1. Introduction
Cancer continues to pose a substantial global health threat despite advancements in diagnosis and treatment [
1]. In a recent update in 2020, there were an estimated 19.3 million new cases and 10.0 million cancer-related deaths [
2], an increase from 18.1 million cases and 9.6 million deaths in 2018 [
3]. This rise can be attributed to factors such as population growth, increased exposure to risk factors like smoking and obesity, and changing reproductive patterns due to economic development and urbanization. Lung cancer is the most frequently diagnosed and deadliest, followed closely by breast cancer. Breast cancer is the most common cancer in women worldwide, with over 2.3 million new cases in 2020, significantly contributing to cancer-related mortality [
4]. Breast cancer is now recognized as a diverse group of diseases with distinct clinical behaviors, molecular components, risk factors, prognostic markers, and responses to treatment. Molecular classification relies on markers like estrogen receptors (ER), progesterone receptors (PR) and HER2 and Ki67 proliferation rate [
5]. As a result, breast cancer has been categorized into five subtypes: luminal A, luminal B, triple-negative, and two HER2-positive types. Breast cancers with HER2 overexpression constitute 15–25% of cases, being aggressive and challenging to treat. Trastuzumab was approved by the FDA in 1998 and demonstrated a 37% relative improvement in overall survival with an increase of about 9% in the probability of 10-year OS when combined with chemotherapy [
6]. Mutations in PIK3R1, activating PI3K/Akt/mTOR, drive trastuzumab resistance, especially in HER2-overexpressing breast cancer [
7]. Approximately 70% of HER2-positive breast cancer patients develop resistance to trastuzumab within a year of treatment initiation, despite initial responsiveness [
8].
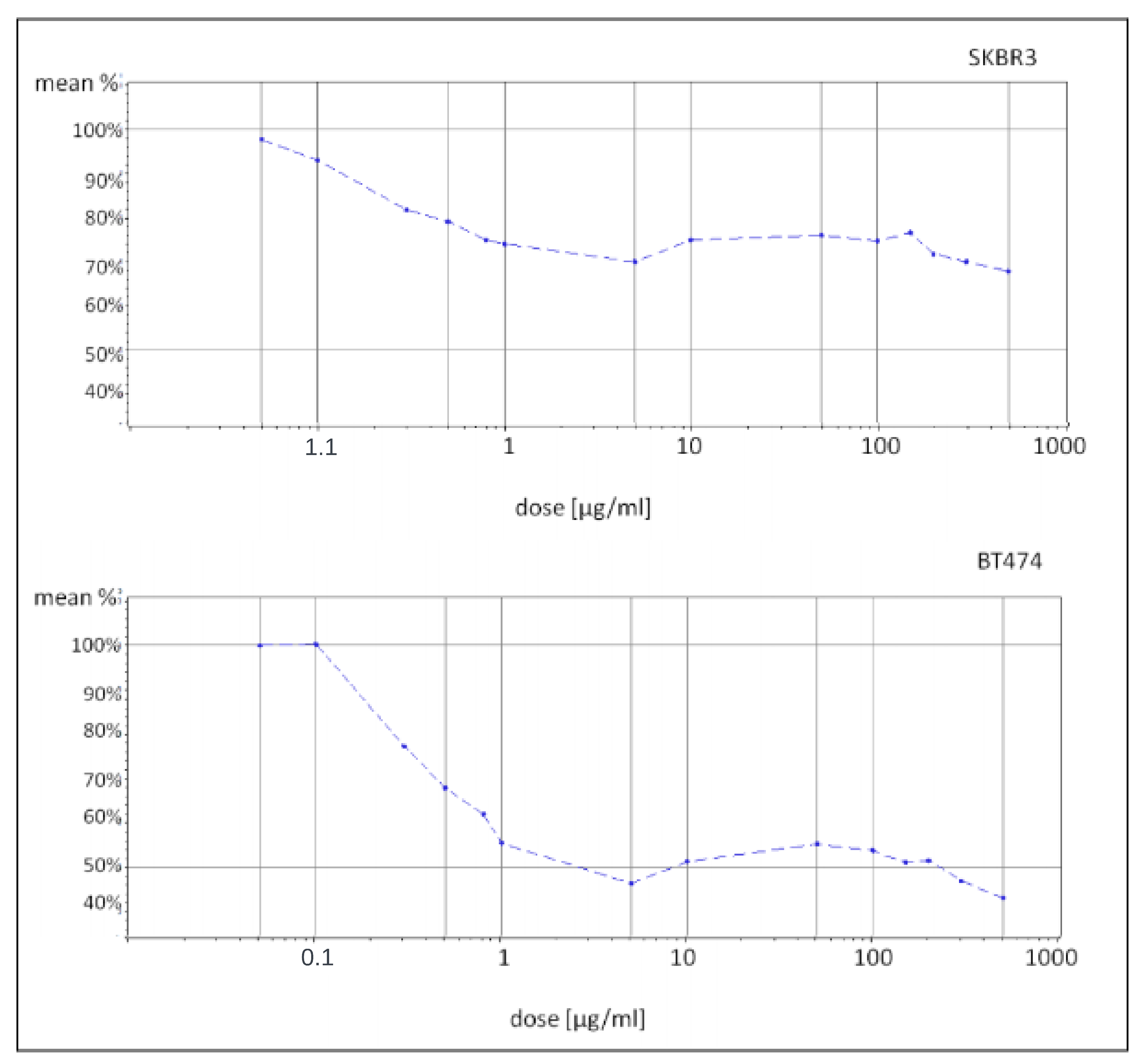
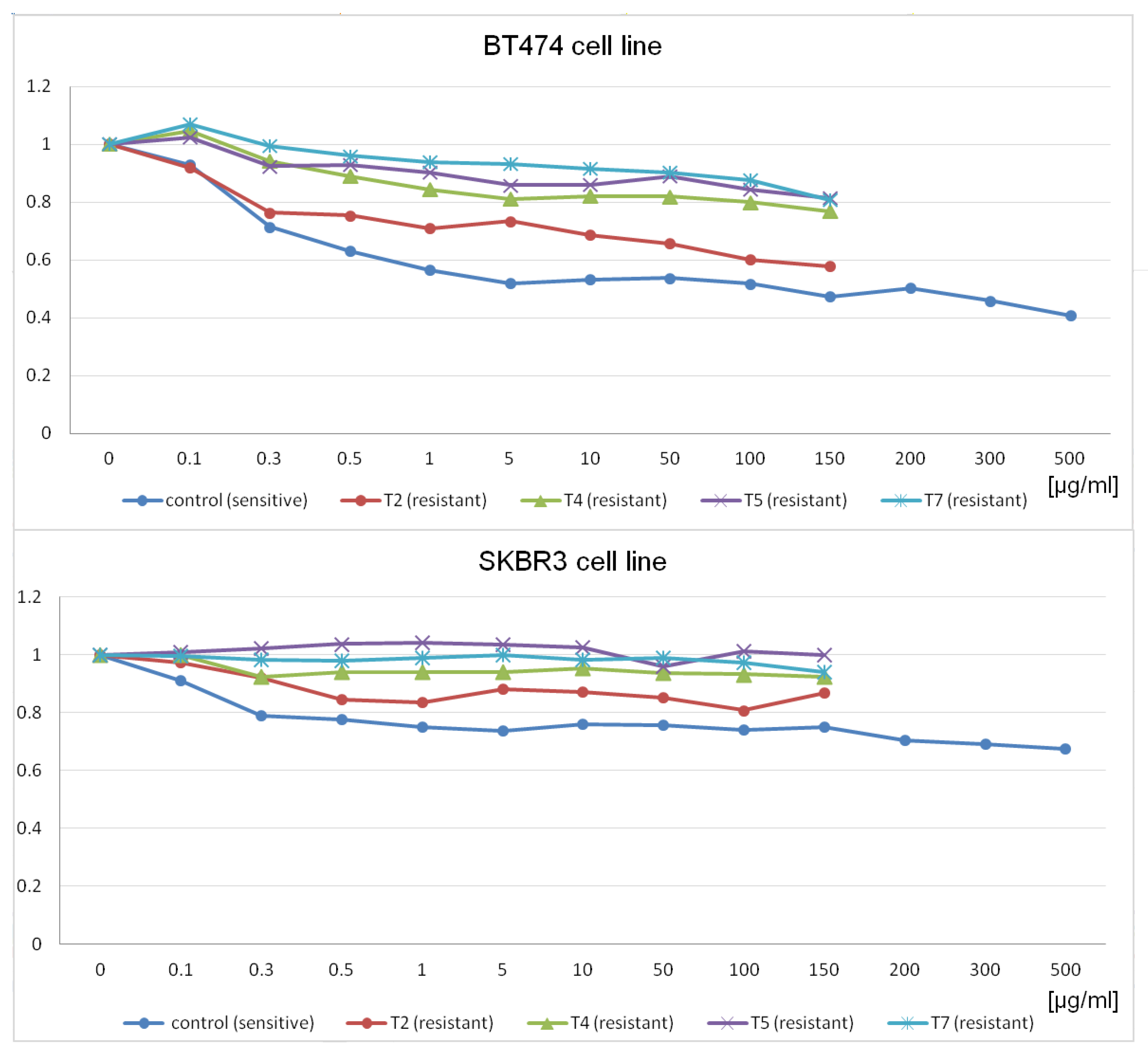
In this context, the present study aimed to comprehensively investigate the molecular mechanisms underlying trastuzumab resistance using a two-fold approach. Initially, well-established breast cancer cell lines, namely SKBR3 and BT474 [
9], were used to mimic the in vitro conditions representative of HER2-positive breast cancer. Optimal trastuzumab treatment doses were determined for each cell line based on their respective proliferation rates in response to a wide range of medication dosages. Subsequently, both cell lines were continuously cultivated with a steady dosage of trastuzumab over several months, leading to the development of trastuzumab-resistant cell lines. To capture the dynamic changes occurring during the acquisition of resistance, six distinct time points were selected for further analysis, encompassing the progression from the untreated cell line at the outset to the fully resistant cell line at the termination of the experiment. Concurrently, a second stage of the study involved the extraction of nucleic acids from the aforementioned cell lines for subsequent high-throughput microarray experiments targeting gene and microRNA expressions. The resulting expression data were subjected to comprehensive bioinformatics analyses to unravel the intricate molecular mechanisms underpinning the development of trastuzumab resistance. Through this approach, we focused on the 25 genes and microRNAs with the most statistically significant changes in expression levels across time, implicated in the development of trastuzumab resistance, and demonstrated crucial roles in protein–protein interactions. Notably, among the identified genes and microRNAs,
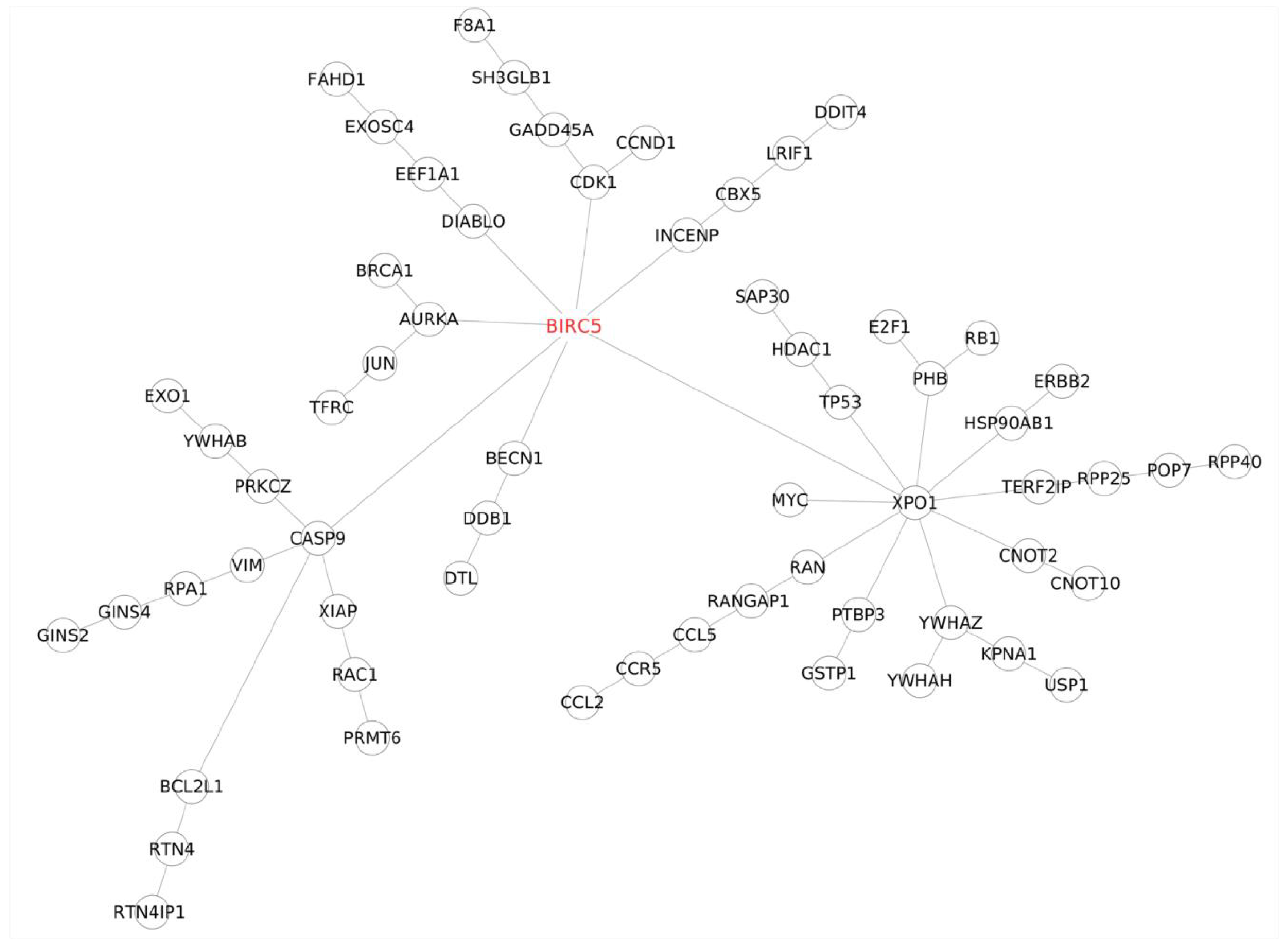
BIRC5,
E2F1,
TFRC, and
USP1 emerged as the top candidates influencing trastuzumab resistance, while miR-574-3p, miR-4530, miR-8485, and miR-197-3p were identified as the key microRNAs regulating this process. In-depth investigations using the prediction by the miRDB database [
10] highlighted the miR-4701-5p targeting
TFRC and the targeting of
E2F1 and
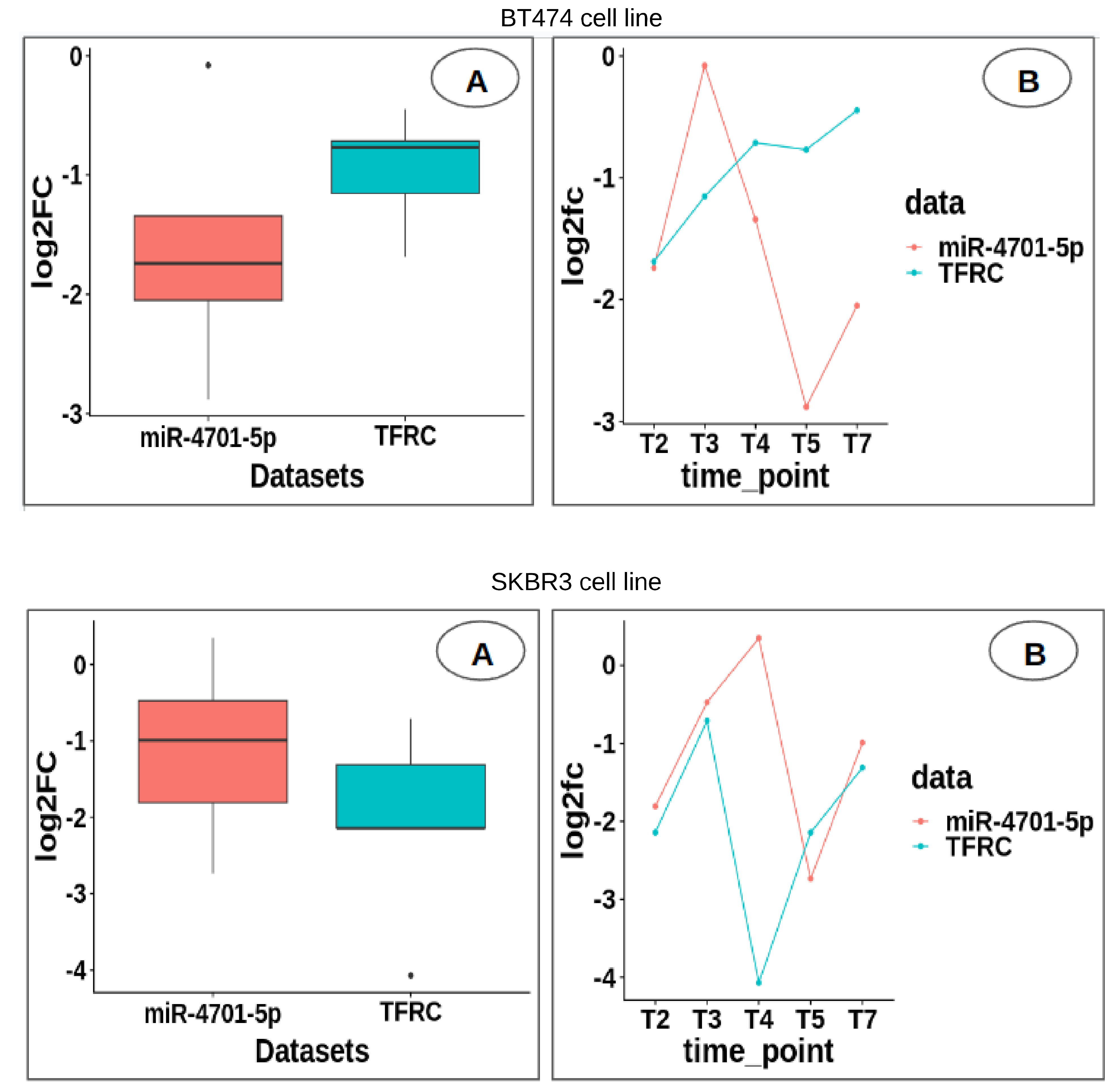
USP1 by hsa-miR-8485. Moreover, the expression revealed substantial upregulation of miR-4701-5p compared to
TFRC in the SKBR3 cell line, providing crucial insights into the intricate regulatory mechanisms governing trastuzumab resistance. Overall, the findings of this study shed light on the critical molecular players and pathways driving trastuzumab resistance in HER2-positive breast cancer, offering valuable insights into potential therapeutic targets and strategies for overcoming treatment challenges associated with this aggressive subtype of breast cancer.
4. Discussion
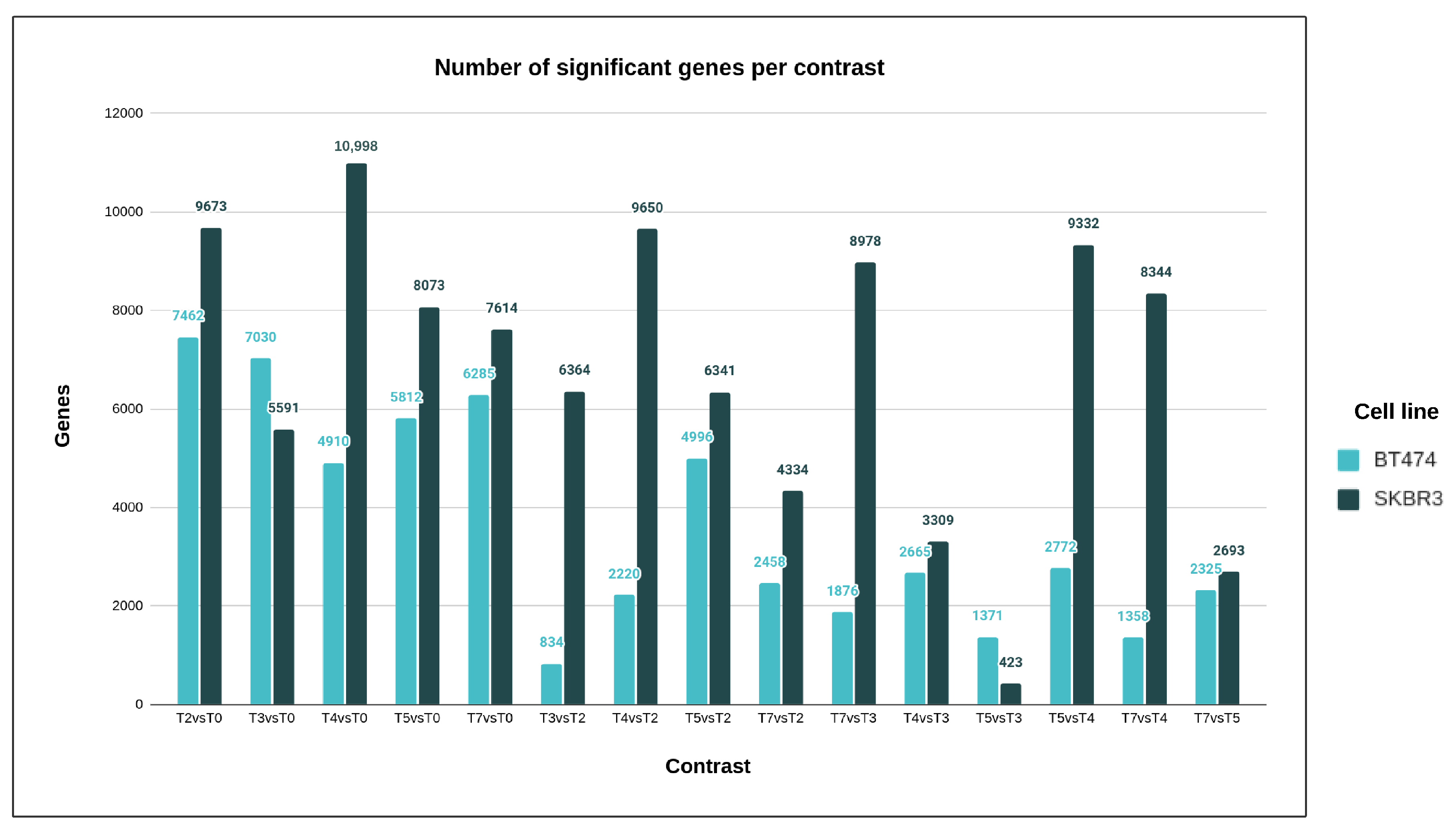
The primary aim of this research was to delve into the molecular mechanisms responsible for developing resistance to trastuzumab, a drug used in breast cancer treatment. Traditional studies on Herceptin resistance mainly focused on identifying differences between resistant and sensitive cells [
26,
27,
28]. However, this approach has limitations because cells can actively adapt to changing environments and treatment conditions. To address this, we proposed a novel approach combining a longitudinal investigation of in vitro resistance development with high-throughput microarray technology. Our comprehensive analysis of gene expression data encompassed over 34,000 genes and identified 8873 and 13,891 genes significantly contributing to trastuzumab resistance in two breast cancer cell lines, BT474 and SKBR3, with 5675 genes common to both cell lines. Further analysis grouped these genes into various molecular functions and biological processes using Gene Ontology terms. Additionally, we identified significant signaling pathways through KEGG Pathway analysis, with eight pathways commonly affected in both cell lines. Our study highlights the complexity of trastuzumab resistance development, emphasizing the need for a multi-dimensional understanding of the process. This research contributes valuable insights into potential targets for more effective breast cancer treatments [
29,
30]. In both cell lines, four genes—
BIRC5,
E2F1,
USP1, and
TFRC—emerged as highly significant transcripts within the top 25.
BIRC5 and
E2F1 exhibited a distinct expression pattern throughout the study. They initially decreased upon drug exposure and then gradually increased, mirroring the gradual development of drug resistance. This pattern aligns with existing research on
BIRC5’s role in trastuzumab resistance, where the overactive PI3K/Akt [
31] pathway leads to survivin overexpression, contributing to resistance [
32]. In patients with HER2-overexpressing breast cancer, higher pretreatment survivin RNA levels correlated with poorer responses to trastuzumab, indicating
BIRC5’s involvement in primary Herceptin resistance [
32].
E2F1, a member of the E2F transcription factor family, shares similarities with survivin (BIRC5) in contributing to trastuzumab resistance.
E2F1 regulates cell growth and apoptosis, often activated in response to DNA damage [
33]. Fanconi anemia DNA repair pathway emerged as crucial in trastuzumab resistance through KEGG pathway analysis.
E2F1 can also activate
BIRC5, a known resistance factor.
E2F1 is linked to HER2 signaling and trastuzumab actions, with research showing its involvement in proliferative breast tumors. Interestingly,
E2F1 expression levels rise during trastuzumab exposure, indicating its potential role in drug resistance through independent HER2 pathway activation. While the roles of
BIRC5 and
E2F1 in drug resistance in HER2 breast cancer are understood, the involvement of
USP1 and
TRFC remains unclear.
USP1 has been linked to cancer development and metastasis but not yet to trastuzumab resistance. Interestingly,
USP1 inhibitors have shown promise in leukemia treatment. Suppressing
USP1 leads to the degradation of the ID1 transcription factor, crucial for cancer progression. The high representation of
USP1 in molecular function and biological process gene ontology terms suggests its significant role in trastuzumab resistance mechanisms. Unlike
USP1, current scientific literature does not mention
TFRC’s role in Herceptin resistance development. Recent research has revealed
TFRC’s involvement in various cancer-related signaling pathways, notably the endocytosis pathway, which is significant in trastuzumab resistance. While
TFRC’s contribution to Gene Ontology terms is less pronounced than
USP1, it plays a role in the “response to drug” biological process. This study emphasizes the importance of further investigating both genes in trastuzumab resistance development, as their correlation with drug resistance is not yet clear, and their molecular functions in the context of cancer progression and trastuzumab resistance require deeper exploration.
Understanding the molecular mechanisms behind resistance to Herceptin is critical. This study identified several genes, such as BIRC5, BRCA1, RB1, ERBB2, and others, as significant players in trastuzumab resistance, supporting previous research. New candidate genes like E2F1, USP1, and TFRC were also highlighted. Some genes, like IGF2BP1 and GSTP1, showed potential involvement but require further confirmation. Surprisingly, this study did not confirm the involvement of certain previously reported genes in resistance. The findings suggest that primary Herceptin resistance may be linked to alterations in downstream components of HER2 signaling pathways or antiapoptotic proteins, rather than HER2 receptor activity itself. In contrast, acquired resistance may involve changes at the receptor level, such as epitope masking or upregulation of receptor components. Some intrinsic resistance mechanisms may overlap with acquired resistance, aligning with earlier research. Overall, this study underscores the complexity of trastuzumab resistance involving both HER2-dependent and independent pathways. Our study significantly advances our understanding of trastuzumab resistance by identifying new molecular contributors and genetic pathways. This knowledge informs future cancer genetics and molecular medicine research, potentially leading to effective adjuvant therapies. However, clinical validation is crucial, as preclinical and in vitro findings may not always translate. Additionally, we uncovered unexplored long non-coding RNAs and proteins, offering opportunities for further basic research.
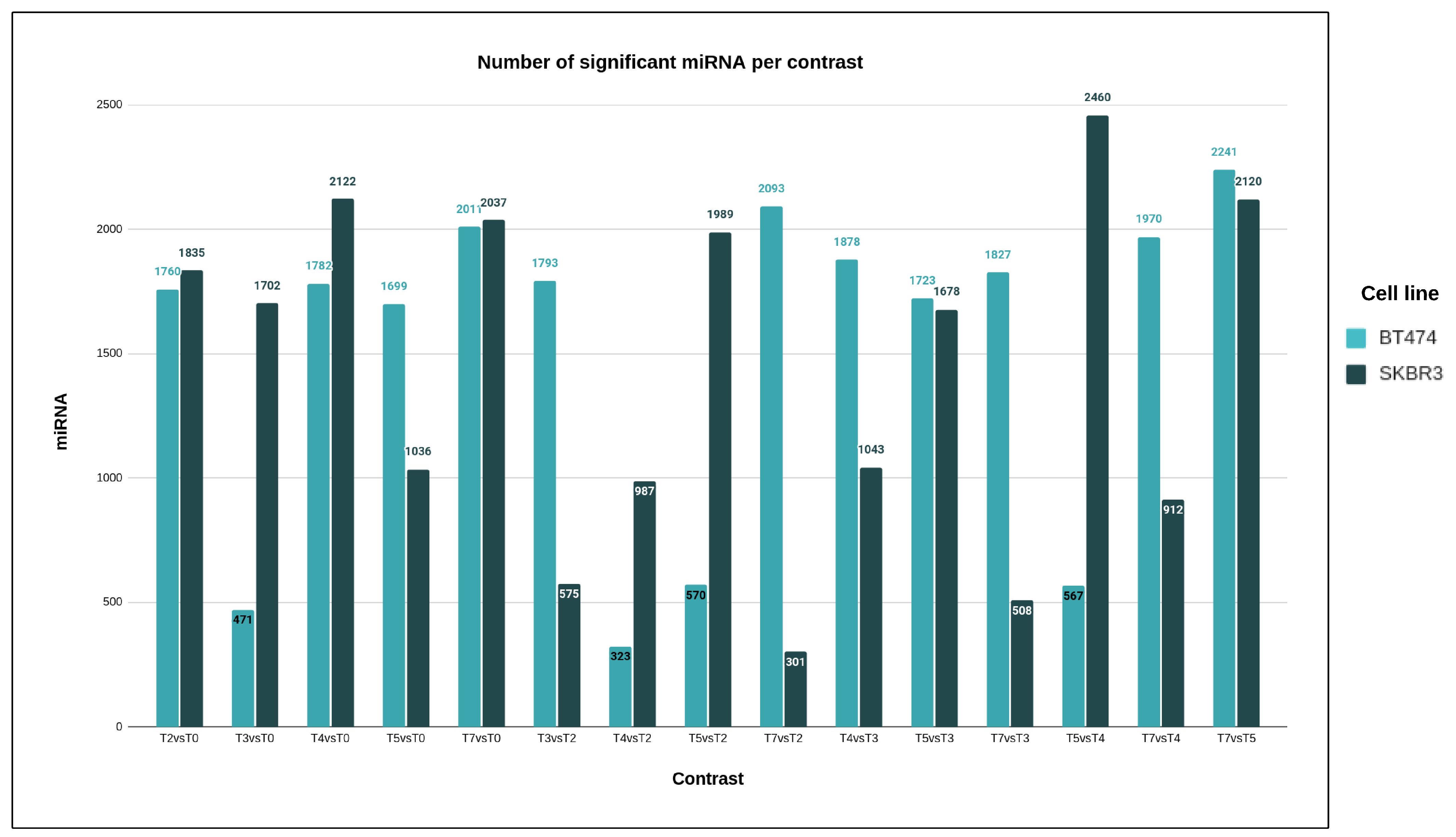
This study unequivocally demonstrates the substantial role of microRNA molecules in developing resistance to trastuzumab. In both cell lines, more than 99.6% of the tested human microRNAs exhibited statistically significant expression changes during the development of drug resistance, even when applying stringent statistical criteria. Notably, using a more rigorous threshold for medical purposes revealed a high percentage of significant microRNAs, emphasizing the specificity of microRNA action. MicroRNA molecules play crucial roles in cellular processes such as proliferation, development, metabolism, differentiation, and apoptosis [
34,
35]. Their ability to target multiple mRNAs due to imperfect matching and their widespread regulatory influence on protein-coding genes further highlight their complexity [
34]. Despite constituting only a small portion of the human genome, microRNAs are predicted to regulate a substantial portion of protein-coding genes. Four microRNAs, specifically hsa-miR-574-3p, hsa-miR-4530, hsa-miR-8485, and hsa-miR-197-3p, ranked among the top 25 most significant microRNAs in both cell lines, underscoring their significant involvement in the development of trastuzumab resistance. In silico analysis was conducted to identify potential targets of specific microRNAs (miRNAs) associated with Herceptin resistance in breast cancer cell lines. For hsa-miR-574-3p, TargetScan predicted potential targets such as
RAC1,
BIRC5,
E2F1,
PMP22, and
EGFR [
36]. Luciferase reporter assays confirmed direct regulation of
RAC1 and
EGFR by miR-574-3p, both implicated in Herceptin resistance [
37]. Similarly, miR-4530 showed potential targets, including FOXO1, MAPK4, and AKT predicted by miRDB [
10] whereas Target Scan predicted
BIRC5,
TFRC,
HER2,
ESR2, and AURKA [
36], some of which are involved in HER2 signaling and were important in Herceptin resistance. Another miRNA, hsa-miR-8485, was found to potentially target genes like
BIRC5,
E2F1,
USP1,
RAC1,
EPHA2,
PTEN, and
CCND1 [
10,
36], where,
E2F1 and
USP1, being from the top four, were highly significant in both cell lines. Additionally, miR-4701-5p was identified as targeting TFRC [
10], which was highly significant in both cell lines. These findings provide insights into the molecular mechanisms underlying Herceptin resistance in breast cancer. The Hsa-miR-197-3p, a microRNA, plays a crucial role in cancer progression, particularly in breast, bladder, and thyroid cancers. Multiple studies emphasize the influence of long non-coding RNAs (ncRNAs) on regulating miR-197-3p expression. LIFR-AS1 inhibits cell proliferation, migration, and invasion in breast cancer by repressing miR-197 [
38]. Similar results were observed in bladder [
39] and thyroid cancers [
40], where miR-197-3p downregulation led to decreased cell proliferation, migration, and invasion due to the actions of specific ncRNAs. Notably, miR-197-3p is linked to the PTEN/PI3K-Akt pathway [
40], which plays a key role in HER2 signaling. This suggests that miR-197-3p might be involved in an alternative pathway compensating for trastuzumab’s therapeutic effects, a drug targeting HER2. Additionally, miR-197 targets
MAPK1, a gene associated with trastuzumab resistance. Overexpressing miR-197 can reverse drug resistance by inhibiting
MAPK1, as seen in gastric cancer cells [
41]. Overall, miR-197-3p has a significant role in trastuzumab resistance development, potentially through its regulation of
MAPK1 and involvement in alternative signaling pathways [
41]. Furthermore, miR-197-3p directly regulates other genes implicated in trastuzumab resistance, including
FOXJ2 [
42],
MTHFD1 [
43],
RAN [
44],
TUSC2 [
45], and
FUS1 [
46], though their specific roles in drug resistance and cancer progression require further investigation. These findings support the hypothesis that miR-197-3p is a key player in developing resistance to trastuzumab, a critical drug in breast cancer treatment.
In summary, our study suggests that nearly all known human microRNAs may play a role in developing resistance to the drug trastuzumab. We identified four microRNAs that are particularly important in both biological cell lines studied. Two of these microRNAs are confirmed to be involved in either HER2 signaling or drug resistance, supporting our findings’ reliability. The other two highly significant microRNAs, which have limited existing information, were identified through computational analysis as potential regulators of genes associated with trastuzumab resistance. However, further research is needed to validate these hypotheses and understand the underlying mechanisms.
5. Conclusions
The current study conducted high-throughput microarray experiments to investigate the intricate dynamics of gene and microRNA expression changes during the development of trastuzumab resistance in two prominent breast cancer cell lines, BT474 and SKBR3. The analysis of a pool of 34,000 genes revealed distinct patterns of differential expression, with 8874 and 13,892 genes implicated in resistance development in BT474 and SKBR3, respectively. Remarkably, our findings highlighted the significant involvement of key genes, including BIRC5, E2F1, USP1, and TFRC, which are crucial players in both cell lines, particularly within the context of HER2 signaling. Moreover, the identification of novel contributors to Herceptin resistance, such as IGF2BP1, GSTM3, RASD1, KLK11, GSTP1, YWHAH, DTL, DOLK, NACC2, DDIT, and DNAJA3, among the top 25 significant genes, suggests the existence of previously unrecognized mechanisms in drug resistance. Importantly, our research also underscored the role of established genes, including BIRC5, E2F1, BRCA1, RB1, ERBB2, EPHA2, IGFBP3, ADAM10, FOXM1, RAC1, MYC, CCND1, PTEN, TP53, MAP2K4, and PI3KCA in contributing to resistance. Notably, protein–protein interaction analysis illuminated the pivotal roles of BIRC5, E2F1, and RB1 as central hubs within networks linked to Herceptin resistance. Furthermore, the significant impact of long non-coding RNAs and microRNAs in this resistance context was evident, indicating their potential as vital regulators in the process. Gene Ontology analysis highlighted enriched molecular functions such as receptor binding, protein kinase activities, and DNA replication, while biological processes encompassed crucial aspects like cell cycle regulation, apoptosis, and DNA damage repair. Pathway analysis brought to light 9 and 75 affected networks in BT474 and SKBR3, respectively, with the convergence of eight common pathways, notably including cell cycle and p53 signaling. Notably, our investigation revealed HER2-dependent and independent resistance mechanisms, thereby ruling out the involvement of epitope masking and other ERBB receptors. Noteworthy complexities observed in SKBR3 possibly arose from disparities in the cancer stage, considering the primary vs. metastatic distinction. Intriguingly, our study highlighted the significant role of microRNAs in Herceptin resistance, with hsa-miR-574-3p, hsa-miR-4530, hsa-miR-8485, and hsa-miR-197-3p emerging as critical contributors, including some previously unreported microRNAs specific to each cell line. These comprehensive findings shed light on the multifaceted landscape of trastuzumab resistance in breast cancer, providing valuable insights for the development of more effective therapeutic strategies and personalized treatment approaches. Admittedly, the findings have been obtained based on an analysis of a limited amount of material (that included, due to resource constraints, only two independent cell-line replicates at each time point). Evaluation of their credibility should take this aspect of the study into account. In this respect, further validation of the findings would be important. For instance, an in vivo validation and analysis of gene expression at the protein level could be used to ensure the accuracy and comprehensiveness of the microarray-based findings. An investigation of the expression of identified DEGs in samples of patients included in clinical databases and of the association with patients’ outcomes could shed light on the relevance of the study’s findings to clinical practice. These extensions are left for future research.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}