
MRAP2 Inhibits β-Arrestin-2 Recruitment to the Prokineticin Receptor 2
 ,
,  and
and 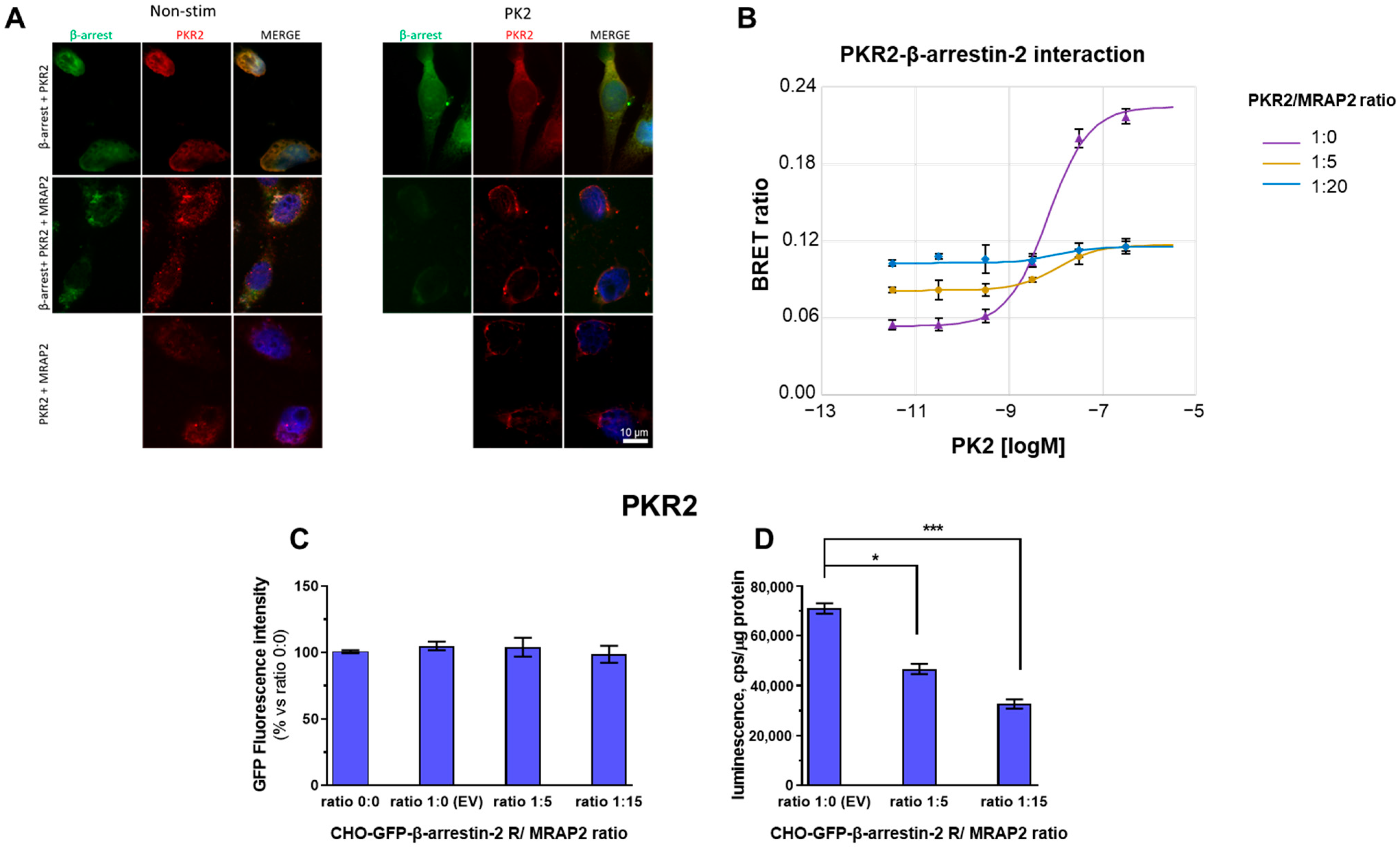{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Expression Constructs
2.3. Expression and Purification of Recombinant Proteins in E. coli
2.4. Glutathione S-Transferase (GST) Pull-Down
2.5. Cell Cultures, Transfection and Stimulation
2.6. Ligand Production
2.7. Crosslinking
2.8. Immunoprecipitation
2.9. Western Blotting
2.10. Bioluminescence Resonance Energy Transfer (BRET) Assay
2.11. Immunofluorescence
2.12. Data Analysis
3. Results
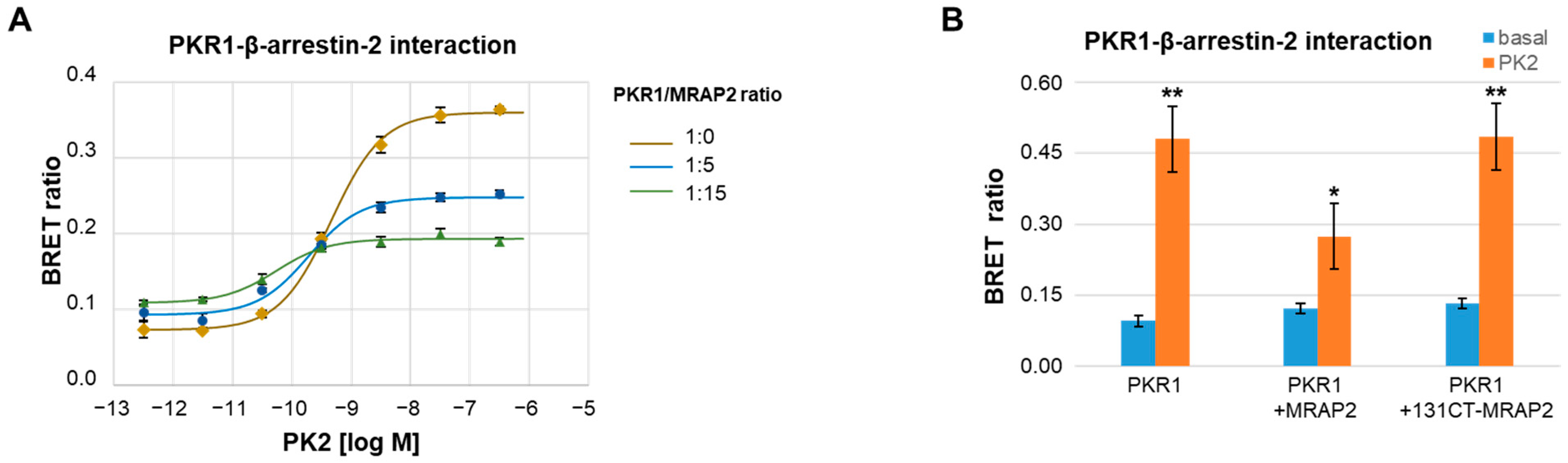
3.1. MRAP2 Inhibits PK2-Stimulated β-Arrestin-2 Recruitment to PKRs
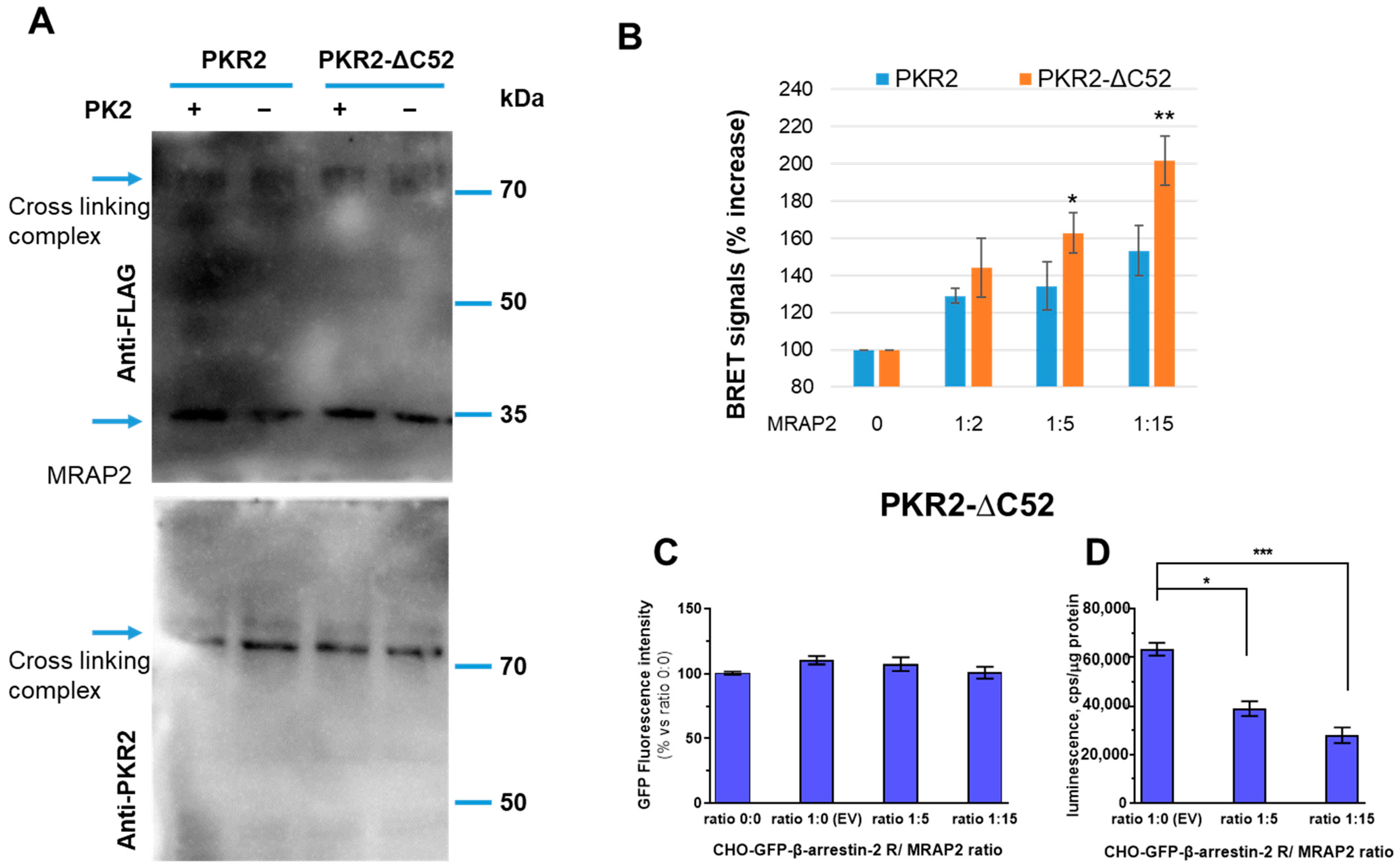
3.2. Role of PKR2 C-Terminal Region in MRAP2 Modulation of β-Arrestin-2 Recruitment
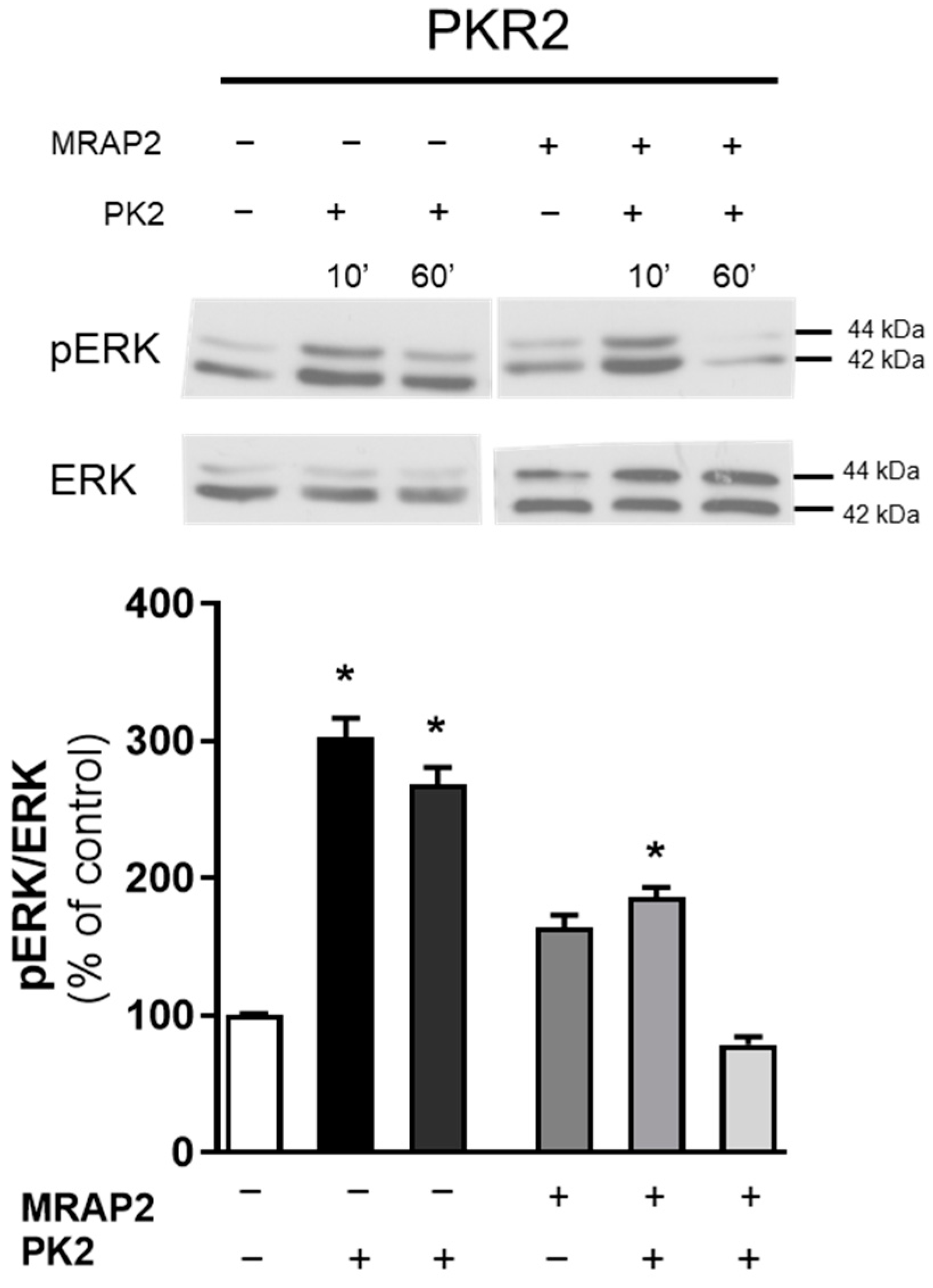
3.3. MRAP2 Inhibits β-Arrestin–Dependent Erk Activation
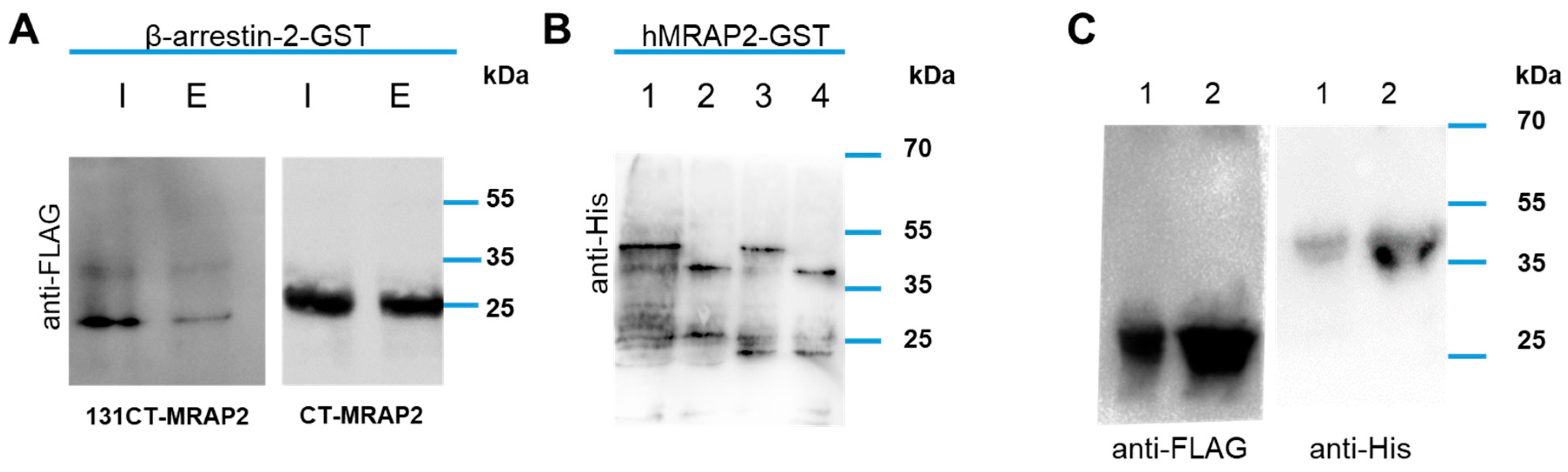
3.4. MRAP2 Physical Interact with β-Arrestin-2
4. Discussion
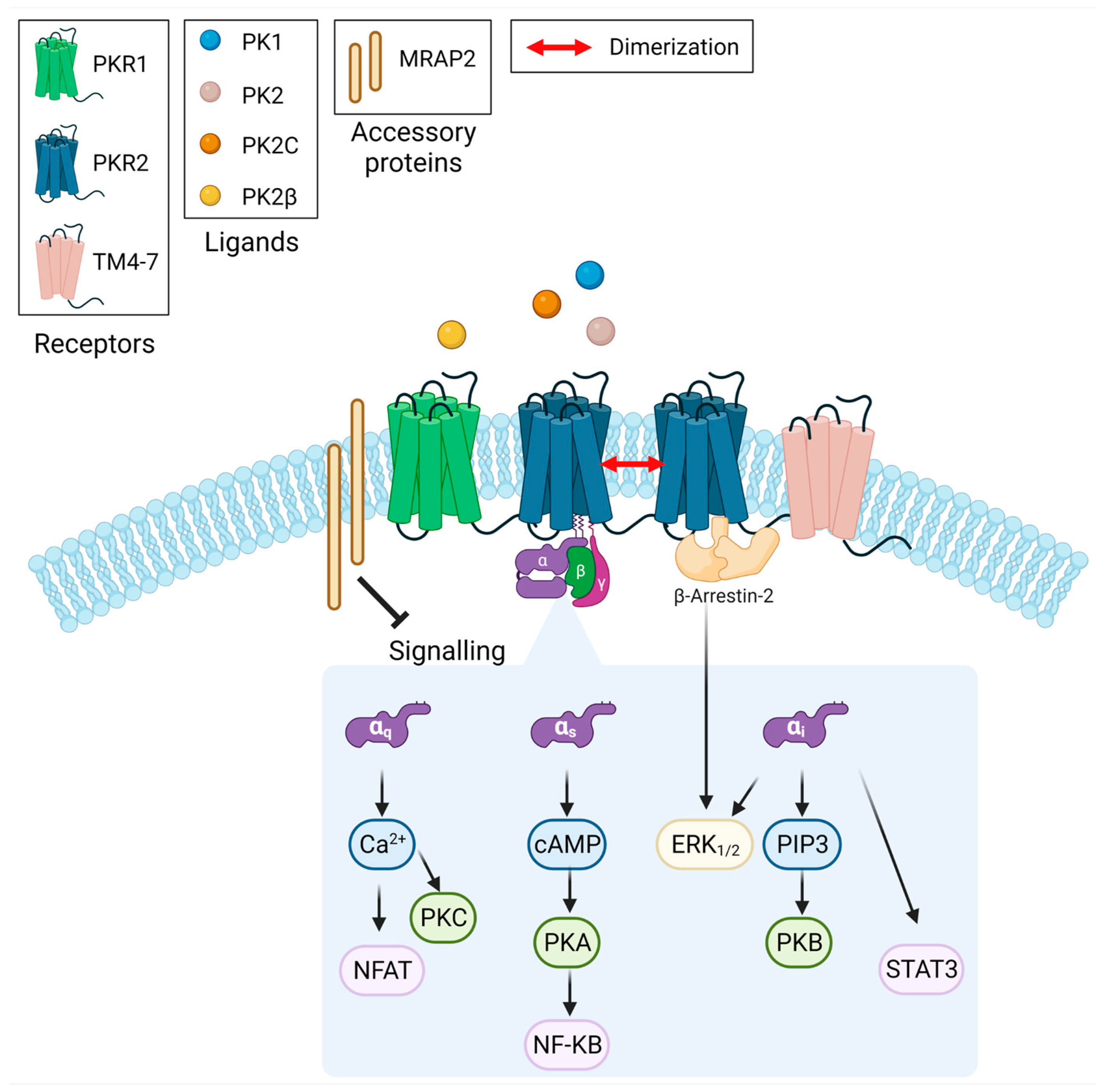
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luttrell, L.M.; Lefkowitz, R.J. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, J. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef]
- Kaser, A.; Winklmayr, M.; Lepperdinger, G.; Kreil, G. The AVIT protein family. Secreted 449 cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. 2003, 4, 469–473. [Google Scholar] [CrossRef]
- Vincenzi, M.; Kremić, A.; Jouve, A.; Lattanzi, R.; Miele, R.; Benharouga, M.; Alfaidy, N.; Migrenne-Li, S.; Kanthasamy, A.G.; Porcionatto, M.; et al. Therapeutic Potential of Targeting Prokineticin Receptors in Diseases. Pharmacol. Rev. 2023, 75, 1167–1199. [Google Scholar] [CrossRef]
- Negri, L.; Ferrara, N. The Prokineticins: Neuromodulators and Mediators of Inflammation and Myeloid Cell-Dependent Angiogenesis. Physiol. Rev. 2018, 98, 1055–1082. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 2006, 417, 405–410. [Google Scholar] [CrossRef]
- Starnes, A.N.; Jeff, R.; Jones, J.R. Inputs and Outputs of the Mammalian Circadian Clock. Biology 2023, 12, 508. [Google Scholar] [CrossRef]
- Xin, H.; Lu, R.; Lee, H.; Zhang, W.; Zhang, C.; Deng, J.; Liu, Y.; Shen, S.; Wagner, K.U.; Forman, S.; et al. G-protein-coupled receptor agonist BV8/-prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem. 2016, 288, 13842–13849. [Google Scholar] [CrossRef]
- Qu, X.; Zhuang, G.; Yu, L.; Meng, G.; Ferrara, N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: Key role of Stat3 signaling. J. Biol. Chem. 2012, 287, 19574–19584. [Google Scholar] [CrossRef]
- LeCouter, J.; Zlot, C.; Tejada, M.; Peale, F.; Ferrara, N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate 4hematopoiesis and hematopoietic cell mobilization. Proc. Natl. Acad. Sci. USA 2004, 101, 16813–16818. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Trypanosoma cruzi trans-sialidase inducesSTAT3 and ERK activation by prokineticin receptor 2 binding. Cell Biochem. Funct. 2021, 39, 326–334. [Google Scholar] [CrossRef]
- Yin, W.; Liu, H.; Peng, Z.; Chen, D.; Li, J.; Li, J.-D. Mechanisms that underlie the internalization and extracellular signal regulated kinase 1/2 activation by PKR2 receptor. Cell. Signal. 2014, 26, 1118–1124. [Google Scholar] [CrossRef]
- Casella, I.; Ambrosio, C. Prokineticin receptors interact unselectively with several G protein subtypes but bind selectively to beta-arrestin 2. Cell. Signal. 2021, 83, 110000. [Google Scholar] [CrossRef]
- Chen, J.; Kuei, C.; Sutton, S.; Wilson, S.; Yu, J.; Kamme, F.; Mazur, C.; Lovenberg, T.; Liu, C. Identification and pharmacological characterization of prokineticin 2 beta as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 2005, 67, 2070–2076. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Vincenzi, M.; Fullone, M.R.; Miele, R. Identification and Characterization of a New Splicing Variant of Prokineticin 2. Life 2022, 12, 248. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Identification and characterization of Prokineticin receptor 2 splicing TM4-7 variant and its modulation in an animal model of Alzheimer’s disease. Neurpeptides 2019, 73, 49–56. [Google Scholar] [CrossRef]
- Iwasa, T.; Matsuzaki, T.; Tungalagsuvd, A.; Munkhzaya, M.; Kawami, T.; Yamasaki, M.; Murakami, M.; Kato, T.; Kuwahara, A.; Yasui, T.; et al. Changes in the responsiveness of hypothalamic PK2 and PKR1 gene expression to fasting in developing male rats. Int. J. Dev. Neurosci. 2014, 38, 87–90. [Google Scholar] [CrossRef]
- Magnan, C.; Migrenne-Li, S. Pleiotropic effects of prokineticin 2 in the control of energy metabolism. Biochimie 2021, 186, 73–81. [Google Scholar] [CrossRef]
- Gardiner, J.V.; Bataveljic, A.; Patel, N.A.; Bewick, G.A.; Roy, D.; Campbell, D.; Greenwood, H.C.; Murphy, K.G.; Hameed, S.; Jethwa, P.H.; et al. Prokineticin 2 is a hypothalamic neuropeptide that potently inhibits food intake. Diabetes 2010, 59, 397–406. [Google Scholar] [CrossRef]
- Hinkle, P.M.; Sebag, J.A. Structure and function of the Melanocortin2 receptor accessory protein (MRAP). Mol. Cell. Endocrinol. 2009, 300, 25–31. [Google Scholar] [CrossRef]
- Wang, M.; Lyu, J.; Zhang, C. Single transmembrane GPCR modulating proteins: Neither single nor simple. Protein Cell 2023, XX, pwad035. [Google Scholar] [CrossRef]
- Baron, M.; Maillet, J.; Huyvaert, M.; Dechaume, A.; Boutry, R.; Loiselle, H.; Durand, E.; Toussaint, B.; Vaillant, E.; Philippe, J.; et al. Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat. Med. 2019, 25, 1733–1738. [Google Scholar] [CrossRef]
- Webb, T.; Chan, L.; Cooray, S.N.; Cheetham, M.; Chapple, P.; Clark, A.J.L. Distinct Melanocortin 2 Receptor Accessory Protein Domains Are Required for Melanocortin 2 Receptor Interaction and Promotion of Receptor Trafficking. Endocrinology 2009, 150, 720–726. [Google Scholar] [CrossRef]
- Wang, M.; Zhai, Y.; Lei, X.; Xu, J.; Jiang, B.; Kuang, Z.; Zhang, C.; Liu, S.; Bian, S.; Yang, X.M.; et al. Determination of the Interaction and Pharmacological Modulation of MCHR1 Signaling by the C-Terminus of MRAP2 Protein. Front. Endocrinol. 2022, 13, 848728. [Google Scholar] [CrossRef]
- Chen, V.; Bruno, A.E.; Britt, L.L.; Hernandez, C.C.; Gimenez, L.E.; Peisley, A.; Cone, R.D.; Millhauser, G.L. Membrane orientation and oligomerization of the melanocortin receptor accessory protein 2. J. Biol. Chem. 2020, 295, 16370–16379. [Google Scholar] [CrossRef]
- Chan, L.F.; Webb, T.R.; Chung, T.T.; Meimaridou, E.; Cooray, S.N.; Guasti, L.; Chapple, J.P.; Egertová, M.; Elphick, M.R.; Cheetham, M.E.; et al. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc. Natl. Acad. Sci. USA 2009, 106, 6146–6151. [Google Scholar] [CrossRef]
- Berruien, N.N.A.; Smith, C.L. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene 2021, 757, 144949. [Google Scholar] [CrossRef]
- Chaly, A.L.; Srisai, D.; Gardner, E.E.; Sebag, J.A. The Melanocortin Receptor Accessory Protein 2 promotes food intake through inhibition of the Prokineticin Receptor-1. eLife 2016, 5, e12397. [Google Scholar] [CrossRef]
- Yin, T.C.; Mittal, A.; Buscaglia, P.; Li, W.; Sebag, J.A. Activation of amygdala prokineticin receptor 2 neurons drives the anorexigenic activity of the neuropeptide PK2. J. Biol. Chem. 2023, 299, 102814. [Google Scholar] [CrossRef]
- Lattanzi, R.; Severini, C.; Miele, R. Prokineticin 2 in cancer-related inflammation. Cancer Lett. 2022, 546, 215838. [Google Scholar] [CrossRef]
- Désaubry, L.; Kanthasamy, A.G.; Nebigil, C.G. Prokineticin signaling in heart-brain developmental axis: Therapeutic options for heart and brain injuries. Pharm. Res. 2020, 160, 105190. [Google Scholar] [CrossRef]
- Dodé, C.; Rondard, P. PROK2/PROKR2 Signaling and Kallmann Syn-drome. Front. Endocrinol. 2013, 4, 19. [Google Scholar] [CrossRef]
- Gordon, R.; Neal, M.L.; Luo, J.; Langley, M.R.; Harischandra, D.S.; Panicker, N.; Charli, A.; Jin, H.; Anantharam, V.; Woodruff, T.M.; et al. Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat. Commun. 2016, 1, 12932. [Google Scholar] [CrossRef]
- Nebigil, C.G. Prokineticin Is a New Linker between Obesity and Cardiovascular Diseases. Front. Cardiovasc. Med. 2017, 4, 20. [Google Scholar] [CrossRef]
- Cheng, X.-X.; Li, M.-Q.; Peng, T. Novel Insights into Prokineticin 1 Role in Pregnancy-related Diseases. Int. J. Med. Sci. 2024, 21, 27–33. [Google Scholar] [CrossRef]
- Lattanzi, R.; Miele, R. Non-Peptide Agonists and Antagonists of the Prokineticin Receptors. Curr. Issues Mol. Biol. 2022, 44, 6323–6332. [Google Scholar] [CrossRef]
- Lattanzi, R.; Casella, I.; Fullone, M.R.; Maftei, D.; Miele, R. Mapping the interaction site for β-arrestin-2 in the prokineticin 2 receptor. Cell Signal. Submitted.
- Fullone, M.R.; Maftei, D.; Vincenzi, M.; Lattanzi, R.; Miele, R. Identification of Regions Involved in the Physical Interaction between Melanocortin Receptor Accessory Protein 2 and Prokineticin Receptor 2. Biomolecules 2022, 12, 474. [Google Scholar] [CrossRef]
- Reed, S.E.; Staley, E.M.; Mayginnes, J.P.; Pintel, D.J.; Tullis, G.E. Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J. Virol. Methods 2006, 138, 85–98. [Google Scholar] [CrossRef]
- Sbai, Q.; Monnier, C.; Dodé, C.; Pin, J.-P.; Hardelin, J.-P.; Rondard, P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014, 28, 3734–3744. [Google Scholar] [CrossRef]
- Pulvermuller, A.; Maretzki, D.; Rudnicka-Nawrot, M.; Clay Smith, W.C.; Palczewski, K.; Hofmann, K.P. Functional Differences in the Interaction of Arrestin and Its Splice Variant, p44, with Rhodopsin. Biochemistry 1997, 36, 9253–9260. [Google Scholar] [CrossRef]
- Christopoulos, A.; Kenakin, T. G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol. Ther. 2006, 110, 465–502. [Google Scholar] [CrossRef]
- Kotliar, I.B.; Lorenzen, E.; Schwenk, J.M.; Hay, D.L.; Sakmar, T.P. Elucidating the interactome of G protein-coupled receptors an receptor activity-modifying proteins. Pharmacol. Rev. 2023, 75, 1–34. [Google Scholar] [CrossRef]
- Rouault, A.A.; Lee, A.A.; Sebag, J.A. Regions of MRAP2 required for the inhibition of orexin and prokineticin receptor signaling. Biochim. Biophys. Acta 2017, 1864, 2322–2329. [Google Scholar] [CrossRef]
- Fullone, M.R.; Maftei, D.; Vincenzi, M.; Lattanzi, R.; Miele, R. Arginine 125 Is an Essential Residue for the Function of MRAP2. Int. J. Mol. Sci. 2022, 23, 9853. [Google Scholar] [CrossRef]
- Srisai, D.; Yin, T.C.; Lee, A.A.; Rouault, A.A.; Pearson, N.A.; Grobe, J.L.; Sebag, J.A. MRAP2 regulates ghrelin receptor signaling and hunger sensing. Nat. Commun. 2017, 8, 713. [Google Scholar] [CrossRef]
- Rouault, A.A.J.; Rosselli-Murai, L.K.; Hernandez, C.C.; Gimenez, L.E.; Tall, G.G.; Sebag, J.A. The GPCR accessory protein MRAP2 regulates both biased signaling and constitutive activity of the ghrelin receptor GHSR1a. Sci. Signal. 2020, 13, eaax4569. [Google Scholar] [CrossRef]
- Rouault, A.A.J.; Buscaglia, P.; Sebag, J.A. MRAP2 inhibits β-arrestin recruitment to the ghrelin receptor by preventing GHSR1a phosphorylation. J. Biol. Chem. 2022, 298, 102057. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Wisler, J.W.; Rockman, H.A.; Lefkowitz, R.J. Biased G protein-coupled receptor signaling: Changing the paradigm of drug discovery. Circulation 2018, 137, 2315–2317. [Google Scholar] [CrossRef] [PubMed]
- Wess, J.; Oteng, A.-B.; Rivera-Gonzalez, O.; Gurevich, E.V.; Gurevich, V.V. β-Arrestins: Structure, Function, Physiology, and Pharmacological Perspectives. Pharmacol. Rev. 2023, 75, 854–884. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Xia, F.; Li, Z.; Shen, C.; Yang, Z.; Hou, H.; Sun, S.; Feng, Y.; Yong, X.; Tian, X.; et al. Structure, function and drug discovery of GPCR signaling. Mol. Biomed. 2023, 4, 46. [Google Scholar] [CrossRef]
- Kahsai, A.W.; Shah, K.S.; Shim, P.J.; Lee, M.A.; Shreiber, B.N.; Schwalb, A.M.; Zhang, X.; Kwon, H.Y.; Huang, L.-Y.; Soderblom, E.J.; et al. Signal transduction at GPCRs: Allosteric activation of the ERK MAPK by β-arrestin. Proc. Natl. Acad. Sci. USA 2023, 120, e2303794120. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.; Jiang, B.; Zhai, Y.; Zheng, J.; Yang, L.; Tai, X.; Li, Y.; Fu, S.; Xu, J.; et al. Identification of MRAP protein family as broad-spectrum GPCR modulators. Clin. Transl. Med. 2022, 12, e1091. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattanzi, R.; Casella, I.; Fullone, M.R.; Maftei, D.; Vincenzi, M.; Miele, R. MRAP2 Inhibits β-Arrestin-2 Recruitment to the Prokineticin Receptor 2. Curr. Issues Mol. Biol. 2024, 46, 1607-1620. https://doi.org/10.3390/cimb46020104
Lattanzi R, Casella I, Fullone MR, Maftei D, Vincenzi M, Miele R. MRAP2 Inhibits β-Arrestin-2 Recruitment to the Prokineticin Receptor 2. Current Issues in Molecular Biology. 2024; 46(2):1607-1620. https://doi.org/10.3390/cimb46020104
Chicago/Turabian StyleLattanzi, Roberta, Ida Casella, Maria Rosaria Fullone, Daniela Maftei, Martina Vincenzi, and Rossella Miele. 2024. "MRAP2 Inhibits β-Arrestin-2 Recruitment to the Prokineticin Receptor 2" Current Issues in Molecular Biology 46, no. 2: 1607-1620. https://doi.org/10.3390/cimb46020104