
Identification of Potential Therapeutic Targets on the Level of DNA/mRNAs, Proteins and Metabolites: A Systematic Mapping Review of Scientific Texts’ Fragments from Open Targets

Abstract
:1. Introduction
2. Methods
- The biological entity has evidence of existence at the protein level.
- Annotation belongs to the following hierarchy groups: (a) general-annotation; generic-function, (b) general-annotation, (c) general-annotation; generic-interaction, (d) general-annotation; medical.
- The quality of annotation is designated as “GOLD”.
- The annotation does not belong to the following categories (a) caution, (b) domain-info, (c) sequence-caution, (d) variant-info.
- Annotation has no negative evidence.
- Quality of evidence is designated as “GOLD”.
- The type of evidence is “publication”.
- Terms describing the evidence are (a) physical interaction evidence used in manual assertion, (b) experimental evidence used in manual assertion, (c) direct assay evidence used in manual assertion, and (d) experimental evidence.
- PubMed ID exists for the publication.
- neXtProt ID for the biological entity has a one-to-one relationship to the Ensemble gene ID, which is used in Open Targets.
- Terms describing experimental studies conducted on the nucleic acids (DNA or RNA) level.
- Terms describing experimental studies conducted on the protein level.
- Terms describing experimental studies conducted on the metabolite level.
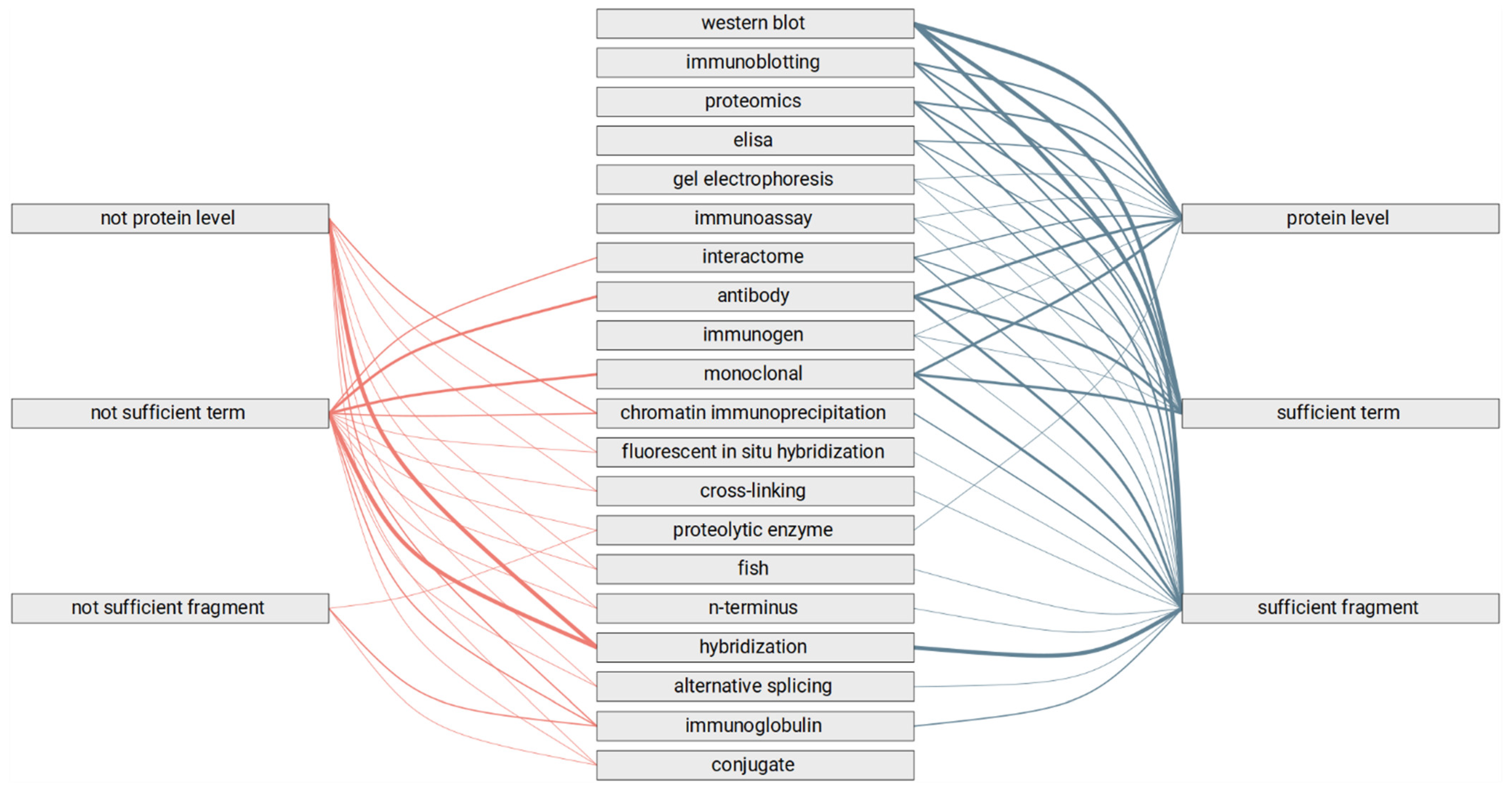
- Does the text fragment allow us to judge the category of the experimental method?
- Are highlighted terms sufficient to judge the category of experimental method?
- Is the specific level of the study mentioned in the text fragment?
3. Results and Discussion
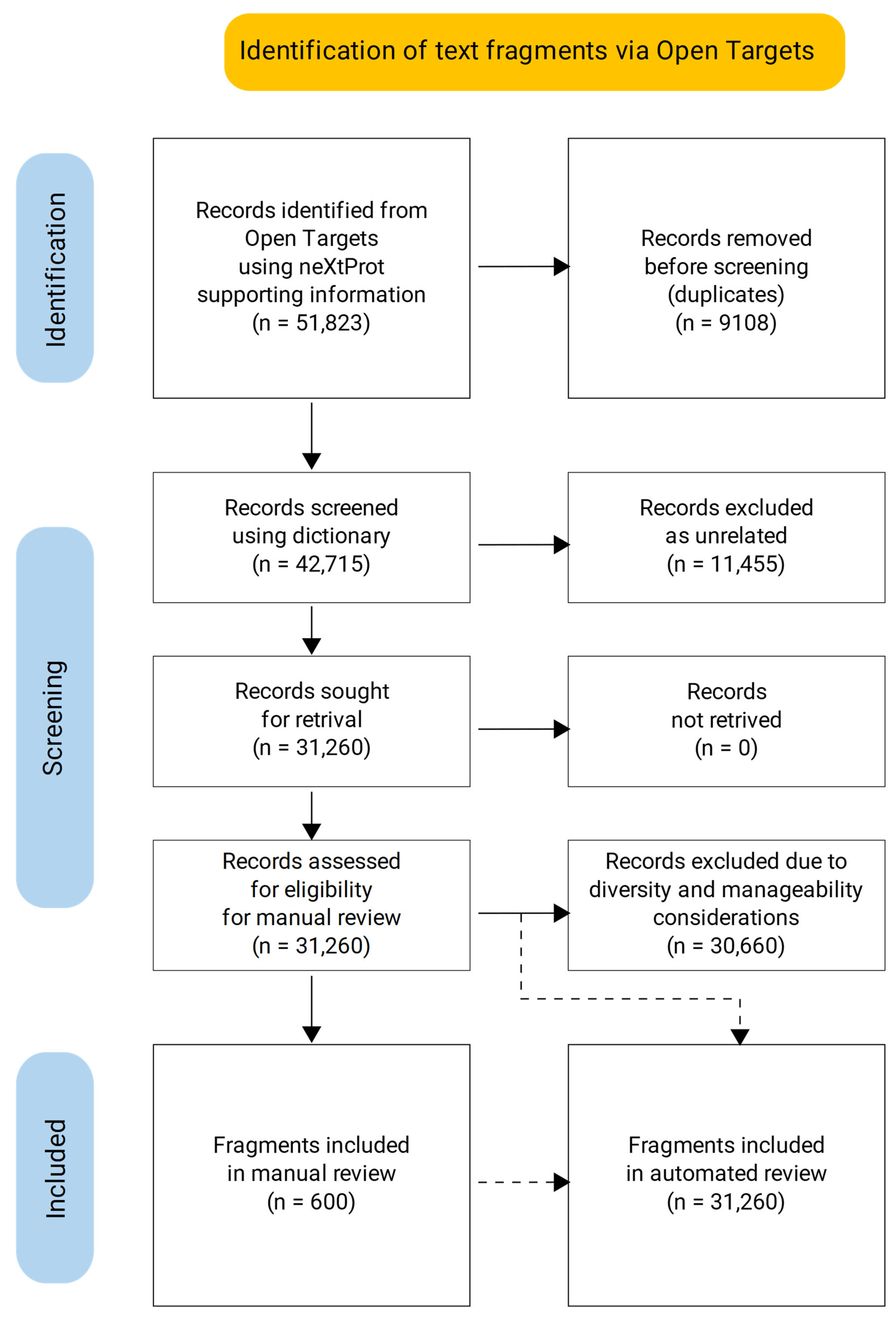
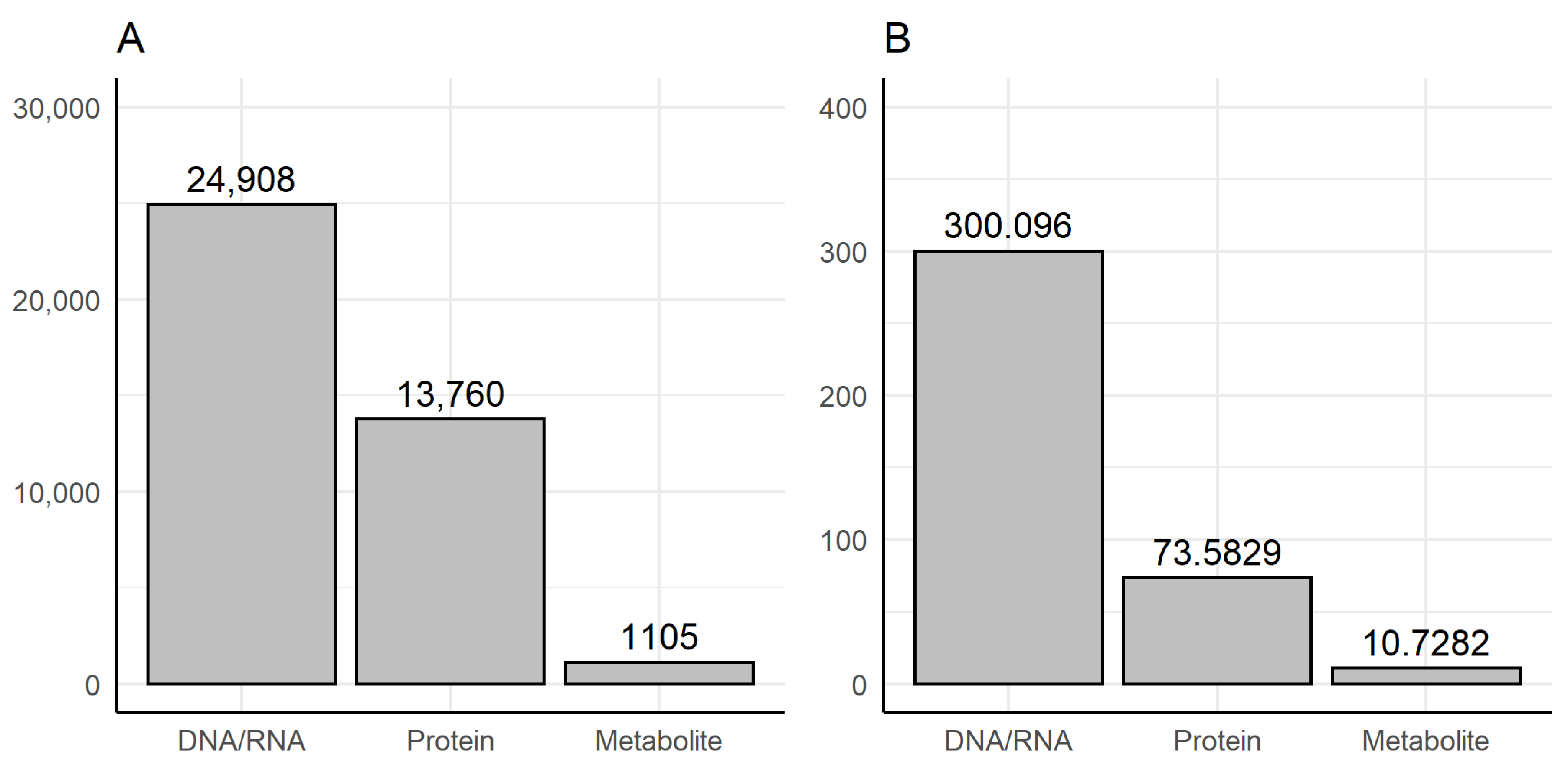
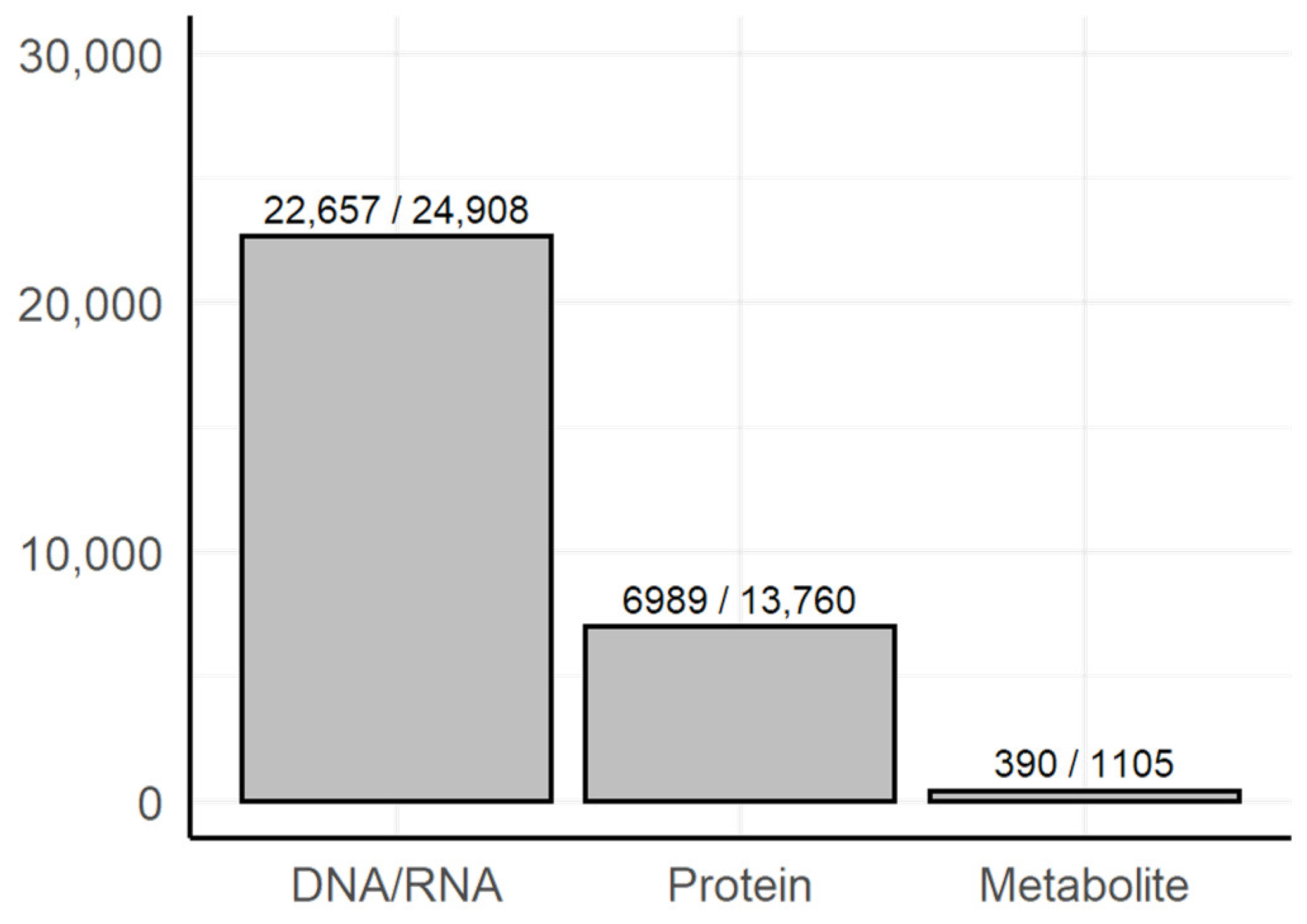
3.1. Dictionary-Based Screening
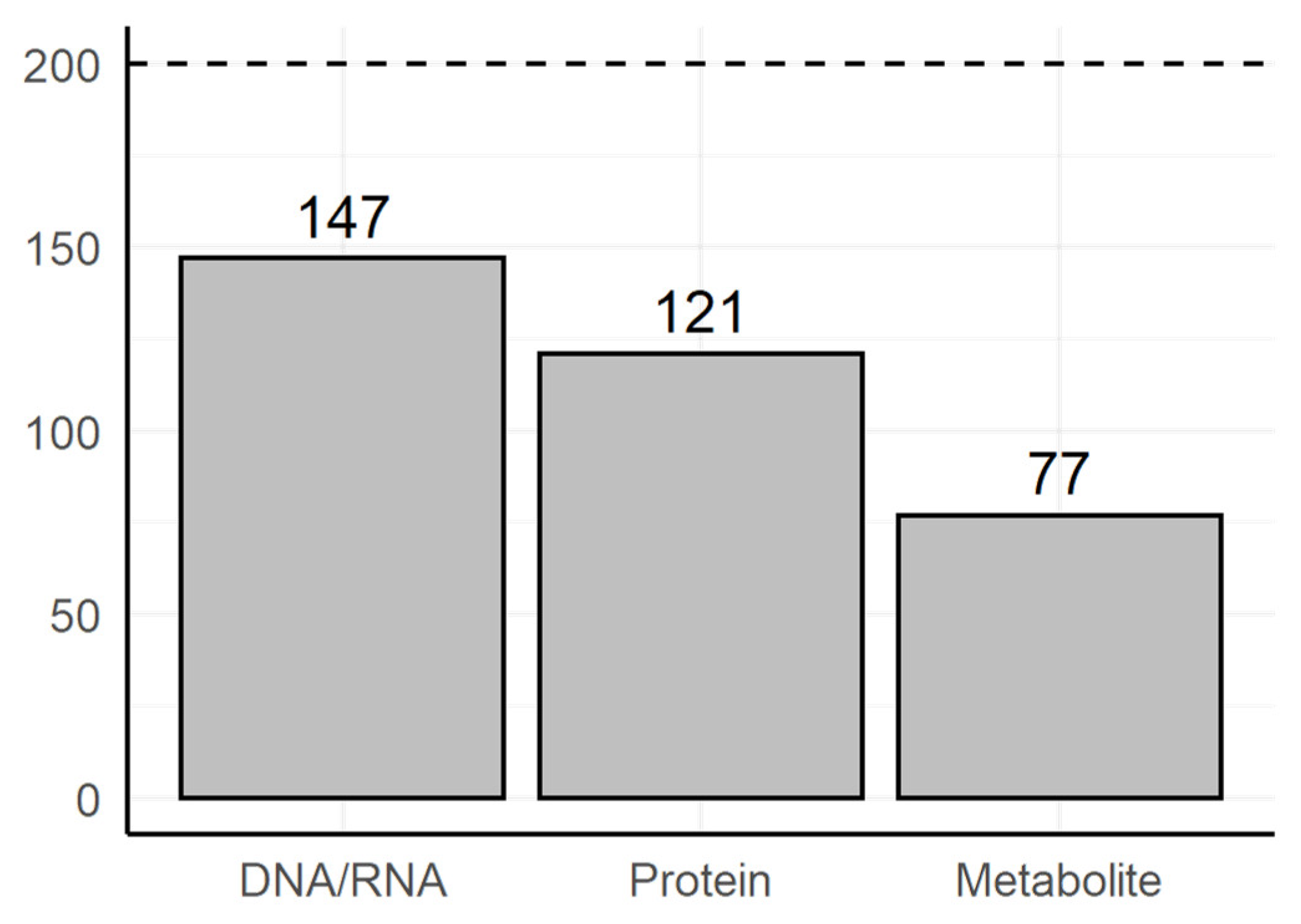
3.2. Manual Review of the Selected Text Fragments
- Does the text fragment allow us to judge the category of the experimental method?
- Are highlighted terms sufficient to judge the category of experimental method?
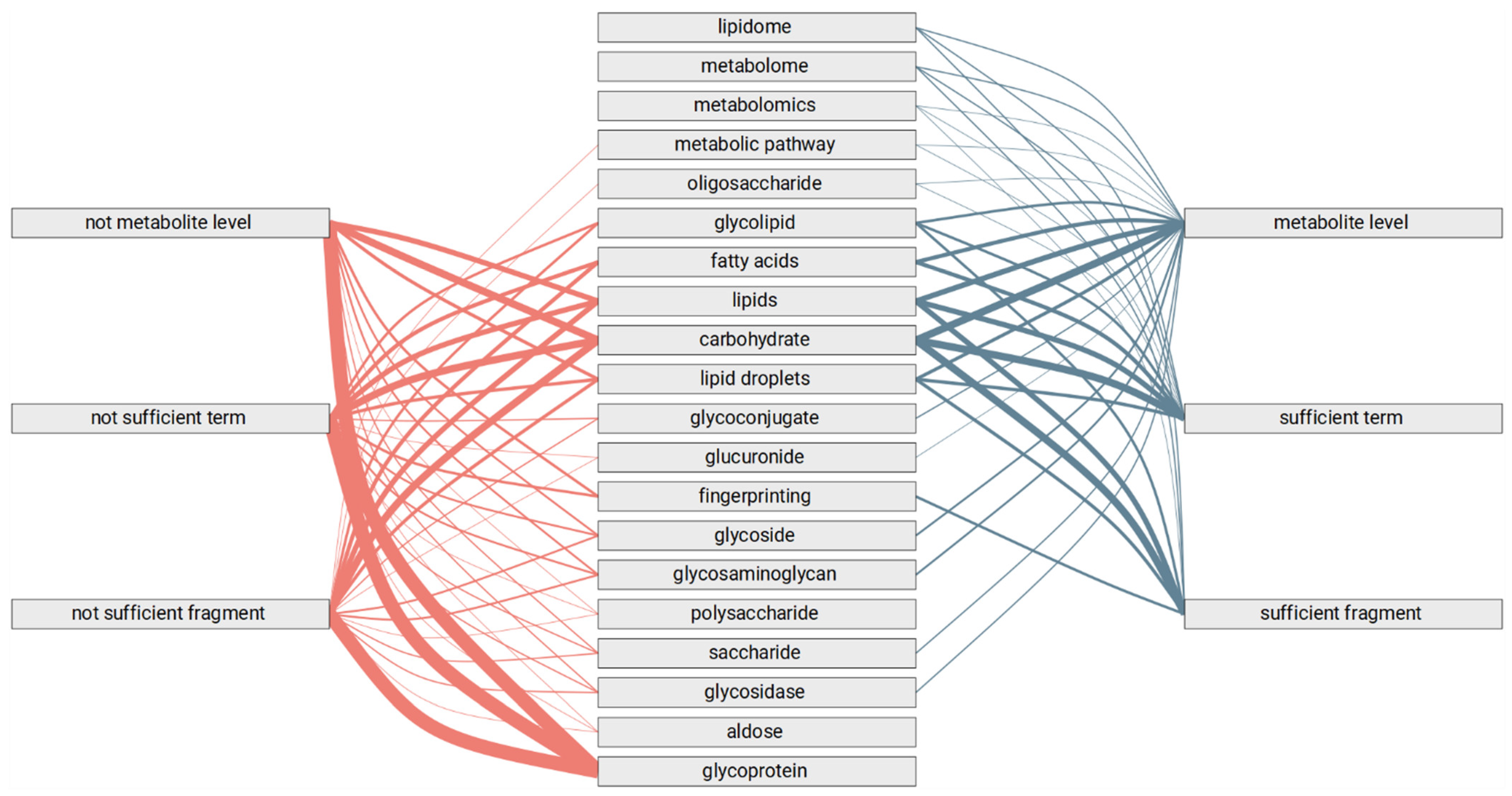
3.3. Automated Review of the Text Fragments
- Methods of genomics and transcriptomics are still the most accessible and mature ones. They allow monitoring of the molecules both in high-throughput and targeted modes. Thus, such methods are the most often used.
- The number of biological macromolecules associated with diseases via metabolites is limited (the macromolecule should be principally related to some metabolite and studied enough for this relationship to be known).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Imming, P.; Sinning, C.; Meyer, A. Drugs, Their Targets and the Nature and Number of Drug Targets. Nat. Rev. Drug Dis. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Ackloo, S.; Al Chawaf, A.; Al-Lazikani, B.; Antolin, A.; Baell, J.B.; Beck, H.; Beedie, S.; Betz, U.A.K.; Bezerra, G.A.; et al. Target 2035--Update on the Quest for a Probe for Every Protein. RSC Med. Chem. 2022, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting Systematic Drug-Target Identification and Prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, H.; Yang, H.; Li, M.; Xie, Z.; Li, W. Computational Resources Associating Diseases with Genotypes, Phenotypes and Exposures. Brief. Bioinform. 2019, 20, 2098–2115. [Google Scholar] [CrossRef]
- Zheng, C.J.; Han, L.Y.; Yap, C.W.; Ji, Z.L.; Cao, Z.W.; Chen, Y.Z. Therapeutic Targets: Progress of Their Exploration and Investigation of Their Characteristics. Pharmacol. Rev. 2006, 58, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Grant, M.J.; Booth, A. A Typology of Reviews: An Analysis of 14 Review Types and Associated Methodologies. Health Info. Libr. J. 2009, 26, 91–108. [Google Scholar] [CrossRef]
- Cooper, I.D. What Is a “Mapping Study?”. J. Med. Libr. Assoc. JMLA 2016, 104, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Pineau, C.; Packer, N.H.; Cristea, I.M.; Lindskog, C.; Weintraub, S.T.; Orchard, S.; Roehrl, M.H.A.; et al. The 2022 Report on the Human Proteome from the HUPO Human Proteome Project. J. Proteome Res. 2022, 22, 1024–1042. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. Rev. Panam. Salud Publica/Pan. Am. J. Public Health 2022, 46, 105906. [Google Scholar] [CrossRef]
- Foster, E.D.; Deardorff, A. Open Science Framework (OSF). J. Med. Libr. Assoc. 2017, 105, 203. [Google Scholar] [CrossRef] [Green Version]
- Zahn-Zabal, M.; Michel, P.A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; De Laval, V.R.; et al. The NeXtProt Knowledgebase in 2020: Data, Tools and Usability Improvements. Nucleic Acids Res. 2020, 48, D328–D334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuda, J.; Bowater, R.P.; Fojta, M.; Gauglitz, G.; Glatz, Z.; Hapala, I.; Havliš, J.; Kilar, F.; Kilar, A.; Malinovská, L.; et al. Terminology of Bioanalytical Methods (IUPAC Recommendations 2018). Pure Appl. Chem. 2018, 90, 1121–1198. [Google Scholar] [CrossRef] [Green Version]
- Pogodin, P.V.; Ilgisonis, E.V.; Tarasova, O.A.; Kiseleva, O.I.; Filimonov, D.A.; Ponomarenko, E.A. Tcstf: Tool for Categorization of Short Text Fragments. In Proceedings of the XXXVIII Symposium of Bioinformatics and Computer-Aided Drug Discovery, Online, 24–26 May 2022; p. 51. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Temple Lang, D. RCurl: General Network (HTTP/FTP/...) Client Interface for R, Version 1.98–1.12. 2022. Available online: https://cran.r-project.org/web/packages/RCurl/index.html (accessed on 9 April 2023).
- Ooms, J. The Jsonlite Package: A Practical and Consistent Mapping Between JSON Data and R Objects. arXiv 2014. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef] [Green Version]
- Ilgisonis, E.V.; Pogodin, P.V.; Kiseleva, O.I.; Tarbeeva, S.N.; Ponomarenko, E.A. Evolution of Protein Functional Annotation: Text Mining Study. J. Pers. Med. 2022, 12, 479. [Google Scholar] [CrossRef] [PubMed]
- Wolters, M.A. A Genetic Algorithm for Selection of Fixed-Size Subsets with Application to Design Problems. J. Stat. Softw. 2015, 68, 1–18. [Google Scholar] [CrossRef]
- Benoit, K.; Watanabe, K.; Wang, H.; Nulty, P.; Obeng, A.; Müller, S.; Matsuo, A. Quanteda: An R Package for the Quantitative Analysis of Textual Data. J. Open Source Softw. 2018, 3, 774. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Caret: Classification and Regression Training; Version 6.0-94; Astrophysics Source Code Library: Harvard, MA, USA, 2022. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 9783319242774. [Google Scholar]
- Iannone, R. DiagrammeR: Graph/Network Visualization, Version 1.0.9. 2022. Available online: https://cran.r-project.org/web/packages/DiagrammeR/DiagrammeR.pdf (accessed on 9 April 2023).
- Bartlett, J.M.S.; Stirling, D. A Short History of the Polymerase Chain Reaction. Methods Mol. Biol. 2003, 226, 3–6. [Google Scholar] [CrossRef]
- Westermeier, R.; Marouga, R. Protein Detection Methods in Proteomics Research. Biosci. Rep. 2005, 25, 19–32. [Google Scholar] [CrossRef]
- Dzieciatkowska, M.; Hill, R.; Hansen, K.C. GeLC-MS/MS Analysis of Complex Protein Mixtures. Methods Mol. Biol. 2014, 1156, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motone, K.; Nivala, J. Not If but When Nanopore Protein Sequencing Meets Single-Cell Proteomics. Nat. Methods 2023, 20, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Seydel, C. Single-Cell Metabolomics Hits Its Stride. Nat. Methods 2021, 18, 1452–1456. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of the Text Fragments in TCSTF | Biological Entity | Text Fragment | Category |
|---|---|---|---|
| 72 | FTL | In our patients with undetectable serum ferritin levels, idiopathic generalized seizures, and atypical RLS, we screened all exons encoding the FTL gene for neuroferritinopathy-associated mutations. As a result, we detected a G > T nucleotide substitution (G310T) in exon 3 (Figure 1A). | DNA/RNA level |
| 1 | DOCK8 | Combined immunodeficiency associated with DOCK8 mutations. | DNA/RNA level |
| 3 | IL23R | A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. | DNA/RNA level |
| 153 | GLI2 | We describe three novel heterozygous frameshift or nonsense GLI2 mutations, predicting truncated proteins lacking the activator domain associated with IGHD or combined pituitary hormone deficiency and ectopic posterior pituitary lobe without HPE. In addition, the GLI2 coding region of patients with isolated GH deficiency (IGHD) or combined pituitary hormone deficiency was amplified by PCR using intronic primers and sequenced. | DNA/RNA level |
| 90 | IDH2 | Subsequent studies revealed that IDH mutations were extremely rare in primary (de novo) glioblastomas but were common in recurrent glioblastomas developing secondary to low-grade tumors, which frequently have IDH mutations [5]. These findings suggested that IDH mutations are an early event in gliomagenesis and persist during the progression to recurrent glioblastomas. In addition, somatic heterozygous IDH1 or IDH2 mutations have frequently been detected in glioma/glioblastomas by genome-wide mutation searches [3,4]. | DNA/RNA level |
| 184 | SRPK1 | Lysates were separated by SDS-PAGE, transferred to nitrocellulose, and subjected to Western blot analysis using anti-SRPK1 (A), anti-SRPK2 (B), anti-topoisomerase I: arthritis foundation/CDC reference sera (C), and anti-cdc2 (CDK1) (mouse monoclonal) (D). | Protein level |
| 152 | Spexin | Spexin is a Novel Human Peptide that Reduces Adipocyte Uptake of Long Chain Fatty Acids and Causes Weight Loss in Rodents with Diet-induced Obesity Spexin is a novel human peptide that reduces adipocyte uptake of long-chain fatty acids and causes weight loss in rodents with diet-induced obesity. Spexin is a novel hormone involved in weight regulation, potentially for obesity therapy. A commercial immunoassay allowed us to examine possible relationships between circulating levels of Spexin and those of known obesity-related adipokines in human sera. | Protein level |
| 123 | GGA2 | As recent studies have shown GGA1 and GGA3 protein level alterations in postmortem samples of AD patients, we also compared the expression of GGA2 in 26 temporal lobe samples obtained from control and AD patients. Some AD patients showed altered GGA2 levels compared with matched controls. (D) GGA2 levels were analyzed by Western blot in postmortem temporal lobe samples of AD and controlled patients (MADRC). | Protein level |
| 97 | NBS1 | To test whether NBS1 indeed interacts with the mTOR/Rictor/SIN1 complex, co-immunoprecipitation assays using extracts from a lung cancer cell line H1299 were used. | Protein level |
| 43 | prostatic acid phosphatase | In semen, proteolytic peptide fragments from prostatic acid phosphatase can form amyloid fibrils termed SEVI (semen-derived enhancer of viral infection). | Protein level |
| 43 | FIT2 | Miranda etAI-nonASCII- al4 found that mice with adiposeaI-nonASCII-A-nonASCII-A<90>specific FIT2 deficiency developed severe, progressive lipodystrophy with fatty liver, tissue macrophage infiltration, and insulin resistance, with few but abnormally large lipid droplets on histology. | Metabolite level |
| 19 | CCDC3 | Thus, we decided first to determine if CCDC3 could affect lipid metabolism in hepatic cancer cells by performing a metabolomics analysis. | Metabolite level |
| 108 | alpha-N-acetylgalactosaminidase | The degradation of blood group glycolipid A-6-2 (GalNAc(alpha1-->3) [Fuc alpha1-->2]Gal(beta1-->4)GlcNAc(beta1-->3)Gal(beta1-->4)Glc(beta1-->1’)C er, IV2-alpha-fucosyl-IV3-alpha-N-acetylgalactosaminylneolact otetraosylceramide), tritium-labeled in its ceramide moiety, was studied in situ, in skin fibroblast cultures from normal controls, from patients with defects of lysosomal alpha-N-acetylgalactosaminidase, and patients with other lysosomal storage diseases. | Metabolite level |
| 84 | NNMT | To address this problem, we employed an untargeted metabolomics approach18, where metabolomes from NNMT-OE and GFP-OE renal carcinoma (769P), ovarian cancer (OVCAR3), and melanoma (MUM2C) cells were comparatively analyzed by an HPLC-Q-TOF-MS system operating in the broad mass scanning mode (m/z range of 50aI-nonASCII-A-nonASCII-A-nonASCII-1200 Da). | Metabolite level |
| 167 | DDHD2 | In line with the function of DDHD2 in lipid metabolism and its role in the CNS, an abnormal lipid peak indicating accumulation of lipids was detected with cerebral magnetic resonance spectroscopy, which provides an applicable diagnostic biomarker that can distinguish the DDHD2 phenotype from other complex HSP phenotypes. | Metabolite level |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pogodin, P.V.; Kiseleva, O.I.; Ilgisonis, E.V. Identification of Potential Therapeutic Targets on the Level of DNA/mRNAs, Proteins and Metabolites: A Systematic Mapping Review of Scientific Texts’ Fragments from Open Targets. Curr. Issues Mol. Biol. 2023, 45, 3406-3418. https://doi.org/10.3390/cimb45040223
Pogodin PV, Kiseleva OI, Ilgisonis EV. Identification of Potential Therapeutic Targets on the Level of DNA/mRNAs, Proteins and Metabolites: A Systematic Mapping Review of Scientific Texts’ Fragments from Open Targets. Current Issues in Molecular Biology. 2023; 45(4):3406-3418. https://doi.org/10.3390/cimb45040223
Chicago/Turabian StylePogodin, Pavel V., Olga I. Kiseleva, and Ekaterina V. Ilgisonis. 2023. "Identification of Potential Therapeutic Targets on the Level of DNA/mRNAs, Proteins and Metabolites: A Systematic Mapping Review of Scientific Texts’ Fragments from Open Targets" Current Issues in Molecular Biology 45, no. 4: 3406-3418. https://doi.org/10.3390/cimb45040223