
Precision Medicine in Erythropoietin Deficiency and Treatment Resistance: A Novel Approach to Management of Anaemia in Chronic Kidney Disease
 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Overview of Pharmacogenetics
3. Impact of Iron Deficiency and Treatment
4. Erythropoietin
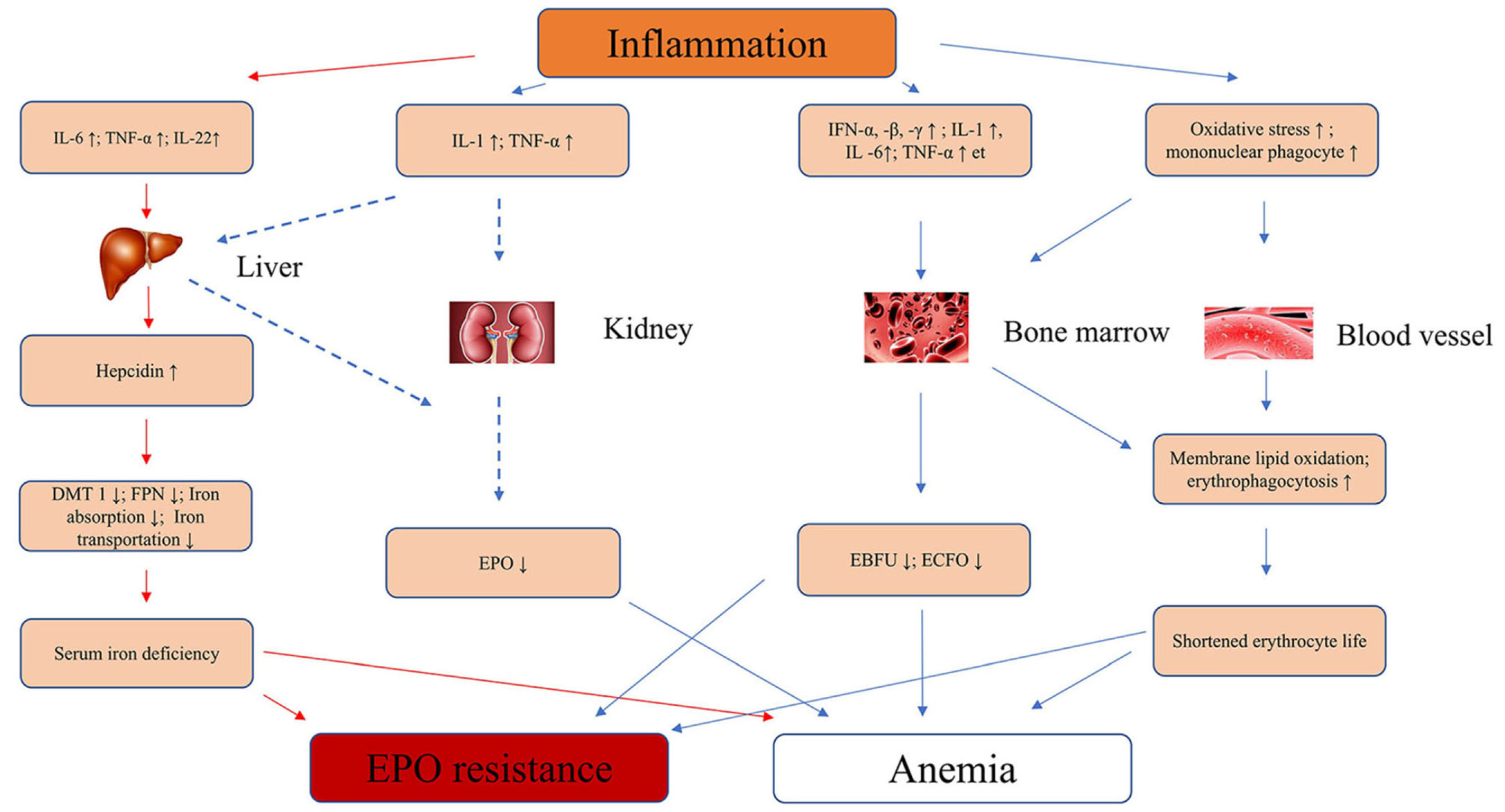
5. Pathophysiology of Inflammation and Linked Genetic Factors
6. Erythropoietin Treatment and Resistance
7. Precision Medicine in EPO Deficiency and Treatment Resistance
8. Genetic Factors of EPO Deficiency and Treatment Resistance

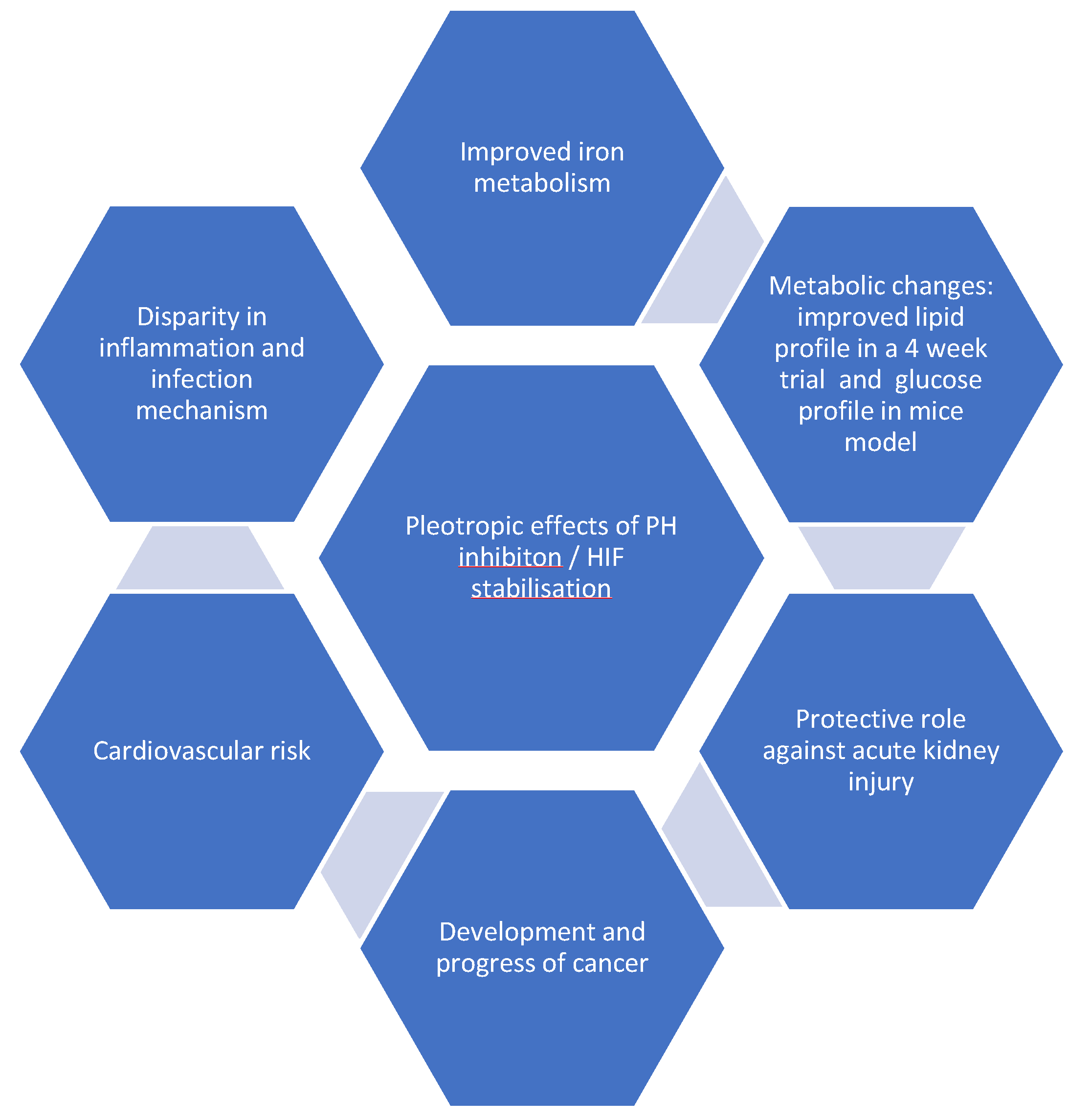
9. Hypoxia-Inducible Factor Prolyl Hydroxylase (HIF-PH) Inhibitors
10. Future Treatment Approaches
11. Potential Approach from the Perspective of Drug Design and Development to Addressing Treatment Resistance in Erythropoietin Deficiency Anaemia: Receptor Modification
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bonomini, M.; Del Vecchio, L.; Sirolli, V.; Locatelli, F. New Treatment Approaches for the Anemia of CKD. Am. J. Kidney Dis. 2016, 67, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drüeke, T.B.; Massy, Z.A. Erythropoiesis-Stimulating Agents and Mortality. J. Am. Soc. Nephrol. 2019, 30, 907–908. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Ho, Y.; Tarng, D.-C. Iron Therapy in Chronic Kidney Disease: Days of Future Past. Int. J. Mol. Sci. 2021, 22, 1008. [Google Scholar] [CrossRef]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Fishbane, S.; El-Shahawy, M.A.; Pecoits-Filho, R.; Pham Van, B.; Houser, M.T.; Frison, L.; Little, D.J.; Guzman, N.J.; Pergola, P.E. OLYMPUS: A Phase 3, Randomized, Double-Blind, Placebo-Controlled, International Study of Roxadustat Efficacy in Patients with Non-Dialysis-Dependent (NDD) CKD and Anemia [Abstract TH-OR023]. J. Am. Soc. Nephrol. 2019, 30, 6. [Google Scholar]
- Barratt, J.; Andric, B.; Tataradze, A.; Schömig, M.; Reusch, M.; Valluri, U.; Mariat, C. Roxadustat for the treatment of anaemia in chronic kidney disease patients not on dialysis: A Phase 3, randomized, open-label, active-controlled study (DOLOMITES). Nephrol. Dial. Transplant. 2021, 36, 1616–1628. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Ueno, Y.; Kinugawa, K. Impact of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor on Renal Function in Patient with Heart Failure. J. Cardiovasc. Dev. Dis. 2021, 8, 189. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factor–prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int. Suppl. 2021, 11, 8–25. [Google Scholar] [CrossRef]
- Scott, S.A. Personalizing medicine with clinical pharmacogenetics. Genet. Med. 2011, 13, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.T.; Lopez, D. Pharmacogenetics: An Important Part of Drug Development with A Focus on Its Application. Int. J. Biomed. Investig. 2018, 1, 111. [Google Scholar]
- Lee, H.H.; Ho, R.H. Interindividual and interethnic variability in drug disposition: Polymorphisms in organic anion transporting polypeptide 1B1 (OATP1B1; SLCO1B1). Br. J. Clin. Pharmacol. 2017, 83, 1176–1184. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Xu, G. A Novel Choice to Correct Inflammation-Induced Anemia in CKD: Oral Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat. Front. Med. 2020, 7, 393. [Google Scholar] [CrossRef]
- Ueda, N.; Takasawa, K. Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease. Nutrients 2018, 10, 1173. [Google Scholar] [CrossRef] [Green Version]
- Camaschella, C. Iron deficiency. Blood 2019, 133, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campodonico, J.; Nicoli, F.; Motta, I.; Migone De Amicis, M.; Bonomi, A.; Cappellini, M.; Agostoni, P. Prognostic role of transferrin saturation in heart failure patients. Eur. J. Prev. Cardiol. 2021, 28, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Kopyt, N.P. Oral Ferric Maltol for the Treatment of Iron-Deficiency Anemia in Patients With CKD: A Randomized Trial and Open-Label Extension. Am. J. Kidney Dis. 2021, 78, 846–856.e841. [Google Scholar] [CrossRef] [PubMed]
- Tolkien, Z.; Stecher, L.; Mander, A.P.; Pereira, D.I.A.; Powell, J.J. Ferrous Sulfate Supplementation Causes Significant Gastrointestinal Side-Effects in Adults: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0117383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Allen, S.; Kopyt, N.; Pergola, P. Iron Replacement Therapy with Oral Ferric Maltol: Review of the Evidence and Expert Opinion. J. Clin. Med. 2021, 10, 4448. [Google Scholar] [CrossRef]
- Barish, C.F.; Koch, T.; Butcher, A.; Morris, D.; Bregman, D.B. Safety and Efficacy of Intravenous Ferric Carboxymaltose (750 mg) in the Treatment of Iron Deficiency Anemia: Two Randomized, Controlled Trials. Anemia 2012, 2012, 172104. [Google Scholar] [CrossRef] [Green Version]
- Charytan, C.; Bernardo, M.V.; Koch, T.A.; Butcher, A.; Morris, D.; Bregman, D.B. Intravenous ferric carboxymaltose versus standard medical care in the treatment of iron deficiency anemia in patients with chronic kidney disease: A randomized, active-controlled, multi-center study. Nephrol. Dial. Transplant. 2013, 28, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Boots, J.M.M.; Quax, R.A.M. High-Dose Intravenous Iron with Either Ferric Carboxymaltose or Ferric Derisomaltose: A Benefit-Risk Assessment. Drug Saf. 2022, 45, 1019–1036. [Google Scholar] [CrossRef] [PubMed]
- Pollock, R.F.; Biggar, P. Indirect methods of comparison of the safety of ferric derisomaltose, iron sucrose and ferric carboxymaltose in the treatment of iron deficiency anemia. Expert Rev. Hematol. 2020, 13, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koury, M.J.; Haase, V.H. Anaemia in kidney disease: Harnessing hypoxia responses for therapy. Nat. Rev. Nephrol. 2015, 11, 394–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolze, I.; Berchner-Pfannschmidt, U.; Freitag, P.; Wotzlaw, C.; Rössler, J.; Frede, S.; Acker, H.; Fandrey, J. Hypoxia-inducible erythropoietin gene expression in human neuroblastoma cells. Blood 2002, 100, 2623–2628. [Google Scholar] [CrossRef]
- Koury, M.J.; Bondurant, M.C. Maintenance by erythropoietin of viability and maturation of murine erythroid precursor cells. J. Cell. Physiol. 1988, 137, 65–74. [Google Scholar] [CrossRef]
- Molineux, G.; Foote, M.A.; Elliott, S.G. Erythropoietins and Erythropoiesis: Molecular, Cellular, Preclinical, and Clinical Biology; Birkhäuser: Basel, Switzerland, 2005. [Google Scholar]
- Nangaku, M.; Eckardt, K.-U. Pathogenesis of Renal Anemia. Semin. Nephrol. 2006, 26, 261–268. [Google Scholar] [CrossRef]
- Souma, T.; Yamazaki, S.; Moriguchi, T.; Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Abe, M.; Kiyomoto, H.; Ito, S.; et al. Plasticity of Renal Erythropoietin-Producing Cells Governs Fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1599–1616. [Google Scholar] [CrossRef] [Green Version]
- Baer, A.N.; Dessypris, E.N.; Goldwasser, E.; Krantz, S.B. Blunted erythropoietin response to anaemia in rheumatoid arthritis. Br. J. Haematol. 1987, 66, 559–564. [Google Scholar] [CrossRef]
- Miller, C.B.; Jones, R.J.; Piantadosi, S.; Abeloff, M.D.; Spivak, J.L. Decreased Erythropoietin Response in Patients with the Anemia of Cancer. N. Engl. J. Med. 1990, 322, 1689–1692. [Google Scholar] [CrossRef]
- La Ferla, K.; Reimann, C.; Jelkmann, W.; Hellwig-Bürgel, T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-κB. FASEB J. 2002, 16, 1–17. [Google Scholar] [CrossRef]
- Roach, K.M.; Duffy, S.M.; Coward, W.; Feghali-Bostwick, C.; Wulff, H.; Bradding, P. The K+ Channel KCa3.1 as a Novel Target for Idiopathic Pulmonary Fibrosis. PLoS ONE 2014, 8, e85244. [Google Scholar] [CrossRef]
- Batmunkh, C.; Krajewski, J.; Jelkmann, W.; Hellwig-Bürgel, T. Erythropoietin production: Molecular mechanisms of the antagonistic actions of cyclic adenosine monophosphate and interleukin-1. FEBS Lett. 2006, 580, 3153–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttinger, E.P. TGF-β in Renal Injury and Disease. Semin. Nephrol. 2007, 27, 309–320. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.C.K.; Zhang, H.; Kong, Y.-Z.; Tan, J.-J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced Glycation End-Products Induce Tubular CTGF via TGF-β–Independent Smad3 Signaling. J. Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential Role of Smad3 in Angiotensin II–Induced Vascular Fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef]
- Huang, X.R.; Chung, A.C.K.; Zhou, L.; Wang, X.J.; Lan, H.Y. Latent TGF-β1 Protects Against Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M. Inflammatory Mediators and Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies; Liu, B.-C., Lan, H.-Y., Lv, L.-L., Eds.; Springer: Singapore, 2019; pp. 381–406. [Google Scholar]
- Petreski, T.; Piko, N.; Ekart, R.; Hojs, R.; Bevc, S. Review on Inflammation Markers in Chronic Kidney Disease. Biomedicines 2021, 9, 182. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F. Innate Immunity System in Patients With Cardiovascular and Kidney Disease. Circ. Res. 2023, 132, 915–932. [Google Scholar] [CrossRef]
- Sharples, E.J.; Varagunam, M.; Sinnott, P.J.; McCloskey, D.J.; Raftery, M.J.; Yaqoob, M.M. The Effect of Proinflammatory Cytokine Gene and Angiotensin-Converting Enzyme Polymorphisms on Erythropoietin Requirements in Patients on Continuous Ambulatory Peritoneal Dialysis. Perit. Dial. Int. 2006, 26, 64–68. [Google Scholar] [CrossRef]
- Yadav, P.; Divvi, V.S.S.R.; Shah, T. Assessment of Cytokine (α-TNF) with Erythropoietin and their Correlation in Pulmonary Tuberculosis with Anaemia. J. Pharm. Res. Int. 2021, 33, 1–9. [Google Scholar] [CrossRef]
- Glas, J.; Török, H.-P.; Schneider, A.; Brünnler, G.; Kopp, R.; Albert, E.D.; Stolte, M.; Folwaczny, C. Allele 2 of the Interleukin-1 Receptor Antagonist Gene Is Associated With Early Gastric Cancer. J. Clin. Oncol. 2004, 22, 4746–4752. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.-H.; Lee, T.-W.; Ihm, C.-G.; Lee, S.-H.; Moon, J.-Y. Polymorphisms in two genes, IL-1B and ACE, are associated with erythropoietin resistance in Korean patients on maintenance hemodialysis. Exp. Mol. Med. 2008, 40, 161–166. [Google Scholar] [CrossRef] [PubMed]
- KDIGO. KDIGO Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 5–14. [Google Scholar]
- Locatelli, F.; Bárány, P.; Covic, A.; De Francisco, A.; Del Vecchio, L.; Goldsmith, D.; Hörl, W.; London, G.; Vanholder, R.; Van Biesen, W.; et al. Kidney Disease: Improving Global Outcomes guidelines on anaemia management in chronic kidney disease: A European Renal Best Practice position statement. Nephrol. Dial. Transplant. 2013, 28, 1346–1359. [Google Scholar] [CrossRef]
- Hayat, A.; Haria, D.; Salifu, M.O. Erythropoietin stimulating agents in the management of anemia of chronic kidney disease. Patient Prefer. Adherence 2008, 2, 195–200. [Google Scholar]
- López-Gómez, J.M.; Portolés, J.M.; Aljama, P. Factors that condition the response to erythropoietin in patients on hemodialysis and their relation to mortality: New strategies to prevent cardiovascular risk in chronic kidney disease. Kidney Int. 2008, 74, S75–S81. [Google Scholar] [CrossRef] [Green Version]
- Rossert, J.; Gassmann-Mayer, C.; Frei, D.; McClellan, W. Prevalence and predictors of epoetin hyporesponsiveness in chronic kidney disease patients. Nephrol. Dial. Transplant. 2007, 22, 794–800. [Google Scholar] [CrossRef]
- Guerrero Riscos, M.Á.; Guerrero-Riscos, M.Á.; Montes Delgado, R.; Montes-Delgado, R.; Seda Guzmán, M.; Seda-Guzmán, M.; Praena-Fernández, J.M.; Praena-Fernández, J.M. Erythropoietin resistance and survival in non-dialysis patients with stage 4–5 chronic kidney disease and heart disease. Nefrología 2012, 32, 343–352. [Google Scholar]
- Xiuling, W.; Jianjun, L.; Ying, Y.; Rong, X.; Lu, W.; Xuedong, W.; Fubin, T. Safety of Weekly Single versus Divided Administration of Moderate-dose Erythropoietin in the Treatment of Maintenance Hemodialysis Patients with Renal Anemia. Chin. Gen. Pract. 2023, 26, 711–717. [Google Scholar]
- Funakoshi, S. Difference in Therapeutic Effects between Roxadustat and Daprodustat, HIF-Ph Inhibitors, Depending on the Blood Type in Hemodialysis (HD) Patients. Blood 2021, 138, 4147. [Google Scholar] [CrossRef]
- Su, K.; Li, Z.; Zhang, L.; Fang, S.; Mao, M.; Sun, Z.; Zhang, X. Tetrahydropyridin-4-ylpicolinoylglycines as novel and orally active prolyl hydroxylase 2 (PHD2) inhibitors for the treatment of renal anemia. Eur. J. Med. Chem. 2022, 238, 114479. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Liu, Y.; Cai, X.; Huang, X.; Fu, W.; Wang, L.; Qiu, L.; Li, J.; Sun, L. Ferroptosis, a new target for treatment of renal injury and fibrosis in a 5/6 nephrectomy-induced CKD rat model. Cell Death Discov. 2022, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Binley, K.; Askham, Z.; Iqball, S.; Spearman, H.; Martin, L.; de Alwis, M.; Thrasher, A.J.; Ali, R.R.; Maxwell, P.H.; Kingsman, S.; et al. Long-term reversal of chronic anemia using a hypoxia-regulated erythropoietin gene therapy. Blood 2002, 100, 2406–2413. [Google Scholar] [CrossRef] [Green Version]
- Lippin, Y.; Dranitzki-Elhalel, M.; Brill-Almon, E.; Mei-Zahav, C.; Mizrachi, S.; Liberman, Y.; Iaina, A.; Kaplan, E.; Podjarny, E.; Zeira, E.; et al. Human erythropoietin gene therapy for patients with chronic renal failure. Blood 2005, 106, 2280–2286. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.K.; Abboud, H.E.; Goddard, K.A.B.; Saad, M.F.; Adler, S.G.; Arar, N.H.; Bowden, D.W.; Duggirala, R.; Elston, R.C.; Hanson, R.L.; et al. Genome-Wide Scans for Diabetic Nephropathy and Albuminuria in Multiethnic Populations: The Family Investigation of Nephropathy and Diabetes (FIND). Diabetes 2007, 56, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Tong, Z.; Yang, Z.; Patel, S.; Chen, H.; Gibbs, D.; Yang, X.; Hau, V.S.; Kaminoh, Y.; Harmon, J.; Pearson, E.; et al. Promoter polymorphism of the erythropoietin gene in severe diabetic eye and kidney complications. Proc. Natl. Acad. Sci. USA 2008, 105, 6998–7003. [Google Scholar] [CrossRef]
- Wang, W.; Koka, V.; Lan, H.Y. Transforming growth factor-β and Smad signalling in kidney diseases. Nephrology 2005, 10, 48–56. [Google Scholar] [CrossRef]
- Santos, E.J.F.; Dias, R.S.C.; Brito Lima, J.F.d.; Salgado Filho, N.; Santos, A.M.d. Erythropoietin resistance in patients with chronic kidney disease: Current perspectives. Int. J. Nephrol. Renov. Dis. 2020, 13, 231–237. [Google Scholar] [CrossRef]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Lieb, M.E.; Menzies, K.; Moschella, M.C.; Ni, R.; Taubman, M.B. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem. Cell Biol. 2002, 80, 421–426. [Google Scholar] [CrossRef]
- Percy, M.J.; Zhao, Q.; Flores, A.; Harrison, C.; Lappin, T.R.J.; Maxwell, P.H.; McMullin, M.F.; Lee, F.S. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 654–659. [Google Scholar] [CrossRef]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-Week Studies of Oral Hypoxia-Inducible Factor–Prolyl Hydroxylase Inhibitor GSK1278863 for Treatment of Anemia. J. Am. Soc. Nephrol. 2016, 27, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, P.; Shukla, D.; Tran, M.G.B.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of Hypoxia Inducible Factor Hydroxylases Protects Against Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2008, 19, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moslehi, J.; Minamishima, Y.A.; Shi, J.; Neuberg, D.; Charytan, D.M.; Padera, R.F.; Signoretti, S.; Liao, R.; Kaelin, W.G. Loss of Hypoxia-Inducible Factor Prolyl Hydroxylase Activity in Cardiomyocytes Phenocopies Ischemic Cardiomyopathy. Circulation 2010, 122, 1004–1016. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.-Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.-R.; et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, J.L.; Duffy, K.J.; Adams, D.F.; Fitch, D.M.; Luo, L.; Pappalardi, M.; Biju, M.; DiFilippo, E.H.; Shaw, T.; Wiggall, K.; et al. Discovery and Preclinical Characterization of GSK1278863 (Daprodustat), a Small Molecule Hypoxia Inducible Factor–Prolyl Hydroxylase Inhibitor for Anemia. J. Pharmacol. Exp. Ther. 2017, 363, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Besarab, A.; Provenzano, R.; Hertel, J.; Zabaneh, R.; Klaus, S.J.; Lee, T.; Leong, R.; Hemmerich, S.; Yu, K.-H.P.; Neff, T.B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol. Dial. Transplant. 2015, 30, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Hao, C.; Liu, B.-C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Flamme, I.; Ellinghaus, P.; Urrego, D.; Krüger, T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS ONE 2017, 12, e0186979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Tanaka, S.; Tanaka, T.; Saito, H.; Ishimoto, Y.; Wakashima, T.; Ueda, M.; Fukui, K.; Shimizu, A.; Inagi, R.; et al. Prolyl Hydroxylase Domain Inhibitor Protects against Metabolic Disorders and Associated Kidney Disease in Obese Type 2 Diabetic Mice. J. Am. Soc. Nephrol. 2020, 31, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kurihara, S.; Anayama, M.; Makino, Y.; Nagasawa, M. Four Cases of Serum Copper Excess in Patients with Renal Anemia Receiving a Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor: A Possible Safety Concern. Case Rep. Nephrol. Dial. 2022, 12, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Qian, J.; Chen, J.; Yu, X.; Mei, C.; Hao, C.; Jiang, G.; Lin, H.; Zhang, X.; Zuo, L.; et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol. Dial. Transplant. 2017, 32, 1373–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvroq (Daprodustat): Japanese Prescribing Information. Available online: https://www.pmda.go.jp/PmdaSearch/iyakuDetail/ResultDataSetPDF/340278_39990D4F1024_1_01 (accessed on 14 August 2020).
- Chertow, G.M.; Pergola, P.E.; Farag, Y.M.K.; Agarwal, R.; Arnold, S.; Bako, G.; Block, G.A.; Burke, S.; Castillo, F.P.; Jardine, A.G.; et al. Vadadustat in Patients with Anemia and Non–Dialysis-Dependent CKD. N. Engl. J. Med. 2021, 384, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Zhang, H.; Guo, Y.; Song, Y.; Tao, Y. Efficacy and Safety of Vadadustat for Anemia in Patients With Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 12, 795214. [Google Scholar] [CrossRef]
- Sugahara, M.; Tanaka, T.; Nangaku, M. Future perspectives of anemia management in chronic kidney disease using hypoxia-inducible factor-prolyl hydroxylase inhibitors. Pharmacol. Ther. 2022, 239, 108272. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Minutolo, R.; De Nicola, L.; Del Vecchio, L. Evolving Strategies in the Treatment of Anaemia in Chronic Kidney Disease: The HIF-Prolyl Hydroxylase Inhibitors. Drugs 2022, 82, 1565–1589. [Google Scholar] [CrossRef]
- Akizawa, T.; Nangaku, M.; Yamaguchi, T.; Koretomo, R.; Maeda, K.; Miyazawa, Y.; Hirakata, H. A Phase 3 Study of Enarodustat in Anemic Patients with CKD not Requiring Dialysis: The SYMPHONY ND Study. Kidney Int. Rep. 2021, 6, 1840–1849. [Google Scholar] [CrossRef]
- Akizawa, T.; Yamada, T.; Nobori, K.; Matsuda, Y.; Hayashi, Y.; Hayasaki, T.; Yamamoto, H. Molidustat for Japanese Patients With Renal Anemia Receiving Dialysis. Kidney Int. Rep. 2021, 6, 2604–2616. [Google Scholar] [CrossRef]
- Joksimovic Jovic, J.; Antic, S.; Nikolic, T.; Andric, K.; Petrovic, D.; Bolevich, S.; Jakovljevic, V. Erythropoietin Resistance Development in Hemodialysis Patients: The Role of Oxidative Stress. Oxidative Med. Cell. Longev. 2022, 2022, 9598211. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Wang, Y.; Sun, L. The Cross-Link between Ferroptosis and Kidney Diseases. Oxidative Med. Cell. Longev. 2021, 2021, 6654887. [Google Scholar] [CrossRef] [PubMed]
- Mravic, M.; He, L.; Kratochvil, H.; Hu, H.; Nick, S.E.; Bai, W.; Edwards, A.; Jo, H.; Wu, Y.; DiMaio, D.; et al. Designed Transmembrane Proteins Inhibit the Erythropoietin Receptor in a Custom Binding Topology. bioRxiv 2023. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Future Treatment Improvement Focus | Author/Year | Country of Study | Study Population, Sample Size (n) | Results | ||
|---|---|---|---|---|---|---|
| Clinical Trials | ||||||
| Comparison between weekly single dose and divided moderate dose of rHuEPO | Xiuling et al. [52] | China | Haemodialyzed (n = 88) | There was no significant difference in terms of medication safety in both groups | ||
| HIF-PH inhibition according to blood group | Funakoshi [53] | Japan | Haemodialyzed (n = 163) | Treatment | Blood group | Efficacy% |
| Roxadustat | A | 47 | ||||
| Daprodustat | O | 55 | ||||
| Basic Experiments | ||||||
| Selective inhibition of PH2 | Su et al. [54] | China | In vivo (n = 25) | Ongoing trial | ||
| Ferroptosis as a potential therapeutic target in CKD | Wang et al. [55] | China | Mouse model (n = 24) | 1. Dysfunctional iron metabolism is an important contributor to ferroptosis. 2. Ferritinophagy was observed among CKD-afflicted rats. 3. Regulation of iron metabolism and TGF-β1/Smad3 pathway can interfere with the progression of renal damage. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yugavathy, N.; Abdullah, B.M.; Lim, S.K.; Abdul Gafor, A.H.B.; Wong, M.G.; Bavanandan, S.; Wong, H.S.; Huri, H.Z. Precision Medicine in Erythropoietin Deficiency and Treatment Resistance: A Novel Approach to Management of Anaemia in Chronic Kidney Disease. Curr. Issues Mol. Biol. 2023, 45, 6550-6563. https://doi.org/10.3390/cimb45080413
Yugavathy N, Abdullah BM, Lim SK, Abdul Gafor AHB, Wong MG, Bavanandan S, Wong HS, Huri HZ. Precision Medicine in Erythropoietin Deficiency and Treatment Resistance: A Novel Approach to Management of Anaemia in Chronic Kidney Disease. Current Issues in Molecular Biology. 2023; 45(8):6550-6563. https://doi.org/10.3390/cimb45080413
Chicago/Turabian StyleYugavathy, Nava, Bashar Mudhaffar Abdullah, Soo Kun Lim, Abdul Halim Bin Abdul Gafor, Muh Geot Wong, Sunita Bavanandan, Hin Seng Wong, and Hasniza Zaman Huri. 2023. "Precision Medicine in Erythropoietin Deficiency and Treatment Resistance: A Novel Approach to Management of Anaemia in Chronic Kidney Disease" Current Issues in Molecular Biology 45, no. 8: 6550-6563. https://doi.org/10.3390/cimb45080413