
Large-Scale Quantitative Proteomic Analysis during Different Stages of Somatic Embryogenesis in Larix olgensis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Sampling for Proteomic Analyses
2.2. Protein Extraction and Trypsin Digestion
2.3. TMT Labeling and High-pH Reversed-Phase Peptide Fractionation
2.4. High-Performance Liquid Chromatography (HPLC) Fractionation Using Easy Nano-Liquid Chromatography (nLC) and LC–MS/MS Analysis
2.5. Protein Identification and Quantitation
2.6. DEP Annotation and Functional Enrichment
2.7. RNA Extraction and RT-qPCR
3. Results
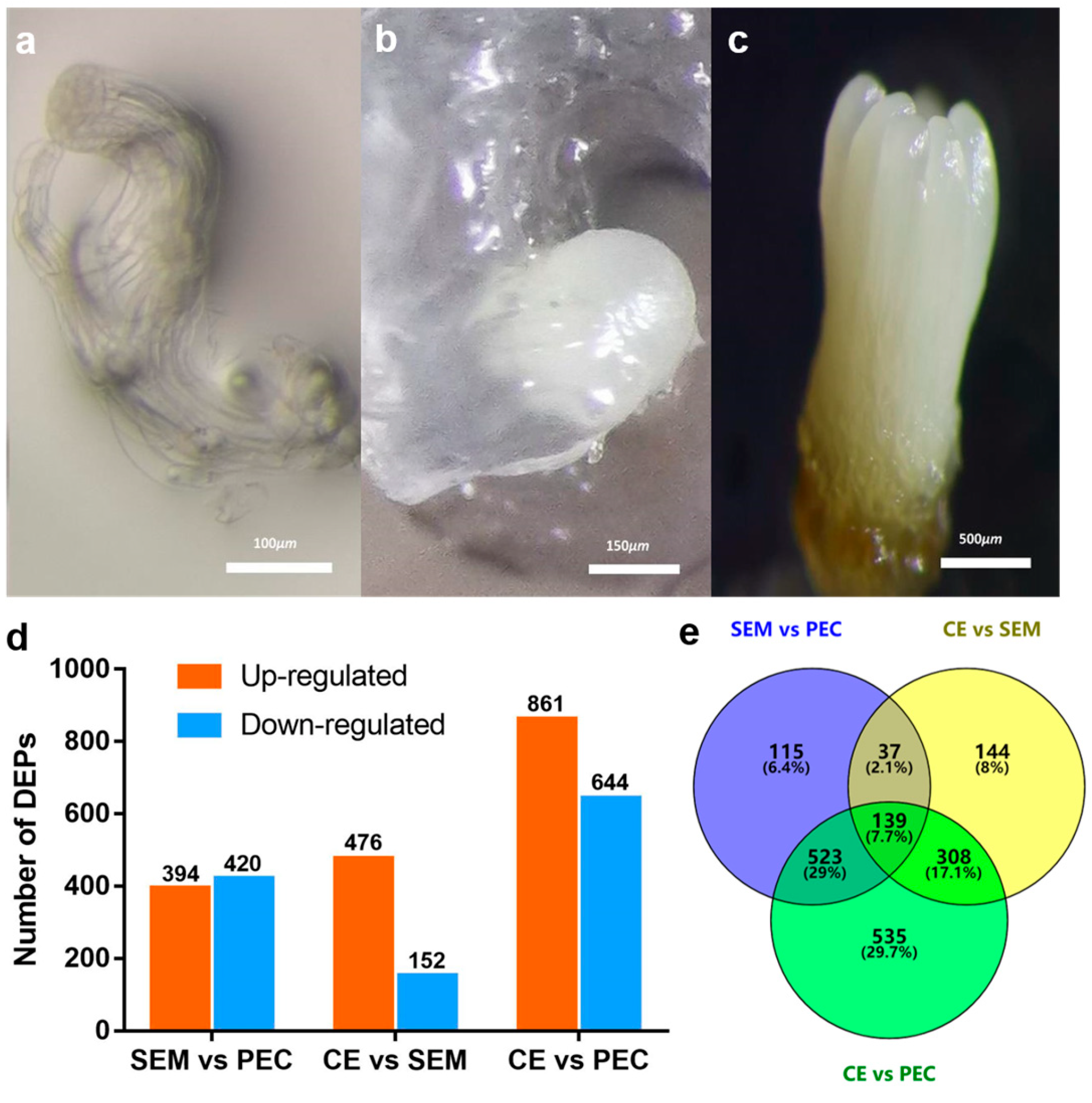
3.1. SE in L. olgensis
3.2. Identification of Proteins in Different Stages of SE via TMT-Based Quantitative Proteomics and DEP Annotation
3.3. Analysis of the Functions of the Major DEPs Involved in SE
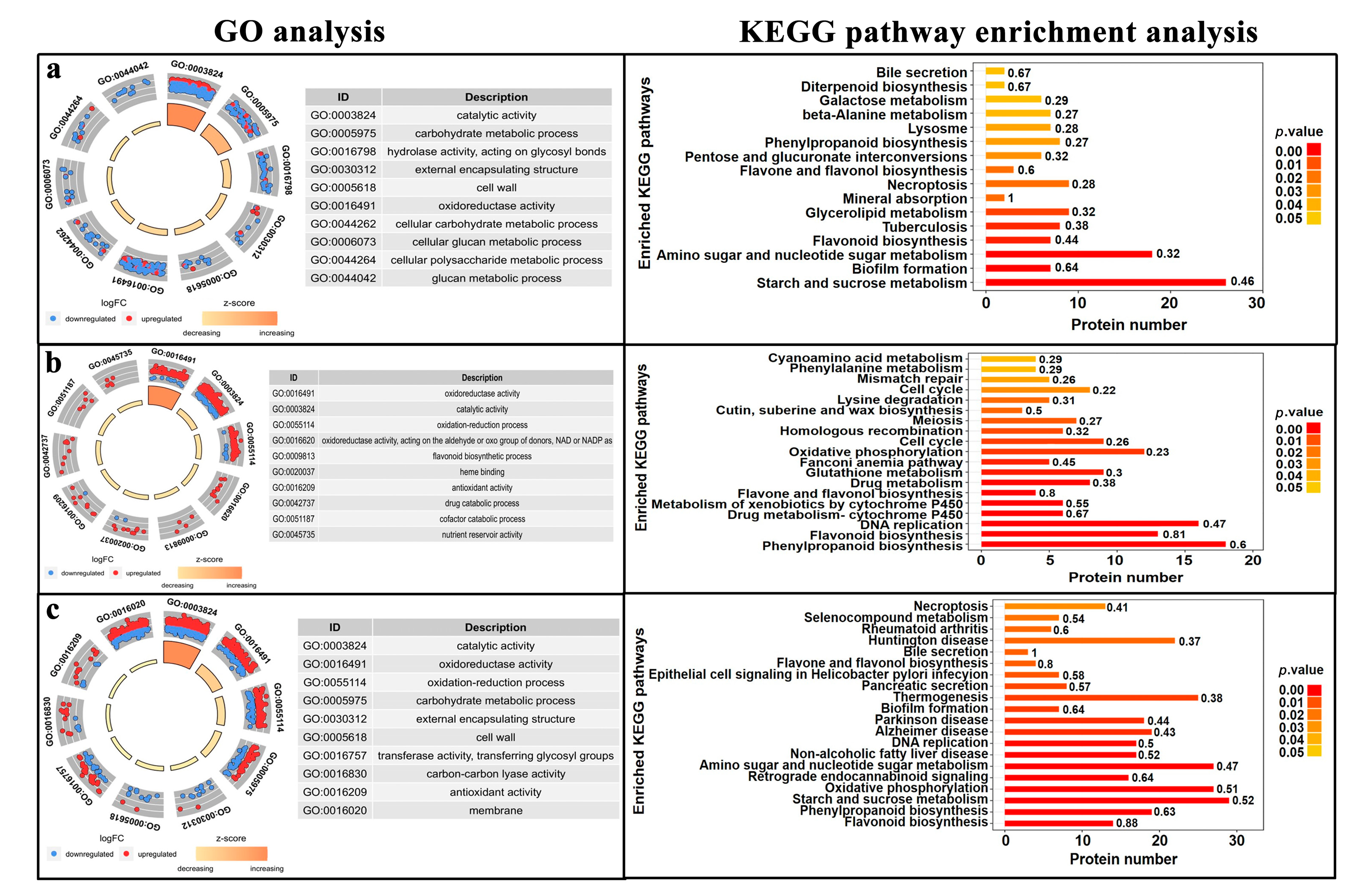
3.3.1. Gene Ontology (GO) Analysis of DEPs
3.3.2. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
3.3.3. Transcription Factors (TFs) Involved in SE
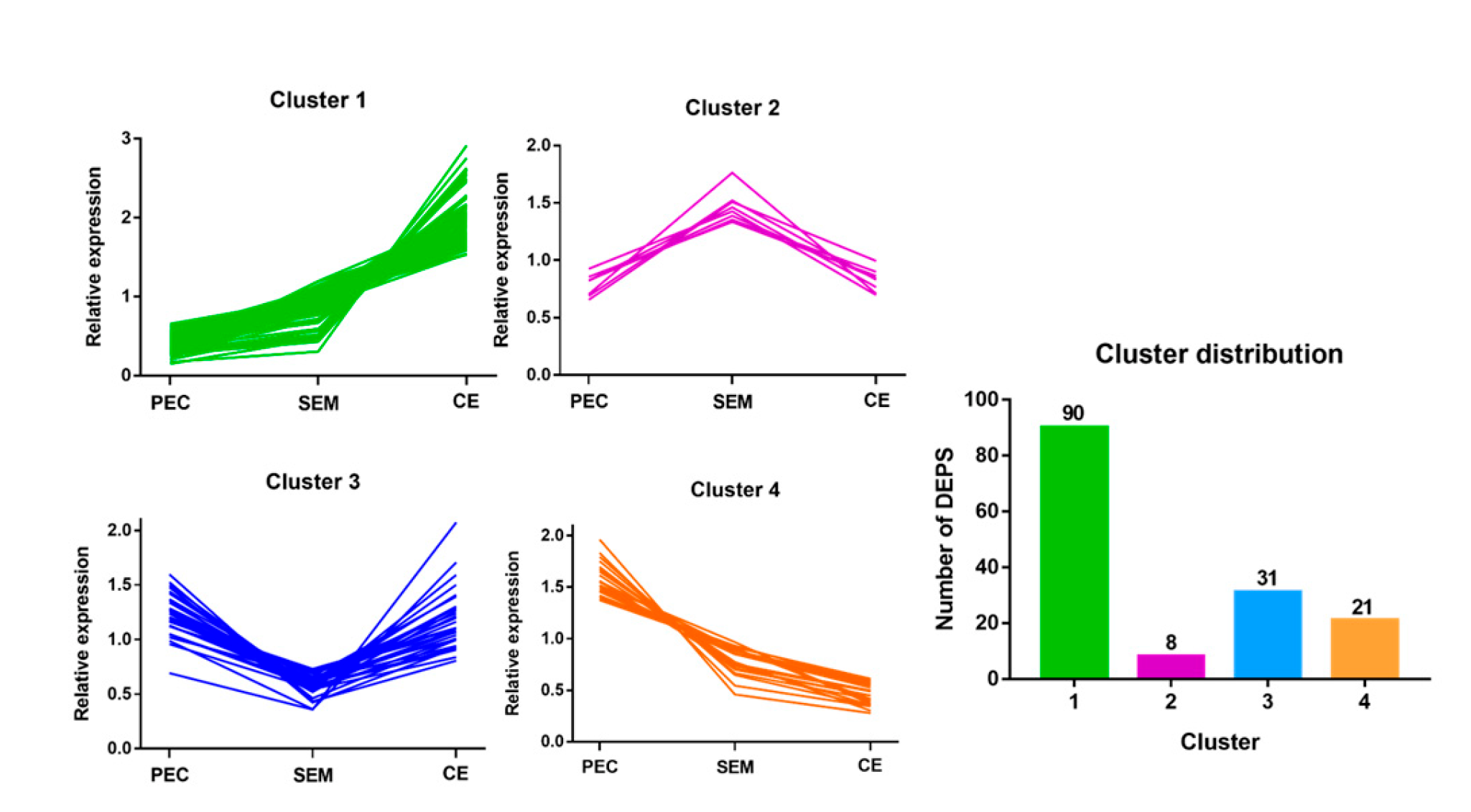
3.4. Hierarchical Cluster Analysis of DEPs at Different Stages of SE
3.5. Correlation Analysis of Transcriptome and Proteome Data
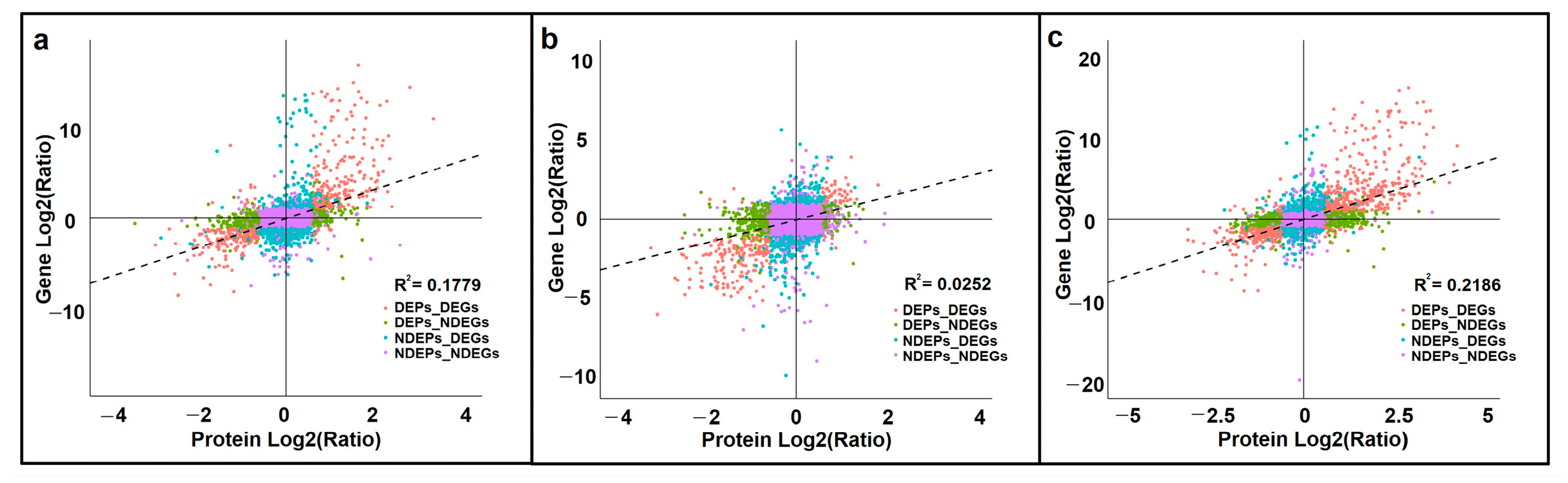
3.5.1. Correlations between Differentially Expressed Genes (DEGs) and DEPs
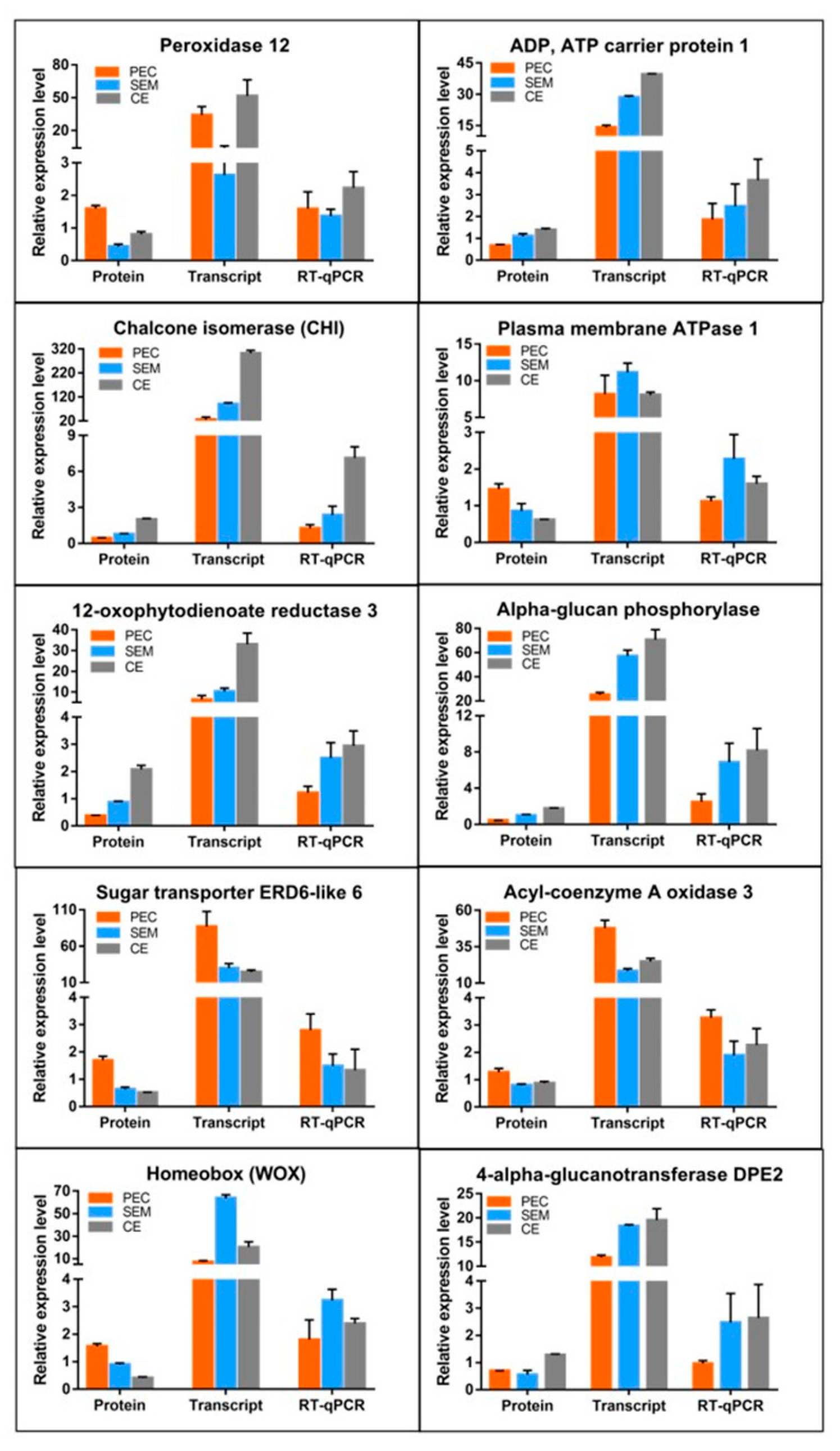
3.5.2. Analysis of the Relative Expression Levels of DEPs in the Proteome and DEGs in the Transcriptome by Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
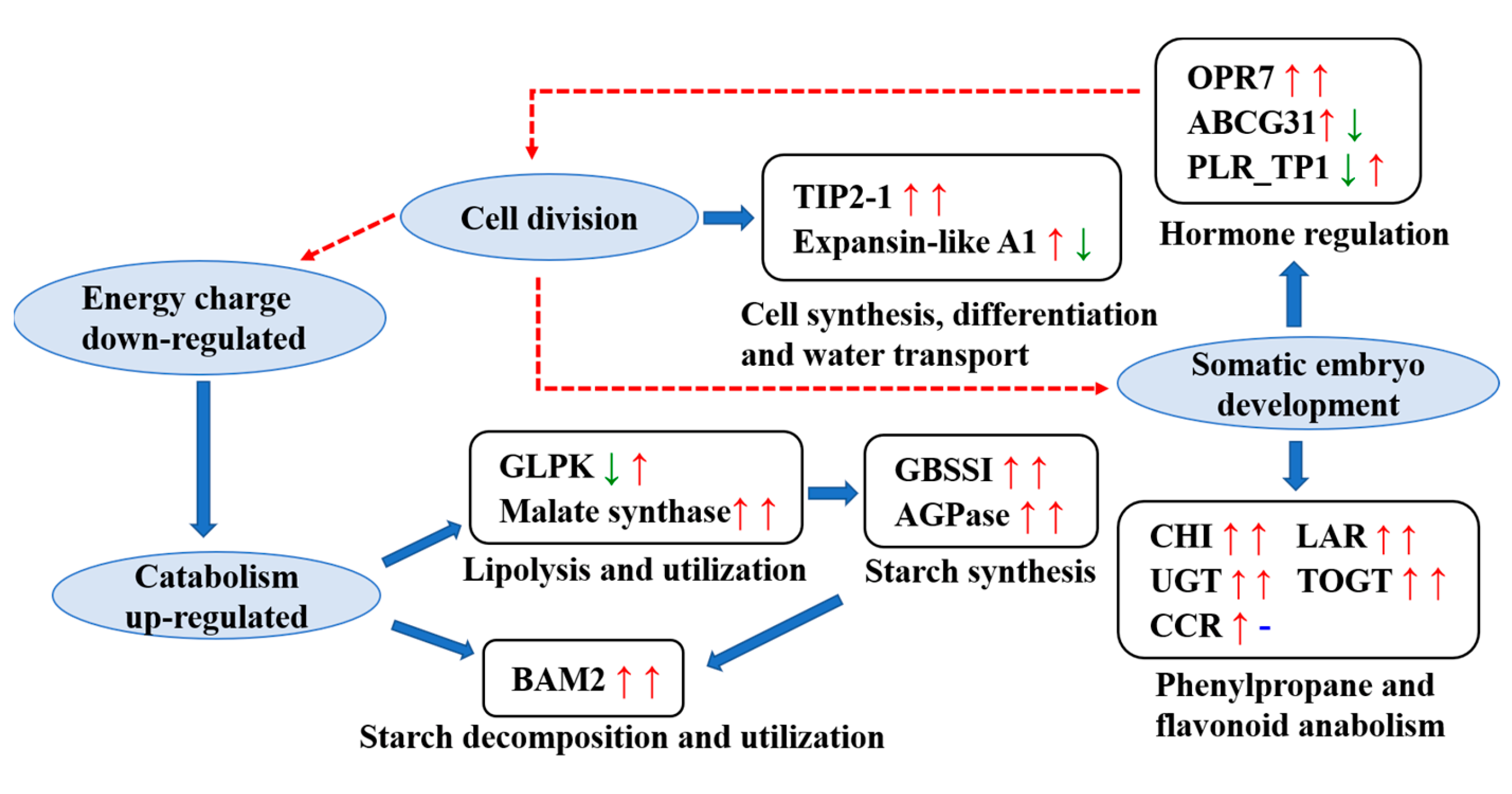
4. Discussion
4.1. DEPs Involved in the Glucose and Lipid Metabolism Pathways
4.2. Main DEPs in Secondary Metabolic Pathways Involved in Stress Resistance
4.3. Major Regulatory DEPs Involved in Phytohormone Signal Transduction and Cell Proliferation
4.4. TF-Related Proteins Involved in SE
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimmerman, J.L. Somatic embryogenesis: A model for early development in higher plants. Plant Cell 1993, 5, 1411–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, X. Regulation of somatic embryogenesis in higher plants. Crit. Rev. Plant Sci. 2010, 29, 36–57. [Google Scholar] [CrossRef]
- Guan, Y.; Li, S.G.; Fan, X.F.; Su, Z.H. Application of somatic embryogenesis in woody plants. Front. Plant Sci. 2016, 7, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.G.; Cheng, W.H.; Tian, W.G.; Li, Y.J.; Liu, F.; Xue, F.; Zhu, Q.H.; Sun, Y.Q.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2018, 96, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Shan, X.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Han, J.; Yuan, Y. iTRAQ-Based quantitative proteomic analysis of embryogenic and non-embryogenic calli derived from a maize (Zea mays L.) inbred line Y. Int. J. Mol. Sci. 2018, 19, 4004. [Google Scholar] [CrossRef] [Green Version]
- Teyssier, C.; Maury, S.; Beaufour, M.; Grondin, C.; Delaunay, A.; Le Metté, C.; Ader, K.; Cadene, M.; Label, P.; Lelu-Walter, M.A. In search of markers for somatic embryo maturation in hybrid larch (Larix × eurolepis): Global DNA methylation and proteomic analyses. Physiol. Plant. 2014, 150, 271–291. [Google Scholar] [CrossRef]
- Lippert, D.; Zhuang, J.; Ralph, S.; Ellis, D.E.; Gilbert, M.; Olafson, R.; Ritland, K.; Ellis, B.; Douglas, C.J.; Bohlmann, J. Proteome analysis of early somatic embryogenesis in Picea glauca. Proteomics 2005, 5, 461–473. [Google Scholar] [CrossRef]
- Fraga, H.P.F.; Vieira, L.N.; Heringer, A.S.; Puttkammer, C.C.; Silveira, V.; Guerra, M.P. DNA methylation and proteome profiles of Araucaria angustifolia (Bertol.) Kuntze embryogenic cultures as affected by plant growth regulators supplementation. Plant Cell Tiss. Organ. Cult. 2016, 125, 353–374. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, S.; Wang, J. Transcriptomic and proteomic analyses of embryogenic tissues in Picea balfouriana treated with 6-benzylaminopurine. Physiol. Plant. 2015, 154, 95–113. [Google Scholar] [CrossRef]
- Jing, D.; Zhang, J.; Xia, Y.; Kong, L.; Ouyang, F.; Zhang, S.; Zhang, H.; Wang, J. Proteomic analysis of stress-related proteins and metabolic pathways in Picea asperata somatic embryos during partial desiccation. Plant Biotechnol. J. 2017, 15, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Eliášová, K.; Konrádová, H.; Dobrev, P.I.; Motyka, V.; Lomenech, A.M.; Fischerová, L.; Lelu-Walter, M.A.; Vondráková, Z.; Teyssier, C. Desiccation as a post-maturation treatment helps complete maturation of Norway spruce somatic embryos: Carbohydrates, phytohormones and proteomic status. Front. Plant Sci. 2022, 13, 823617. [Google Scholar] [CrossRef] [PubMed]
- Gautier, F.; Eliášová, K.; Leplé, J.C.; Vondráková, Z.; Lomenech, A.M.; Le Metté, C.; Label, P.; Costa, G.; Trontin, J.F.; Teyssier, C.; et al. Repetitive somatic embryogenesis induced cytological and proteomic changes in embryogenic lines of Pseudotsuga menziesii [Mirb.]. BMC Plant Biol. 2018, 18, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, H.; Fu, S.; Chen, B.; Sun, W.; Zhang, J.; Zhang, J. An iTRAQ-based proteomics approach to clarify the molecular physiology of somatic embryo development in Prince Rupprecht’s larch (Larix principis-rupprechtii Mayr). PLoS ONE 2015, 10, e0119987. [Google Scholar] [CrossRef] [Green Version]
- Castander-Olarieta, A.; Pereira, C.; Montalbán, I.A.; Mendes, V.M.; Correia, S.; Suárez-Álvarez, S.; Manadas, B.; Canhoto, J.; Moncaleán, P. Proteome-wide analysis of heat-stress in Pinus radiata somatic embryos reveals a combined response of sugar metabolism and translational regulation mechanisms. Front. Plant Sci. 2021, 12, 631239. [Google Scholar] [CrossRef] [PubMed]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Durzan, D.J. Biotechnology of somatic polyembryogenesis and plantlet regeneration in loblolly pine. Nat Biotechnol. 1987, 5, 147–151. [Google Scholar] [CrossRef]
- Winiewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Shi, Y.; Jia, B.; Zhang, Y.; Wang, Q. Evolution and function analysis of glycerol kinase GlpK in Pseudomonasaeruginosa. Biochem. Biophys. Res. Commun. 2023, 645, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Vrinten, P.L.; Nakamura, T. Wheat granule-bound starch synthase I and II are encoded by separate genes that are expressed in different tissues. Plant Physiol. 2000, 122, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Zhao, S.; Jiao, G.; Duan, Y.; Ma, L.; Dong, N.; Lu, F.; Zhu, M.; Shao, G.; Hu, S. OPAQUE3, encoding a transmembrane bZIP transcription factor, regulates endosperm storage protein and starch biosynthesis in rice. Plant Physiol. 2022, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, G.; Sun, T.; Wang, L.; Xu, Q.; Liu, J.; Gao, B. Metabolites and Plant Hormones Related to the Resistance Response to Feeding Stimulation and Leaf Clipping Control in Chinese Pine (Pinus tabuliformis Carr.). Curr. Issues Mol. Biol. 2023, 45, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, J.; Peng, T.; Ni, S.; Feng, Y.; Wang, Q.; Wang, M.; Chu, X.; Fan, Z.; Li, X.; et al. Identification of Chalcone Isomerase Family Genes and Roles of CnCHI4 in Flavonoid Metabolism in Camellia nitidissima. Biomolecules 2022, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yu, K.; Shi, Y.; Wang, J.; Duan, C. Transcription Factor VviMYB86 Oppositely Regulates Proanthocyanidin and Anthocyanin Biosynthesis in Grape Berries. Front. Plant Sci. 2021, 11, 613677. [Google Scholar] [CrossRef]
- Li, P.; Li, Y.J.; Zhang, F.J.; Zhang, G.Z.; Jiang, X.Y.; Yu, H.M.; Hou, B.K. The Arabidopsis UDP-glycosyltransferases UGT79B2 and UGT79B3, contribute to cold, salt and drought stress tolerance via modulating anthocyanin accumulation. Plant J. 2017, 89, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Baltz, R.; Schmitt, C.; Beffa, R.; Fritig, B.; Saindrenan, P. Downregulation of a pathogen-responsive tobacco UDP-Glc:phenylpropanoid glucosyltransferase reduces scopoletin glucoside accumulation, enhances oxidative stress, and weakens virus resistance. Plant Cell 2002, 14, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wu, J.; Lin, L.; Li, P.; Li, S.; Wang, Y.; Li, J.; Sun, Q.; Liang, J.; Wang, Y. Overexpression of Cinnamoyl-CoA reductase 2 in Brassica napus increases resistance to Sclerotinia sclerotiorum by affecting lignin biosynthesis. Front. Plant Sci. 2021, 12, 732733. [Google Scholar] [CrossRef]
- Yan, Y.; Christensen, S.; Isakeit, T.; Engelberth, J.; Meeley, R.; Hayward, A.; Emery, R.J.N.; Kolomiets, M.V. Disruption of OPR7 and OPR8 reveals the versatile functions of jasmonic acid in maize development and defense. Plant Cell 2012, 24, 1420–1436. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, O.; Piqueras, R.; Sánchez-Serrano, J.J.; Solano, R. Ethylene Response Factor1 integrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell 2003, 15, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jofuku, K.D.; Den Boer, B.G.; Van Montagu, M.; Okamuro, J.K. Control of Arabidopsis flower and seed development by the homeotic gene APETALA. Plant Cell 1994, 6, 1211–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghi, L.; Kang, J.; Ko, D.; Lee, Y.; Martinoia, E. The role of ABCG-type ABC transporters in phytohormone transport. Biochem. Soc. Trans. 2015, 43, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Markulin, L.; Corbin, C.; Renouard, S.; Drouet, S.; Gutierrez, L.; Mateljak, I.; Auguin, D.; Hano, C.; Fuss, E.; Lainé, E. Pinoresinol-lariciresinol reductases, key to the lignan synthesis in plants. Planta 2019, 249, 1695–1714. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Loosening of plant cell walls by expansins. Nature 2000, 407, 321–326. [Google Scholar] [CrossRef]
- Maurel, C.; Kado, R.T.; Guern, J.; Chrispeels, M.J. Phosphorylation regulates the water channel activity of the seed-specific aquaporin alpha-TIP. EMBO J. 1995, 14, 3028–3035. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.J.; Hoang, N.V.; Botha, F.C.; Furtado, A.; Marquardt, A.; Henry, R.J. Organ-specific expression of genes associated with the UDP-glucose metabolism in sugarcane (Saccharum spp. hybrids). BMC Genom. 2023, 24, 18. [Google Scholar] [CrossRef]
- Siddique, S.; Sobczak, M.; Tenhaken, R.; Grundler, F.M.W.; Bohlmann, H. Cell wall ingrowths in nematode induced syncytia require UGD2 and UGD. PLoS ONE 2012, 7, e41515. [Google Scholar] [CrossRef] [Green Version]
- Abohatem, M.; Zouine, J.; Hadrami, I.E. Low concentrations of BAP and high rate of subcultures improve the establishment and multiplication of somatic embryos in date palm suspension cultures by limiting oxidative browning associated with high levels of total phenols and peroxidase activities. Sci. Hortic. 2011, 130, 344–348. [Google Scholar] [CrossRef]
- Zhang, Z.; Runions, A.; Mentink, R.A.; Kierzkowski, D.; Karady, M.; Hashemi, B.; Huijser, P.; Strauss, S.; Gan, X.; Ljung, K.; et al. A WOX/Auxin Biosynthesis Module Controls Growth to Shape Leaf Form. Curr. Biol. 2020, 30, 4857–4868. [Google Scholar] [CrossRef]
- Ji, J.; Strable, J.; Shimizu, R.; Koenig, D.; Sinha, N.; Scanlon, M.J. WOX4 promotes procambial development. Plant Physiol. 2010, 152, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshchepkova, E.A.; Omelyanchuk, N.A.; Savina, M.S.; Pasternak, T.; Kolchanov, N.A.; Zemlyanskaya, E.V. Systems biology analysis of the WOX5 gene and its functions in the root stem cell niche. Russ. J. Genet. 2017, 7, 404–420. [Google Scholar] [CrossRef]
- Ueda, M.; Zhang, Z.; Laux, T. Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev. Cell 2011, 20, 264–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Dabi, T.; Weigel, D. Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr. Biol. 2005, 15, 436–440. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, 1522–1527. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, J.; Wang, X.; Liu, W.; Jiang, X.; Gai, Y. Large-Scale Quantitative Proteomic Analysis during Different Stages of Somatic Embryogenesis in Larix olgensis. Curr. Issues Mol. Biol. 2023, 45, 2021-2034. https://doi.org/10.3390/cimb45030130
Hou J, Wang X, Liu W, Jiang X, Gai Y. Large-Scale Quantitative Proteomic Analysis during Different Stages of Somatic Embryogenesis in Larix olgensis. Current Issues in Molecular Biology. 2023; 45(3):2021-2034. https://doi.org/10.3390/cimb45030130
Chicago/Turabian StyleHou, Jiayin, Xuechun Wang, Weifeng Liu, Xiangning Jiang, and Ying Gai. 2023. "Large-Scale Quantitative Proteomic Analysis during Different Stages of Somatic Embryogenesis in Larix olgensis" Current Issues in Molecular Biology 45, no. 3: 2021-2034. https://doi.org/10.3390/cimb45030130