
The Current Status and Future Perspectives of Chimeric Antigen Receptor-Engineered T Cell Therapy for the Management of Patients with Endometrial Cancer

Abstract
:1. Introduction
2. Immunotherapy in the Treatment of Endometrial Cancer
2.1. The Interaction between the Female Endometrium and the Immune System
2.2. Rationale of CAR-T Cell Immunotherapy for the Management of Endometrial Cancer
3. The Molecular Schematics of CAR-T Cells
3.1. Molecular Components and Structure of CAR-T Cells
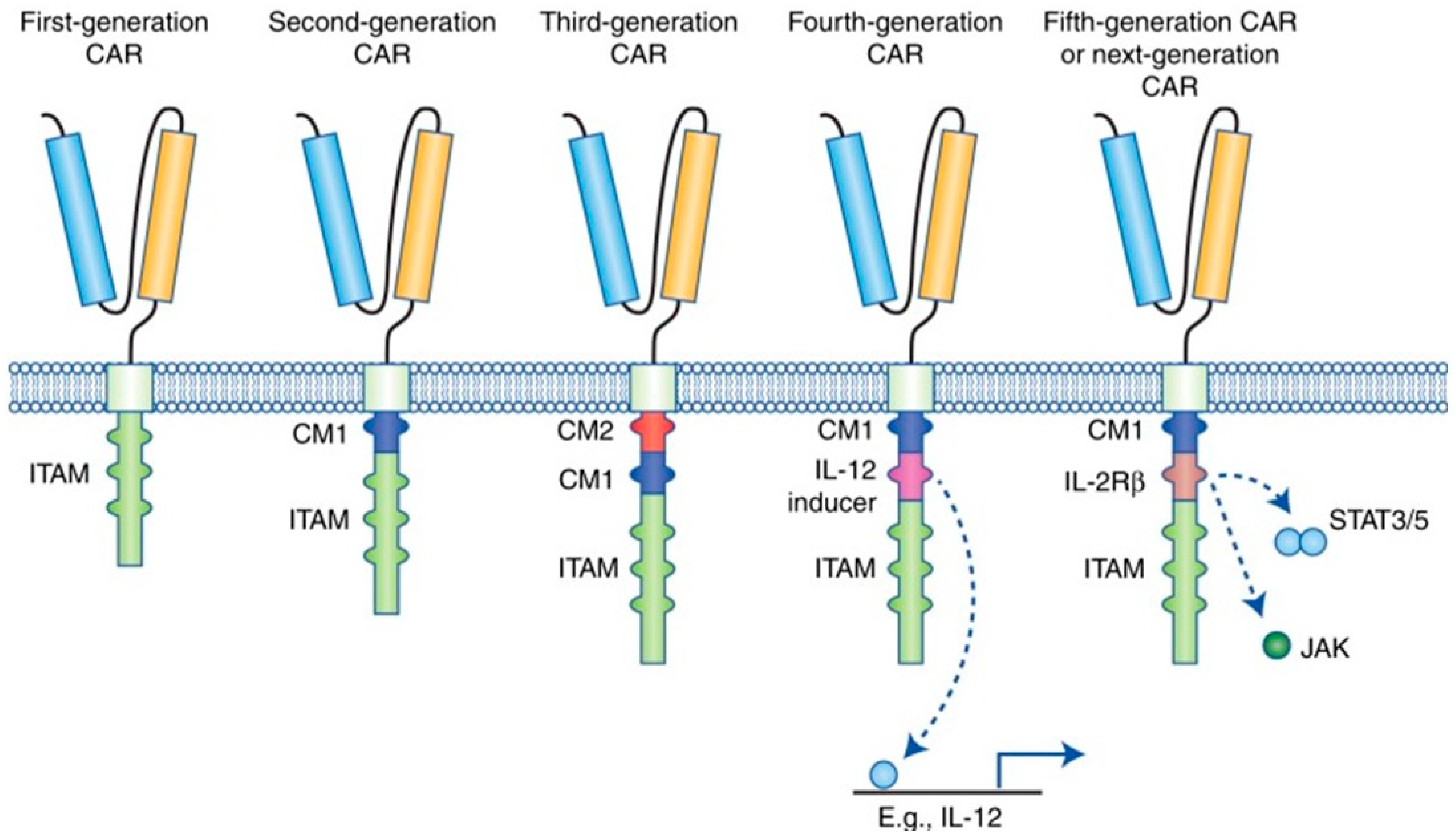
3.2. Molecular Generations of CAR-T Cells
3.2.1. First Generation of CAR-T Cells
3.2.2. Second Generation of CAR-T Cells
3.2.3. Third Generation of CAR-T Cells
3.2.4. Fourth Generation of CAR-T Cells
3.2.5. Fifth Generation/Next Generation of CAR-T Cells

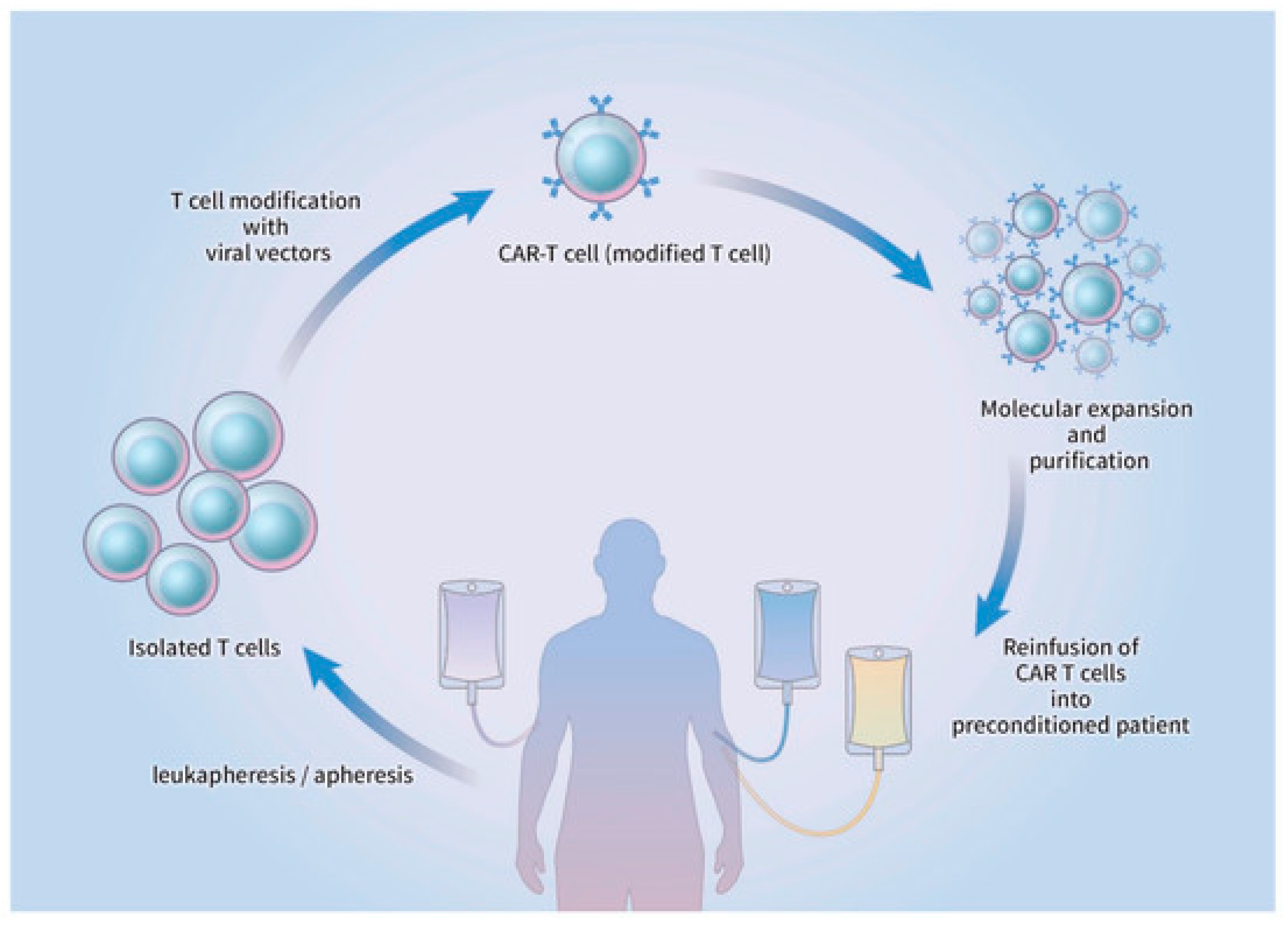
3.3. Preparation and Administration of CAR-T Cell Therapy
4. Molecular Targets for CAR-T Cells in Endometrial Immunotherapy
4.1. Suppressive Protein Phosphatase Type II A (PP2A)
4.2. Human Epidermal Growth Factor Receptor 2
4.3. Androgen Receptor (AR)
5. Current and Ongoing Clinical Trials for CAR-T Cell Immunotherapy in Endometrial Cancer
6. Future Directions for CAR-T Cell Immunotherapy for Endometrial Cancer
7. Adverse Events in CAR-T Cell Therapy
7.1. Cytokine Release Syndrome
7.2. Tumor Lysis Syndrome
7.3. Neurological Toxicities
7.4. “On-Target/Off-Tumor” Toxicity
7.5. Anaphylaxis and Graft-versus-Host Disease (GVHD)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.V.; Rose, P.G. Management Strategies for Recurrent Endometrial Cancer. Expert Rev. Anticancer Ther. 2018, 18, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Schepisi, G.; Casadei, C.; Toma, I.; Poti, G.; Iaia, M.L.; Farolfi, A.; Conteduca, V.; Lolli, C.; Ravaglia, G.; Brighi, N.; et al. Immunotherapy and Its Development for Gynecological (Ovarian, Endometrial and Cervical) Tumors: From Immune Checkpoint Inhibitors to Chimeric Antigen Receptor (CAR)-T Cell Therapy. Cancers 2021, 13, 840. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, A.; Sharma, P.; Poussin, M.; Boesteanu, A.C.; Minutolo, N.G.; Gitto, S.B.; Omran, D.K.; Robinson, M.K.; Adams, G.P.; Simpkins, F.; et al. CAR T Cells Targeting MISIIR for the Treatment of Ovarian Cancer and Other Gynecologic Malignancies. Mol. Ther. 2020, 28, 548–560. [Google Scholar] [CrossRef]
- Barbie, T.U.; Barbie, D.A.; MacLaughlin, D.T.; Maheswaran, S.; Donahoe, P.K. Mullerian Inhibiting Substance inhibits cervical cancer cell growth via a pathway involving p130 and p107. Proc. Natl. Acad. Sci. USA 2003, 100, 15601–15606. [Google Scholar] [CrossRef] [Green Version]
- Masiakos, P.T.; MacLaughlin, D.T.; Maheswaran, S.; Teixeira, J.; Fuller, A.F., Jr.; Shah, P.C.; Kehas, D.J.; Kenneally, M.K.; Dombkowski, D.M.; Ha, T.U.; et al. Human ovarian cancer, cell lines, and primary ascites cells express the human Mullerian inhibiting substance (MIS) type II receptor, bind, and are responsive to MIS. Clin. Cancer Res. 1999, 5, 3488–3499. [Google Scholar] [PubMed]
- Song, J.Y.; Chen, K.Y.; Kim, S.Y.; Kim, M.R.; Ryu, K.S.; Cha, J.H.; Kang, C.S.; MacLaughlin, D.T.; Kim, J.H. The expression of Müllerian inhibiting substance/anti-Müllerian hormone type II receptor protein and mRNA in benign, borderline and malignant ovarian neoplasia. Int. J. Oncol. 2009, 34, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderstraeten, A.; Tuyaerts, S.; Amant, F. The immune system in the normal endometrium and implications for endometrial cancer development. J. Reprod. Immunol. 2015, 109, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.; Moore, M.; Fox, H. Expression of MHC products and leucocyte differentiation antigens in gynaecological neoplasms: An immunohistological analysis of the tumour cells and infiltrating leucocytes. Br. J. Cancer 1985, 52, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiel, D.O.; Ghosh, M.; Fahey, J.V.; Guyre, P.M.; Wira, C.R. Human uterine epithelial cell secretions regulate dendritic cell differentiation and responses to TLR ligands. J. Leukoc. Biol. 2010, 88, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wira, C.R.; Fahey, J.V.; Ghosh, M.; Patel, M.V.; Hickey, D.K.; Ochiel, D.O. Sex hormone regulation of innate immunity in the female reproductive tract: The role of epithelial cells in balancing reproductive potential with protection against sexually transmitted pathogens. Am. J. Reprod. Immunol. 2010, 63, 544–565. [Google Scholar] [CrossRef] [Green Version]
- Wira, C.R.; Grant-Tschudy, K.S.; Crane-Godreau, M.A. Epithelial cells in the female reproductive tract: A central role as sentinels of immune protection. Am. J. Reprod. Immunol. 2005, 53, 65–76. [Google Scholar] [CrossRef]
- Longoria, T.C.; Eskander, R.N. Immunotherapy in endometrial cancer—An evolving therapeutic paradigm. Gynecol. Oncol. Res. Pract. 2015, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.J.; Jones, D.B. An immunohistochemical study of lymphoid tissue in human endometrium. Int. J. Gynecol. Pathol. 1988, 7, 225–235. [Google Scholar] [CrossRef]
- Yeaman, G.R.; Collins, J.E.; Fanger, M.W.; Wira, C.R.; Lydyard, P.M. CD8+ T cells in human uterine endometrial lymphoid aggregates: Evidence for accumulation of cells by trafficking. Immunology 2001, 102, 434–440. [Google Scholar] [CrossRef]
- Yeaman, G.R.; Guyre, P.M.; Fanger, M.W.; Collins, J.E.; White, H.D.; Rathbun, W.; Orndorff, K.A.; Gonzalez, J.; Stern, J.E.; Wira, C.R. Unique CD8+ T cell-rich lymphoid aggregates in human uterine endometrium. J. Leukoc. Biol. 1997, 61, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Meaningful Response to TILs in NSCLC. Cancer Discov. 2021, 11, 2117–2118. [CrossRef] [PubMed]
- Sadelain, M. CD19 CAR T Cells. Cell 2017, 171, 1471. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.D.; Kim, T.J. Chimeric Antigen Receptor-Engineered T Cell Therapy for the Management of Patients with Metastatic Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 640. [Google Scholar] [CrossRef]
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Cao, L.; Xie, J.; Shi, N.; Zhang, Z.; Luo, Z.; Yue, D.; Zhang, Z.; Wang, L.; Han, W.; et al. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: A meta-analysis. Oncotarget 2015, 6, 33961–33971. [Google Scholar] [CrossRef] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Hillerdal, V.; Essand, M. Chimeric antigen receptor-engineered T cells for the treatment of metastatic prostate cancer. BioDrugs 2015, 29, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Plotkin, J.; Jakka, G.; Stewart-Jones, G.; Riviere, I.; Merghoub, T.; Wolchok, J.; Renner, C.; Sadelain, M. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Mol. Ther. Oncolytics 2016, 3, 16023. [Google Scholar] [CrossRef] [Green Version]
- Shank, B.R.; Do, B.; Sevin, A.; Chen, S.E.; Neelapu, S.S.; Horowitz, S.B. Chimeric Antigen Receptor T Cells in Hematologic Malignancies. Pharmacotherapy 2017, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor- and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landoni, E.; Savoldo, B. Treating hematological malignancies with cell therapy: Where are we now? Expert Opin. Biol. Ther. 2018, 18, 65–75. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.L.; Xiu, J.; Chatterjee-Paer, S.; Buckley de Meritens, A.; Burke, W.M.; Tergas, A.I.; Wright, J.D.; Hou, J.Y. Distinct molecular landscapes between endometrioid and nonendometrioid uterine carcinomas. Int. J. Cancer 2017, 140, 1396–1404. [Google Scholar] [CrossRef] [Green Version]
- Remmerie, M.; Janssens, V. PP2A: A Promising Biomarker and Therapeutic Target in Endometrial Cancer. Front. Oncol. 2019, 9, 462. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.S.; Wang, H.; Maggio, D.; Kovach, J.S.; Zhang, Q.; Song, Q.; Marincola, F.M.; Heiss, J.D.; Gilbert, M.R.; Lu, R.; et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 2018, 9, 2126. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Wang, H.; Medina, R.; Zhang, Q.; Xu, C.; Indig, I.H.; Zhou, J.; Song, Q.; Dmitriev, P.; Sun, M.Y.; et al. Inhibition of PP2A with LB-100 Enhances Efficacy of CAR-T Cell Therapy Against Glioblastoma. Cancers 2020, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.; Zanagnolo, V.; Ramirez, N.; Cohn, D.E.; Kelbick, N.; Copeland, L.; Maxwell, G.L.; Fowler, J.M. HER-2 is an independent prognostic factor in endometrial cancer: Association with outcome in a large cohort of surgically staged patients. J. Clin. Oncol. 2006, 24, 2376–2385. [Google Scholar] [CrossRef]
- Black, J.D.; Lopez, S.; Cocco, E.; Bellone, S.; Altwerger, G.; Schwab, C.L.; English, D.P.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; et al. PIK3CA oncogenic mutations represent a major mechanism of resistance to trastuzumab in HER2/neu overexpressing uterine serous carcinomas. Br. J. Cancer 2015, 113, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, D.P.; Bellone, S.; Schwab, C.L.; Bortolomai, I.; Bonazzoli, E.; Cocco, E.; Buza, N.; Hui, P.; Lopez, S.; Ratner, E.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against primary HER2 overexpressing uterine serous carcinoma in vitro and in vivo. Cancer Med. 2014, 3, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, R.; Lopez, S.; Bellone, S.; Cocco, E.; Schwab, C.L.; Black, J.D.; Centritto, F.; Zhu, L.; Bonazzoli, E.; Buza, N.; et al. T-DM1, a novel antibody-drug conjugate, is highly effective against uterine and ovarian carcinosarcomas overexpressing HER2. Clin. Exp. Metastasis 2015, 32, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Tangen, I.L.; Onyango, T.B.; Kopperud, R.; Berg, A.; Halle, M.K.; Øyan, A.M.; Werner, H.M.; Trovik, J.; Kalland, K.H.; Salvesen, H.B.; et al. Androgen receptor as potential therapeutic target in metastatic endometrial cancer. Oncotarget 2016, 7, 49289–49298. [Google Scholar] [CrossRef] [Green Version]
- Reiss, K.A.; Yuan, Y.; Barton, D.; Cushing, D.; Ronczka, A.; Klichinsky, M.; Dees, E.C. A phase 1, first-in-human (FIH) study of adenovirally transduced autologous macrophages engineered to contain an anti-HER2 chimeric antigen receptor (CAR) in subjects with HER2 overexpressing solid tumors. J. Clin. Oncol. 2022, 40, TPS668. [Google Scholar] [CrossRef]
- Zhu, B.; Jia, Q.; Chen, R.; Chen, G.; Zhao, L.; Palmer, N.; Xiang, D.; Chen, F.; Duan, Y.; Wang, H. 568P First-in-human anti-ALPP CAR-T cell immunotherapy for ovarian and endometrial cancer. Ann. Oncol. 2022, 33, S807. [Google Scholar] [CrossRef]
- Hospital, C.P.G. Treatment of Relapsed and/or Chemotherapy Refractory Advanced Malignancies by CART-Meso. 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02580747 (accessed on 20 March 2023).
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Priceman, S.J.; Gerdts, E.A.; Tilakawardane, D.; Kennewick, K.T.; Murad, J.P.; Park, A.K.; Jeang, B.; Yamaguchi, Y.; Yang, X.; Urak, R.; et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology 2018, 7, e1380764. [Google Scholar] [CrossRef]
- Esensten, J.H.; Bluestone, J.A.; Lim, W.A. Engineering Therapeutic T Cells: From Synthetic Biology to Clinical Trials. Annu. Rev. Pathol. 2017, 12, 305–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.; Mazagova, M.; Choi, S.H.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed. Engl. 2016, 55, 7520–7524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.L.; June, C.H. Perspective: Assembly line immunotherapy. Nature 2013, 498, S17. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Jeyarajah, D.R.; Thistlethwaite, J.R., Jr. General aspects of cytokine-release syndrome: Timing and incidence of symptoms. Transpl. Proc. 1993, 25, 16–20. [Google Scholar]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Jhaveri, K.D.; Rosner, M.H. Chimeric Antigen Receptor T Cell Therapy and the Kidney: What the Nephrologist Needs to Know. Clin. J. Am. Soc. Nephrol. 2018, 13, 796–798. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wong, C.W.; Urak, R.; Mardiros, A.; Budde, L.E.; Chang, W.C.; Thomas, S.H.; Brown, C.E.; La Rosa, C.; Diamond, D.J.; et al. CMVpp65 Vaccine Enhances the Antitumor Efficacy of Adoptively Transferred CD19-Redirected CMV-Specific T Cells. Clin. Cancer Res. 2015, 21, 2993–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transpl. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Study Title | Clinical Phase | Identifier | Conditions | Primary Endpoints |
|---|---|---|---|---|
| First-in-human (FIH) study of adenovirally transduced autologous macrophages engineered to contain an anti-HER2 chimeric antigen receptor (CAR) in subjects with HER2 overexpressing tumors | Phase I | NCT04660929 | HER-2 positive Adenocarcinoma HER-2 positive Solid Tumors HER-2 Protein Overexpression HER-2 Gene Amplification | Assessment of the safety and tolerability of CT-0508 Frequency and severity of AEs Assessment of the feasibility of manufacturing CT-0508 |
| First-in-human anti-ALPP CAR-T cell immunotherapy for ovarian and endometrial cancer | Phase I/II | NCT04627740 | Endometrial Cancer Ovarian Cancer | Number of ALPP-positive participants with treatment-related AEs after infusion with anti-ALPP CAR-T cells |
| Treatment of Relapsed and/or Chemotherapy Refractory Advanced Malignancies by CART-meso | Phase I | NCT02580747 | Endometrial Cancer Ovarian Tumor Malignant Mesothelioma Pancreatic Cancer Triple Negative Breast Cancer Other Mesothelin Positive Tumors | Safety and feasibility of CAR-T meso cells Occurrence of AE |
| Grade | Management |
| 1 | Mild: Treated with supportive care such as antipyretic agents, antiemetic agents |
| 2 | Moderate: Requiring IV therapies or parenteral nutrition; some signs of organ dysfunction (i.e., grade 2 Cr or grade 3 LFTs) related to CRS and not attributable to any other condition; hospitalization for management of CRS-related symptoms, including fevers with associated neutropenia |
| 3 | More severe: Hospitalization is required for management of symptoms related to organ dysfunction, including grade 4 LFTs or grade 3 Cr associated with CRS and not attributable to any other conditions; this excludes management of fever or myalgias but includes hypotension treated with IV fluids or low-dose vasopressors, coagulopathy requiring FFP or cryoprecipitate, and hypoxia requiring supplemental O2 (nasal cannula O2, high-flow O2, CPAP, or BiPAP); patients admitted for management of suspected infection owing to fever and/or neutropenia might have grade 2 CRS |
| 4 | Life-threatening complications such as hypotension requiring high-dose vasopressors, hypoxia requiring mechanical ventilation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.-Y.; Kim, T.-J. The Current Status and Future Perspectives of Chimeric Antigen Receptor-Engineered T Cell Therapy for the Management of Patients with Endometrial Cancer. Curr. Issues Mol. Biol. 2023, 45, 3359-3374. https://doi.org/10.3390/cimb45040220
Choi J-Y, Kim T-J. The Current Status and Future Perspectives of Chimeric Antigen Receptor-Engineered T Cell Therapy for the Management of Patients with Endometrial Cancer. Current Issues in Molecular Biology. 2023; 45(4):3359-3374. https://doi.org/10.3390/cimb45040220
Chicago/Turabian StyleChoi, Ji-Young, and Tae-Jin Kim. 2023. "The Current Status and Future Perspectives of Chimeric Antigen Receptor-Engineered T Cell Therapy for the Management of Patients with Endometrial Cancer" Current Issues in Molecular Biology 45, no. 4: 3359-3374. https://doi.org/10.3390/cimb45040220