S.; Tsolou, A.; Kyranas, R.; Koukourakis, M.I. Autophagic flux response and glioblastoma sensitivity to radiation. Cancer Biol. Med. 2018, 15, 260–274. [Google Scholar] [CrossRef]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.L.F.; Hendrickson, E.A.E.A.; Gewirtz, D.A.D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Goehe, R.W.; Di, X.; Hicks, M.A.; Torti, S.V.; Torti, F.M.; Harada, H.; Gewirtz, D.A. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy 2014, 10, 2346–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, D.J.D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D.J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.; Elsayed, Z.; Henderson, S.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef]
- Fitsiou, E.; Soto-Gamez, A.; Demaria, M. Biological functions of therapy-induced senescence in cancer. Semin. Cancer Biol. 2022, 81, 5–13. [Google Scholar] [CrossRef]
- Yoon, J.H.; Ahn, S.G.; Lee, B.H.; Jung, S.H.; Oh, S.H. Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 2012, 83, 747–757. [Google Scholar] [CrossRef]
- Liu, W.; Otkur, W.; Li, L.; Wang, Q.; He, H.; Ye, Y.; Zhang, Y.; Hayashi, T.; Tashiro, S.I.; Onodera, S.; et al. Autophagy induced by silibinin protects human epidermoid carcinoma A431 cells from UVB-induced apoptosis. J. Photochem. Photobiol. B Biol. 2013, 123, 23–31. [Google Scholar] [CrossRef]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA damage response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Tang, B.; Xie, X.; Xiao, Y.F.; Yang, S.M.; Zhang, J.W. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol. Ther. 2015, 16, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Tougeron, D.; Huang, S.; Okamoto, K.; Sinicrope, F.A. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE 2014, 9, e100819. [Google Scholar] [CrossRef]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Zhang, H.; Liu, H.; Han, Y.; Qiu, W.; Li, Z. Inhibiting autophagy flux and DNA repair of tumor cells to boost radiotherapy of orthotopic glioblastoma. Biomaterials 2022, 280, 121287. [Google Scholar] [CrossRef]
- Yu, L.; Tumati, V.; Tseng, S.F.; Hsu, F.M.; Kim, D.N.; Hong, D.; Hsieh, J.T.; Jacobs, C.; Kapur, P.; Saha, D. DAB2IP regulates autophagy in prostate cancer in response to combined treatment of radiation and a DNA-PKcs inhibitor. Neoplasia 2012, 14, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Guo, M.; Ouyang, H.; Li, X.; Cordon-Cardo, C.; Kurimasa, A.; Chen, D.J.; Fuks, Z.; Clifton Ling, C.; Li, G.C. The catalytic subunit of DNA-dependent protein kinase selectively regulates p53-dependent apoptosis but not cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 1584–1588. [Google Scholar] [CrossRef] [Green Version]
- Woo, R.A.; Jack, M.T.; Xu, Y.; Burma, S.; Chen, D.J.; Lee, P.W.K. DNA damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. EMBO J. 2002, 21, 3000–3008. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P. DNA-dependent protein kinase. Int. J. Biochem. Cell Biol. 1997, 29, 935–938. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z.Q. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res. 2011, 1371, 7–15. [Google Scholar] [CrossRef]
- Murad, H.; Alghamian, Y.; Aljapawe, A.; Madania, A. Effects of ionizing radiation on the viability and proliferative behavior of the human glioblastoma T98G cell line. BMC Res. Notes 2018, 11, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Morgan-Lappe, S.E.; Yang, J.; Bockbrader, K.M.; Pamarthy, D.; Thomas, D.; Fesik, S.W.; Sun, Y. Growth inhibition and radiosensitization of glioblastoma and lung cancer cells by small interfering RNA silencing of tumor necrosis factor receptor-associated factor 2. Cancer Res. 2008, 68, 7570–7578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, A.G.; Segura, T.M.; Rintala, A.C.; Rintala-Maki, N.D.; Lee, H. The Generation and Characterization of a Radiation-Resistant Model System to Study Radioresistance in Human Breast Cancer Cells on JSTOR. Radiat. Res. 2001, 156, 739–750. [Google Scholar] [CrossRef]
- Dai, P.L.; Du, X.S.; Hou, Y.; Li, L.; Xia, Y.X.; Wang, L.; Chen, H.X.; Chang, L.; Li, W.H. Different Proteins Regulated Apoptosis, Proliferation and Metastasis of Lung Adenocarcinoma After Radiotherapy at Different Time. Cancer Manag. Res. 2020, 12, 2437–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.V.; Daniel, P.M.; D’Abaco, G.M.; Gogos, A.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Coexpression analysis of CD133 and CD44 identifies proneural and mesenchymal subtypes of glioblastoma multiforme. Oncotarget 2015, 6, 6267–6280. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [Green Version]
- Blahovcova, E.; Richterova, R.; Kolarovszki, B.; Dobrota, D.; Racay, P.; Hatok, J. Apoptosis-related gene expression in tumor tissue samples obtained from patients diagnosed with glioblastoma multiforme. Int. J. Mol. Med. 2015, 36, 1677–1684. [Google Scholar] [CrossRef] [Green Version]
- McKelvey, K.J.; Hudson, A.L.; Donaghy, H.; Stoner, S.P.; Wheeler, H.R.; Diakos, C.I.; Howell, V.M. Differential effects of radiation fractionation regimens on glioblastoma. Radiat. Oncol. 2022, 17, 17. [Google Scholar] [CrossRef]
- Koessinger, A.L.; Cloix, C.; Koessinger, D.; Heiland, D.H.; Bock, F.J.; Strathdee, K.; Kinch, K.; Martínez-Escardó, L.; Paul, N.R.; Nixon, C.; et al. Increased apoptotic sensitivity of glioblastoma enables therapeutic targeting by BH3-mimetics. Cell Death Differ. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Liang, N.; He, Q.; Liu, X.; Sun, H. Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochem. Funct. 2019, 37, 177–184. [Google Scholar] [CrossRef]
- Stagni, V.; Ferri, A.; Cirotti, C.; Barilà, D. ATM Kinase-Dependent Regulation of Autophagy: A Key Player in Senescence? Front. Cell Dev. Biol. 2021, 8, 599048. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 2015, 4, e05289. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Chang, H.; Hu, T.; Wang, J.; Xie, Y.; Cheng, J. The Role and Mechanism of ATM-Mediated Autophagy in the Transition from Hyper-Radiosensitivity to Induced Radioresistance in Lung Cancer under Low-Dose Radiation. Front. Cell Dev. Biol. 2021, 9, 650819. [Google Scholar] [CrossRef]
- Liang, N.; Jia, L.; Liu, Y.; Liang, B.; Kong, D.; Yan, M.; Ma, S.; Liu, X. ATM pathway is essential for ionizing radiation-induced autophagy. Cell. Signal. 2013, 25, 2530–2539. [Google Scholar] [CrossRef]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The Autophagy-Senescence Connection in Chemotherapy: Must Tumor Cells (Self) Eat Before They Sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Tse, K.H.; Chow, H.M.; Gan, Y.; Song, X.; Ma, F.; Qian, Y.X.Y.; She, W.; Herrup, K. ATM loss disrupts the autophagy-lysosomal pathway. Autophagy 2021, 17, 1998–2010. [Google Scholar] [CrossRef]
- Sunderland, P.; Augustyniak, J.; Lenart, J.; Bużańska, L.; Carlessi, L.; Delia, D.; Sikora, E. ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 2020, 190, 111296. [Google Scholar] [CrossRef]
- Fragkos, M.; Beard, P. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS ONE 2011, 6, e22946. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Silva, M.M.B.; Thomé, M.P.; Lenz, G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 2015, 11, 1099–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, T.; As Sobeai, H.M.; Alhoshani, A.; Alhazzani, K.; Almutairi, M.M.; Alotaibi, M. Effect of Autophagy Inhibitors on Radiosensitivity in DNA Repair-Proficient and -Deficient Glioma Cells. Medicina 2022, 58, 889. https://doi.org/10.3390/medicina58070889
Saleh T, As Sobeai HM, Alhoshani A, Alhazzani K, Almutairi MM, Alotaibi M. Effect of Autophagy Inhibitors on Radiosensitivity in DNA Repair-Proficient and -Deficient Glioma Cells. Medicina. 2022; 58(7):889. https://doi.org/10.3390/medicina58070889
Chicago/Turabian StyleSaleh, Tareq, Homood M. As Sobeai, Ali Alhoshani, Khalid Alhazzani, Mashal M. Almutairi, and Moureq Alotaibi. 2022. "Effect of Autophagy Inhibitors on Radiosensitivity in DNA Repair-Proficient and -Deficient Glioma Cells" Medicina 58, no. 7: 889. https://doi.org/10.3390/medicina58070889