
Granulomatosis with Polyangiitis (GPA)—A Multidisciplinary Approach of a Case Report
 , and
, and 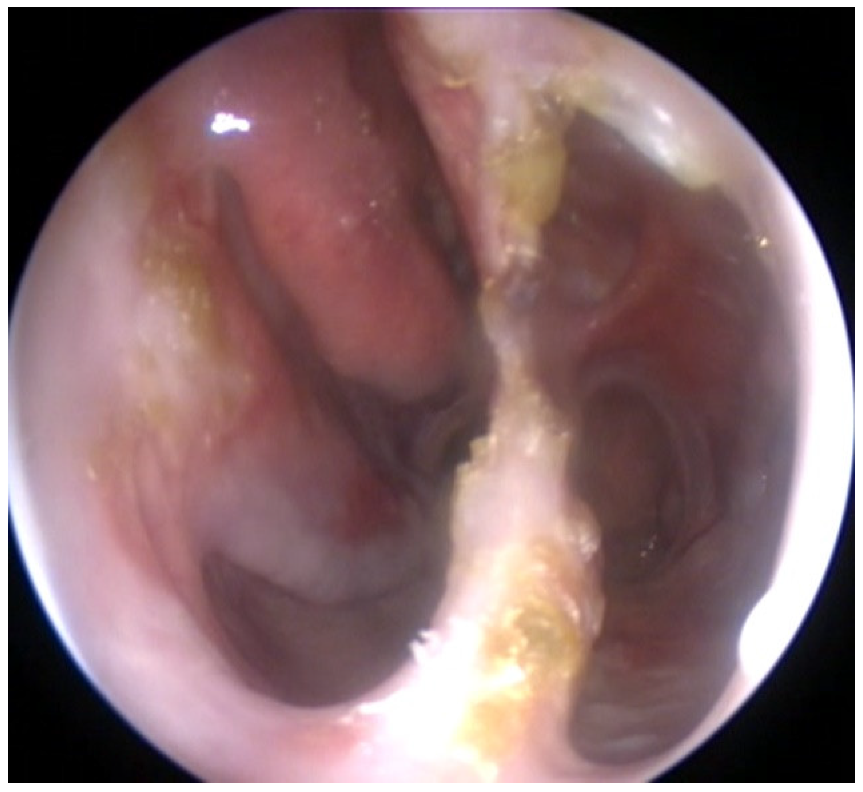{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
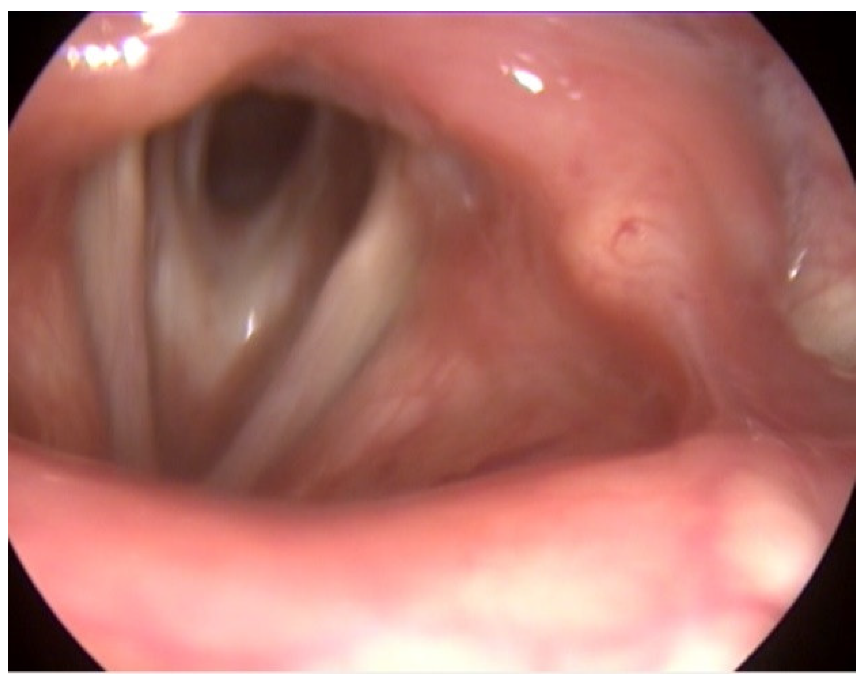
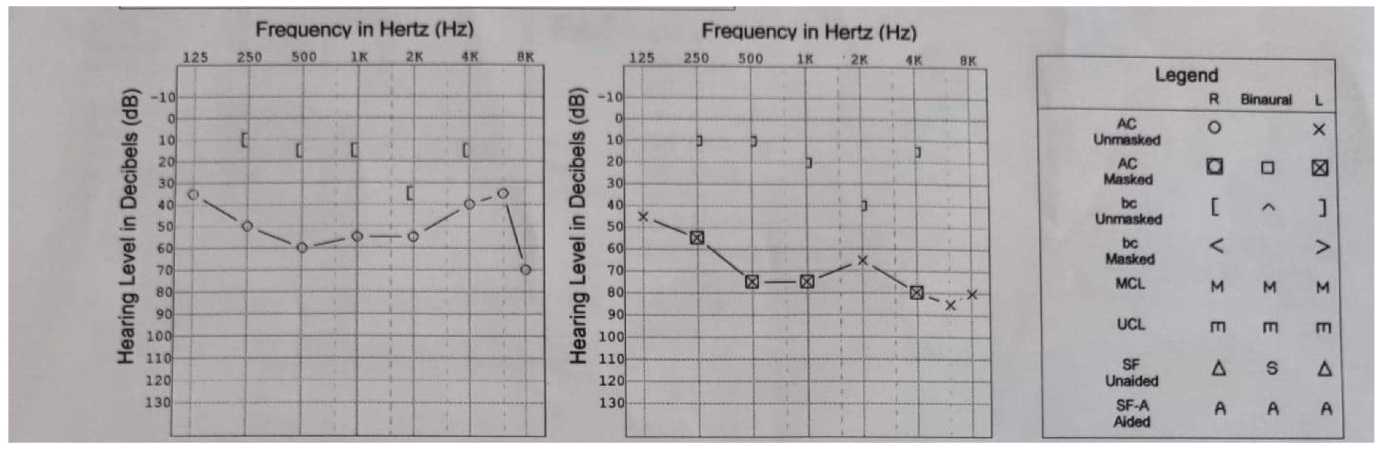
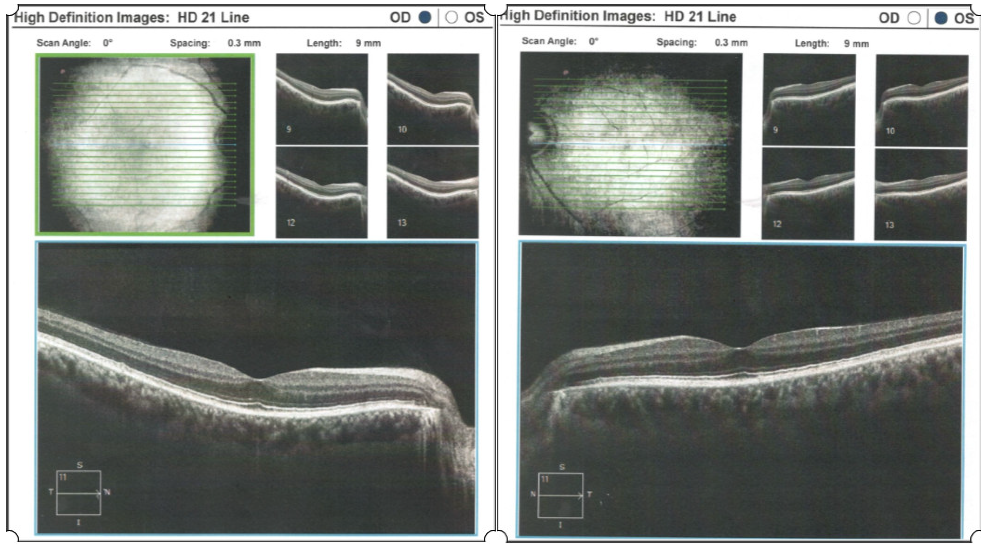
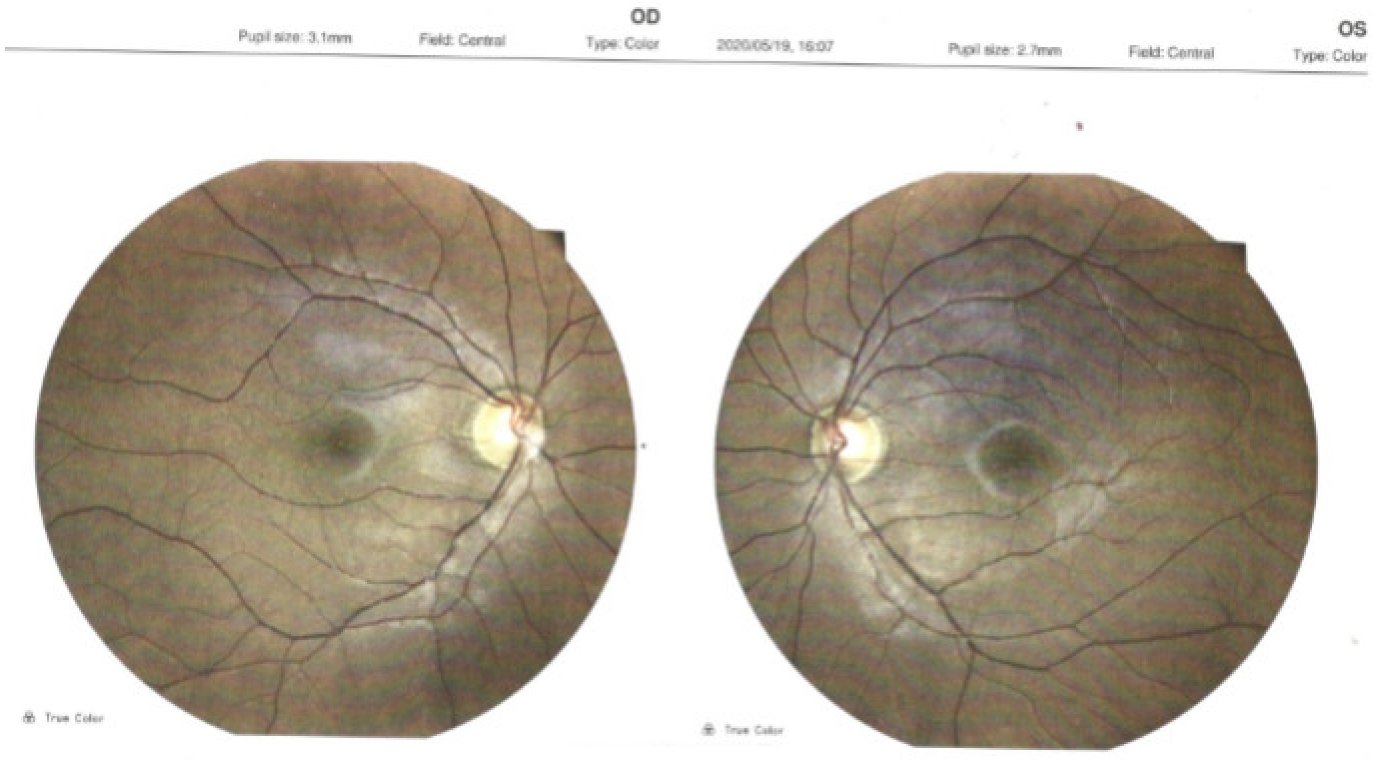
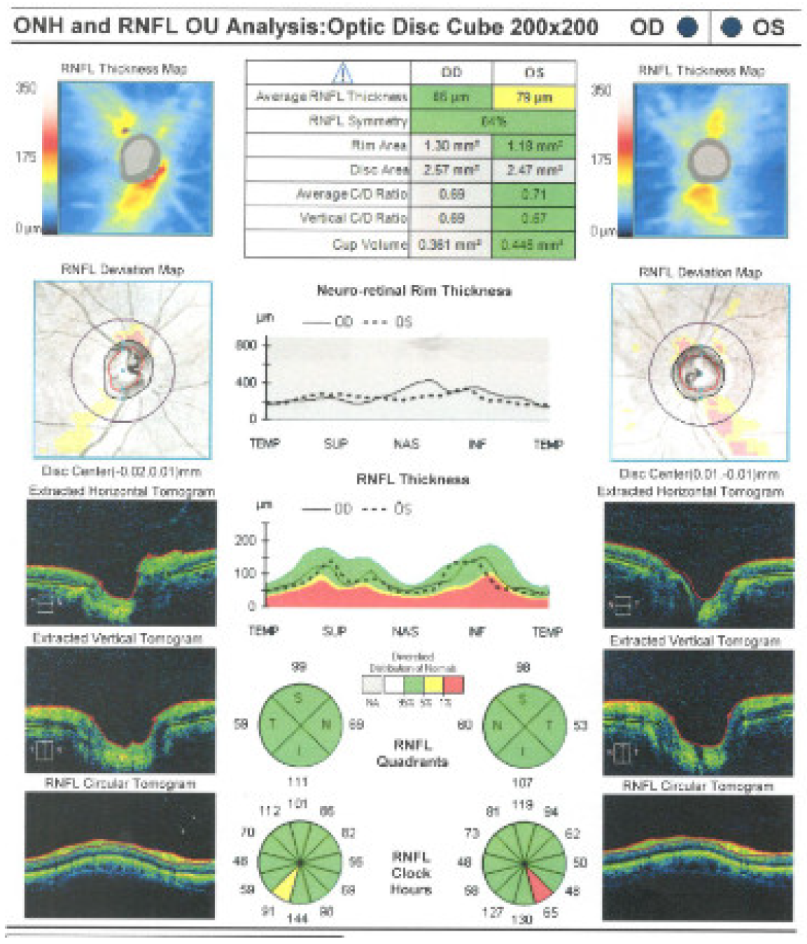
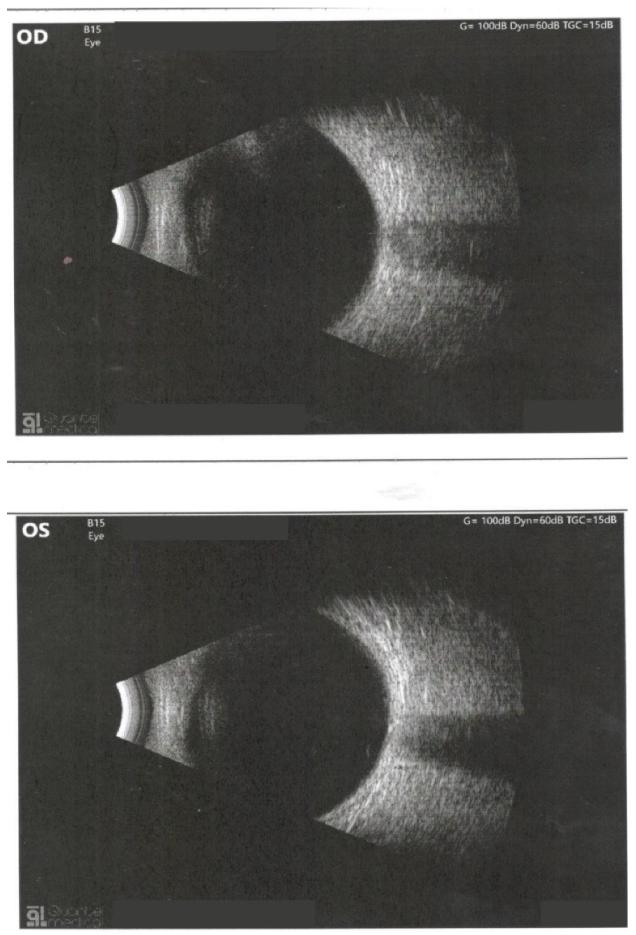
2. Case Report
- Ocular examination
- BCVA (best corrected visual acuity) RE (right eye) = 20/25 (+1.00 dsf × −0.50 dcyl 10°)
- BCVA LE (left eye) = 20/25 (+1.50 dsf × −0.50 dcyl 5°)
- Normal ocular adnexa, normal eye motility.
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Harman, L.E.; Margo, C.E. Wegener’s Granulomatosis. Surv. Ophthalmol. 1998, 42, 458–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.G.; Watts, R.A. Systemic vasculitis: Epidemiology, classification and environmental factors. Ann. Rheum. Dis. 2000, 59, 161–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miloslavsky, E.M.; Specks, U.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St. Clair, E.W.; Tchao, N.K.; et al. Clinical Outcomes of Remission Induction Therapy for Severe Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheum. 2013, 65, 2441–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagnoux, C.; Hogan, S.L.; Chin, H.; Jennette, J.C.; Falk, R.J.; Guillevin, L.; Nachman, P.H. Predictors of treatment resistance and relapse in anti-neutrophil cytoplasmic antibody-associated small-vessel vasculitis: Comparison of two independent cohorts. Arthritis Rheum. 2008, 58, 2908–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegeman, C.A.; Tervaert, J.W.C.; Sluiter, W.J.; Manson, W.L.; DeJong, P.E.; Kallenberg, C.G.M. Association of Chronic Nasal Carriage of Staphylococcus aureus and Higher Relapse Rates in Wegener Granulomatosis. Ann. Intern. Med. 1994, 120, 12–17. [Google Scholar] [CrossRef]
- Yazici, H.; Husby, G.; Watts, R.A. Vasculitis. Bailliere’s Clin. Rheumatol. 1997, 11, 191–450. [Google Scholar] [CrossRef]
- Godman, G.C.; Churg, J. Wegener’s granulomatosis: Pathology and review of the literature. Arch. Pathol. 1954, 6, 533–553. [Google Scholar]
- Lutalo, P.M.K.; D’Cruz, D.P. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J. Autoimmun. 2014, 48–49, 94–98. [Google Scholar] [CrossRef]
- Seo, P.; Stone, J.H. The antineutrophil cytoplasmic antibody-associated vasculitides. Am. J. Med. 2004, 117, 39–50. [Google Scholar] [CrossRef]
- Srouji, I.A.; Andrews, P.; Edwards, C.; Lund, V.J. Patterns of presentation and diagnosis of patients with Wegener’s granulo-matosis: ENT aspects. J. Laryngol. Otol. 2007, 121, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Iannella, G.; Greco, A.; Granata, G.; Manno, A.; Pasquariello, B.; Angeletti, D.; Didona, D.; Magliulo, G. Granulomatosis withpolyangiitis and facial palsy: Literature review and insight in the autoimmune pathogenesis. Autoimmun. Rev. 2016, 15, 621–631. [Google Scholar] [CrossRef]
- Cartin-Ceba, R.; Peikert, T.; Specks, U. Pathogenesis of ANCA-Associated Vasculitis. Curr. Rheumatol. Rep. 2012, 14, 481–493. [Google Scholar] [CrossRef]
- Langford, C.A.; Sneller, M.C.; Hallahan, C.W.; Hoffman, G.S.; Kammerer, W.A.; Talar-Williams, C.; Fauci, A.S.; Lebovics, R.S. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum. 1996, 39, 1754–1760. [Google Scholar] [CrossRef]
- Kornblut, A.D.; Wolff, S.M.; deFries, H.O. Wegener’s granulomatosis. Laryngoscope 1980, 90, 1453–1465. [Google Scholar] [CrossRef] [Green Version]
- Thickett, D.R.; Richter, A.G.; Nathani, N.; Perkins, G.; Harper, L. Pulmonary manifestations of anti-neutrophil cytoplasmic antibody (ANCA)-positive vasculitis. Rheumatology 2006, 45, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Pakrou, N.; Selva, D.; Leibovitch, I. Wegener’s granulomatosis: Ophthalmic manifestations and management. Semin. Arthritis Rheum. 2006, 35, 284–292. [Google Scholar] [CrossRef]
- Robinson, M.R.; Lee, S.S.; Sneller, M.C.; Lerner, R.; Langford, C.A.; Talar-Williams, C.; Cox, T.A.; Chan, C.C.; Smith, J.A. Tarsal-conjunctival disease associated with Wegener’s granulomatosis. Ophthalmology 2003, 110, 1770–1780. [Google Scholar] [CrossRef]
- Hogan, S.L.; Falk, R.J.; Chin, H.; Cai, J.; Jennette, C.E.; Jennette, J.C.; Nachman, P.H. Predictors of Relapse and Treatment Resistance in Antineutrophil Cytoplasmic Antibody-Associated Small-Vessel Vasculitis. Ann. Itern. Med. 2005, 143, 621–631. [Google Scholar] [CrossRef]
- Peng, Y.J.; Fang, P.C.; Huang, W.T. Central retinal artery occlusion in Wegener’s granulomatosis: A case report and review of the literature. Can. J. Ophthalmol. 2004, 39, 785–789. [Google Scholar] [CrossRef]
- Grygiel-Gorniak, B.; Limphaibool, N.; Perkowska, K.; Puszczewicz, M. Clinical manifestations of granulomatosis with polyangiitis: Key considerations and major features. Postgrad. Med. 2018, 130, 581–596. [Google Scholar] [CrossRef]
- Lee, P.Y.; Adil, E.A.; Irace, A.L.; Neff, L.; Son, M.B.F.; Lee, E.Y.; Perez-Atayde, A.; Rahbar, R. The presentation and management of granulomatosis with polyangitis (Wegener’s Granulomatosis) in the pediatric airway: GPA in the Pediatric Airway. Laryngoscope 2017, 127, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Tarabishy, A.B.; Schulte, M.; Papaliodis, G.N.; Hoffman, G.S. Wegener’s granulomatosis: Clinical manifestations, differential diagnosis, and management of ocular and systemic disease. Surv. Ophthamol. 2010, 55, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J. Small-Vessel Vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Straatsma, B.R. Ocular manifestations of Wegener’s granulomatosis. Am. J. Ophthalmol. 1957, 144, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Blaise, P.; Robe-Collignon, N.; Andris, C.; Rakic, J.-M. Wegener’s granulomatosis and posterior ischemic optic neuropathy: Atypical associated conditions. Eur. J. Intern. Med. 2007, 18, 326–327. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Kerr, G.S.; Leavitt, R.Y.; Hallahan, C.W.; Lebovics, R.S.; Travis, W.D.; Rottem, M.; Fauci, A.S. Wegener granulomatosis: Ananalysis of 158 patients. Ann. Intern. Med. 1992, 116, 488–498. [Google Scholar] [CrossRef]
- Loke, Y.K.; Tan, M.H. An unusual case of Wegener’s granulomatosis. Med. J. Malays. 1998, 53, 107–109. [Google Scholar]
- DeSilva, D.J.; Cole, C.; Luthert, P.; Olver, J.M. Masked orbital abscess in Wegener’s granulomatosis. Eye 2007, 21, 246–248. [Google Scholar] [CrossRef] [Green Version]
- Woo, T.L.; Francis, I.C.; Wilcsek, G.A.; Coroneo, M.T.; McNab, A.; Sullivan, T. Australasian orbital and adnexal Wegener’s granulomatosis. Ophthalmology 2001, 108, 1535–1543. [Google Scholar] [CrossRef]
- Ghanem, R.C.; Chang, N.; Aoki, L.; Santo, R.; Matayoshi, S. Vasculitis of the Lacrimal Sac Wallin Wegener Granulomatosis. Ophthalmic Plast. Reconstr. Surg. 2004, 20, 254–257. [Google Scholar] [CrossRef]
- Howe, L.; D’Cruz, D.; Chopdar, A.; Hughes, G. Anterior ischaemic optic neuropathy in Wegener’s granulomatosis. Eur. J. Ophthalmol. 1995, 5, 277–279. [Google Scholar] [CrossRef]
- Boukes, R.J.; deVries-Knoppert, W.A. Lacrimal gland enlargement as one of the ocular manifestations of Wegener’s granulomatosis. Doc. Ophthalmol. 1985, 59, 21–26. [Google Scholar] [CrossRef]
- Kiseleva, T.N.; Grusha, I.O.; Polunina, A.A.; Semenkova, E.N.; Abdurakhmanov, G.A.; Nikol’skaia, G.M. Involvement of lacrimal organs in Wegener’s granulomatosis. Vestn. Oftalmol. 2009, 125, 33–36. [Google Scholar]
- Hibino, M.; Kondo, T. Dacryoadenitis with Ptosis and diplopia as the Initial Presentation of Granulomatosis with Polyangiitis. Intern. Med. 2017, 56, 2649–2653. [Google Scholar] [CrossRef] [Green Version]
- Ismailova, D.S.; Abramova, J.V.; Novikov, P.I.; Grusha, Y.O. Clinical features of different orbital manifestations of granulomatosis with polyangiitis. Graefe’s Archives. Clin. Exp. Ophthalmol. 2018, 256, 1751–1756. [Google Scholar] [CrossRef]
- Fortney, A.C.; Chodosh, J. Conjunctival Ulceration in Recurrent Wegener Granulomatosis. Cornea 2002, 21, 623–624. [Google Scholar] [CrossRef]
- Meier, F.M.; Messmer, E.P.; Bernauer, W. Wegener’s granulomatosis as a cause of cicatrizing conjunctivitis. Br. J. Ophthalmol. 2001, 85, 628. [Google Scholar] [CrossRef] [Green Version]
- Jordan, D.R.; Zafar, A.; Brownstein, S.; Faraji, H. Cicatricial Conjunctival Inflammation with Trichiasis as the Presenting Feature of Wegener Granulomatosis. Ophthalmic Plast. Reconstr. Surg. 2006, 22, 69–71. [Google Scholar] [CrossRef]
- Taylor, S.R.J.; Salama, A.D.; Pusey, C.D.; Lightman, S. Ocular manifestations of Wegener’s granulomatosis. Expert Rev. Ophthalmol. 2007, 2, 91–103. [Google Scholar] [CrossRef]
- Jabs, D.A.; Mudun, A.; Dunn, J.; Marsh, M.J. Episcleritis and scleritis: Clinical features and treatment results. Am. J. Ophthalmol. 2000, 130, 469–476. [Google Scholar] [CrossRef]
- Lamprecht, P.; Lerin-Lozano, C.; Reinhold-Keller, E.; Nölle, B.; Gross, W.L. Retinal artery occlusion in Wegener’s granulomatosis. Rheumatology 2000, 39, 928–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.C.; Boyce, R.L.; Dowd, T.C.; Fordham, J.N. Bilateral central retinal artery occlusion in Wegener’s granulomatosis and α1 antitrypsin deficiency. Br. J. Ophthalmol. 2002, 86, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Khurana, R.N.; Sadda, S.R. Central retinal vein occlusion in Wegener’s granulomatosis without retinal vasculitis. Br. J. Ophthalmol. 2006, 90, 1435–1436. [Google Scholar] [CrossRef] [PubMed]
- Matlach, J.; Freiberg, F.J.; Gadeholt, O.; Göbel, W. Vasculitis like hemorrhagic retinal angiopathy in Wegener’s granulomatosis. BMC Res. Notes 2013, 6, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, A.S.; Kempen, J.H.; Choi, D.; Liesegang, T.L.; Pujari, S.S.; Newcomb, C.; Nussenblatt, R.B.; Rosenbaum, J.T.; Thorne, J.E.; Foster, C.S.; et al. Ocular disease in patients with ANCA-positive vasculitis. J. Ocul. Biol. Dis. Inform. 2009, 3, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.L.; Chavala, S.H.; Ahmed, M.; Chu, D.; Zafirakis, P.; Baltatzis, S.; Ocampo, V.; Foster, C. Ocular manifestations and concepts of systemic vasculitides. Surv. Ophthalmol. 2004, 49, 399–418. [Google Scholar] [CrossRef]
- Samuelson, T.W.; Margo, C.E. Protracted uveitis as the initial manifestation of Wegener’s granulomatosis. Arch. Ophthalmol. 1990, 108, 478–479. [Google Scholar] [CrossRef]
- DuHuong, L.T.; Tran, T.H.; Piette, J.C. Granulomatous uveitis revealing Wegener’s granulomatosis. J. Rheumatol. 2006, 33, 1209–1210. [Google Scholar]
- Sfiniadaki, E.; Tsiara, I.; Theodossiadis, P.; Chatziralli, I. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol. Ther. 2019, 8, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Reinhold-Keller, E.; Fink, C.O.E.; Herlyn, K.; Gross, W.L.; DeGroot, K. High rate of renal relapse in 71 patients with Wegener’s granulomatosis under maintenance of remission with low-dose methotrexate. Arthritis Rheum. 2002, 47, 326–332. [Google Scholar] [CrossRef]
- Comarmond, C.; Cacoub, P. What is the best treatment option for granulomatosis with polyangiitis? Int. J. Clin. Rheumatol. 2015, 10, 227–235. [Google Scholar] [CrossRef]
- Langford, C.A.; Talar-Williams, C.; Barron, K.S.; Sneller, M.C. Use of acyclophosphamide-induction methotrexate-maintenance regimen for the treatment of Wegener’s granulomatosis: Extended follow-up and rate of relapse. Am. J. Med. 2003, 114, 463–469. [Google Scholar] [CrossRef]
- Karia, V.R.; Espinoza, L.R. Risk factors for treatment failures in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Curr. Rheumatol. Rep. 2009, 11, 416–421. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trandafir, C.M.; Balica, N.C.; Horhat, D.I.; Mot, I.C.; Sarau, C.A.; Poenaru, M. Granulomatosis with Polyangiitis (GPA)—A Multidisciplinary Approach of a Case Report. Medicina 2022, 58, 1837. https://doi.org/10.3390/medicina58121837
Trandafir CM, Balica NC, Horhat DI, Mot IC, Sarau CA, Poenaru M. Granulomatosis with Polyangiitis (GPA)—A Multidisciplinary Approach of a Case Report. Medicina. 2022; 58(12):1837. https://doi.org/10.3390/medicina58121837
Chicago/Turabian StyleTrandafir, Cornelia M., Nicolae Constantin Balica, Delia I. Horhat, Ion C. Mot, Cristian A. Sarau, and Marioara Poenaru. 2022. "Granulomatosis with Polyangiitis (GPA)—A Multidisciplinary Approach of a Case Report" Medicina 58, no. 12: 1837. https://doi.org/10.3390/medicina58121837