
Interaction and Collaboration of SP1, HIF-1, and MYC in Regulating the Expression of Cancer-Related Genes to Further Enhance Anticancer Drug Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Specificity Protein 1, Hypoxia-Inducible Factor-1, and MYC as Master Regulators of Cancer
2. What Are SP1, HIF-1, and MYC, and How Do These TFs Benefit Cancer
2.1. SP1: Housekeeping Gene That Regulates Biological Activities
2.2. HIF-1: Functions as a Mediator of Hypoxic Signals
2.3. SP1 and HIF-1
2.4. MYC: A Master Regulator of Cellular Activity
3. Interactions among SP1, HIF-1, and MYC with One Another and Other TFs
3.1. Modulation of SP1, HIF-1, and MYC Activities
3.2. Effect of HIF-1 on SP1 Gene Expression and Vice Versa
3.3. Effects of HIF-1 Compared to the Effects of SP1 on MYC Gene Activities
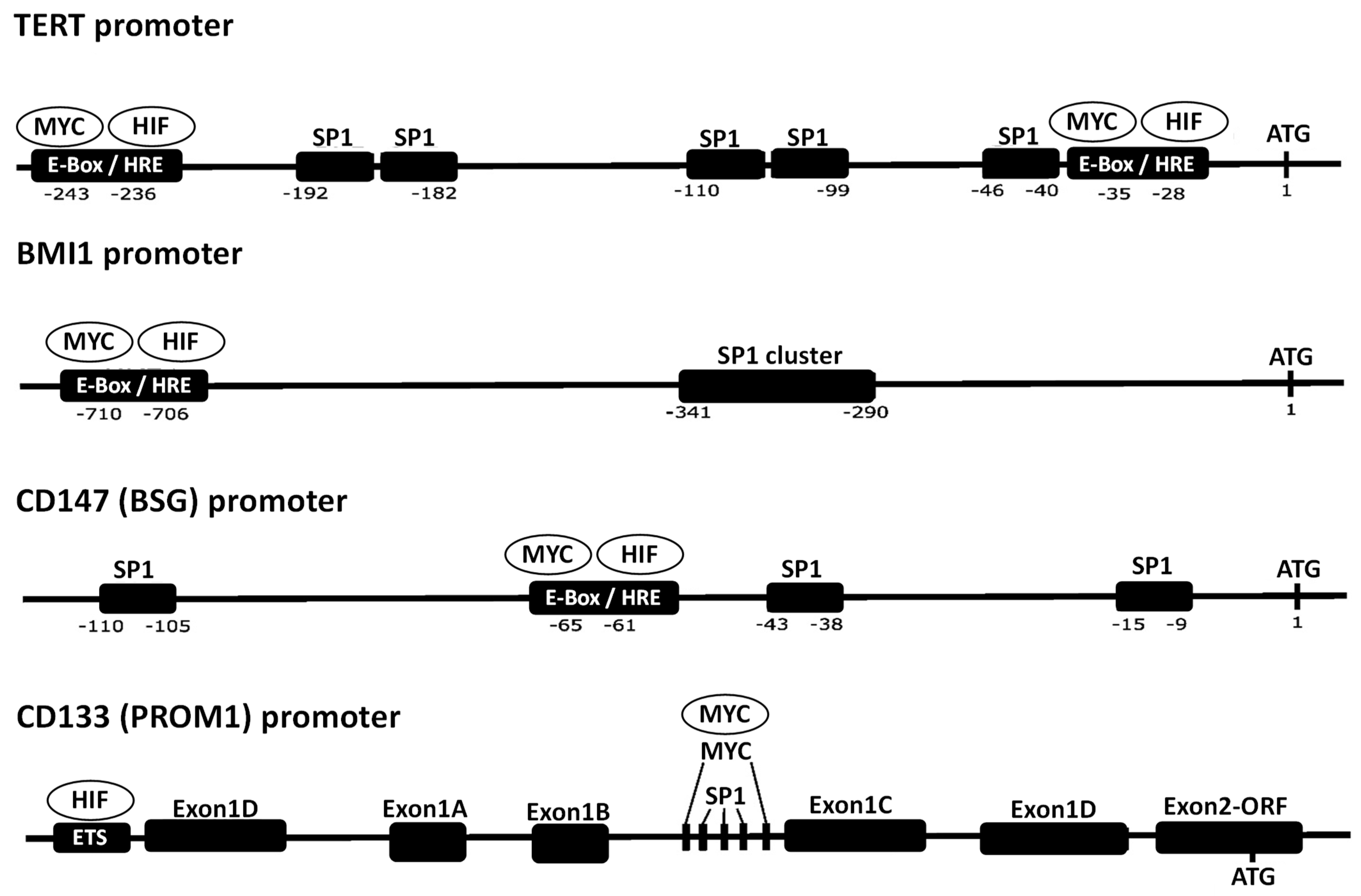
4. Collaboration of SP1, HIF-1, and MYC in Transcriptional Regulation of Their Target Genes
5. Implications of the Interactions and Collaborations of SP1, HIF-1, and MYC in the Development of Anticancer Drugs, and Their Future Perspectives
5.1. SP1, HIF-1, and MYC as Targets for Potential Anticancer Therapies
5.2. Tetramethyl-O-Nordihydroguaiaretic Acid as an Anticancer Drug
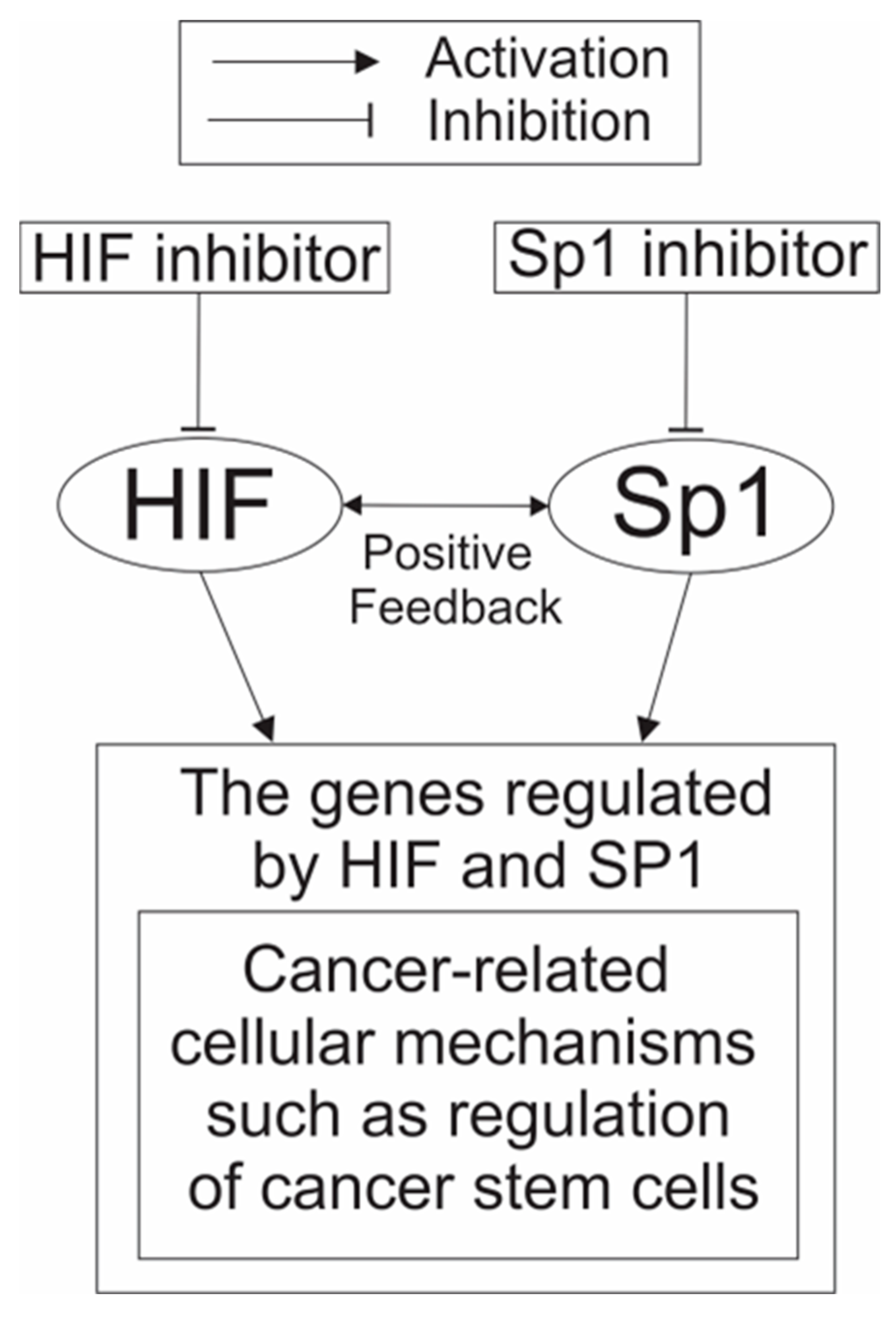
5.3. A Possible Usage of the Combination Treatment of HIF-1 and SP1 Inhibitors as an Anticancer Therapy
5.4. Roles of HIF-1, SP1, and MYC in Normal Cells and the Potential Toxicity of Anticancer Drugs Targeted on These Transcription Factors
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, S.; Filipp, F.V. A network of epigenomic and transcriptional cooperation encompassing an epigenomic master regulator in cancer. NPJ Syst. Biol. Appl. 2018, 1, 24. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Zhao, B.; Xiao, Y.; Hu, S.; Kong, K.; Han, P.; Yue, J.; Deng, Y.; Zhao, Z.; Wu, D.; et al. Understanding the Critical Role of Glycolysis-Related lncRNAs in Lung Adenocarcinoma Based on Three Molecular Subtypes. Biomed Res. Int. 2022, 2022, 7587398. [Google Scholar] [CrossRef] [PubMed]
- Mallik, S.; Qin, G.; Jia, P.; Zhao, Z. Molecular signatures identified by integrating gene expression and methylation in non-seminoma and seminoma of testicular germ cell tumours. Epigenetics 2021, 16, 162–176. [Google Scholar] [CrossRef]
- Sankpal, U.T.; Maliakal, P.; Bose, D.; Kayaleh, O.; Buchholz, D.; Basha, R. Expression of specificity protein transcription factors in pancreatic cancer and their association in prognosis and therapy. Curr. Med. Chem. 2012, 19, 3779–3786. [Google Scholar] [CrossRef] [PubMed]
- Vellingiri, B.; Iyer, M.; Devi Subramaniam, M.; Jayaramayya, K.; Siama, Z.; Giridharan, B.; Narayanasamy, A.; Abdal Dayem, A.; Cho, S.G. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: From Theory to Practice. Int. J. Mol. Sci. 2020, 21, 1153. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Gan, K.; Liu, K.; Xu, B.; Chen, M. SP1 Expression and the Clinicopathological Features of Tumors: A Meta-Analysis and Bioinformatics Analysis. Pathol. Oncol. Res. 2021, 27, 581998. [Google Scholar] [CrossRef]
- Abu, E.L.; Maaty, M.A.; Terzic, J.; Keime, C.; Rovito, D.; Lutzing, R.; Yanushko, D.; Parisotto, M.; Grelet, E.; Namer, I.J.; et al. Hypoxia-mediated stabilization of HIF1A in prostatic intraepithelial neoplasia promotes cell plasticity and malignant progression. Sci. Adv. 2022, 8, eabo2295. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Felsher, D.W. The Myc and Ras Partnership in Cancer: Indistinguishable Alliance or Contextual Relationship? Cancer Res. 2020, 80, 3799–3802. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef]
- Safe, S.; Abbruzzese, J.; Abdelrahim, M.; Hedrick, E. Specificity Protein Transcription Factors and Cancer: Opportunities for Drug Development. Cancer Prev. Res. 2018, 11, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today 2007, 12, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Joshi, E.; Pandya, M.; Desai, U. Current Drugs and Their Therapeutic Targets for Hypoxia-inducible Factors in Cancer. Curr. Protein Pept. Sci. 2023, 24, 447–464. [Google Scholar] [CrossRef]
- Shirai, Y.; Chow, C.C.T.; Kambe, G.; Suwa, T.; Kobayashi, M.; Takahashi, I.; Harada, H.; Nam, J.M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers 2021, 13, 2813. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable: MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef] [PubMed]
- Szpirer, J.; Szpirer, C.; Riviere, M.; Levan, G.; Marynen, P.; Cassiman, J.J.; Wiese, R.; DeLuca, H.F. The Sp1transcription factor gene (SP1) and the 1,25-dihydroxyvitamin D3 receptor gene (VDR) are colocalized on human chromosome arm 12q and rat chromosome 7. Genomics 1991, 11, 168–173. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef]
- Narayan, V.A.; Kriwacki, R.W.; Caradonna, J.P. Structures of zinc finger domains from transcription factor Sp1. Insights into sequence–specific protein–DNA recognition. J. Biol. Chem. 1997, 272, 7801–7809. [Google Scholar] [CrossRef]
- Oka, S.; Shiraishi, Y.; Yoshida, T.; Ohkubo, T.; Sugiura, Y.; Kobayashi, Y. NMR structure of transcription factor Sp1 DNA binding domain. Biochemistry 2004, 43, 16027–16035. [Google Scholar] [CrossRef]
- Nagaoka, M.; Shiraishi, Y.; Sugiura, Y. Selected base sequence outside the target binding site of zinc finger protein Sp1. Nucleic Acids Res. 2001, 29, 4920–4929. [Google Scholar] [CrossRef]
- Cook, T.; Gebelein, B.; Belal, M.; Mesa, K.; Urrutia, R. Three conserved transcriptional repressor domains are a defining feature of the TIEG subfamily of Sp1-like zinc finger proteins. J. Biol. Chem. 1999, 274, 29500–29504. [Google Scholar] [CrossRef] [PubMed]
- Kadonag, J.T.; Courey, A.J.; Ladika, J.; Tjian, R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science 1988, 242, 1566–1570. [Google Scholar] [CrossRef] [PubMed]
- Kadonaga, J.T.; Jones, K.A.; Tjian, R. Promoter specific activation of RNA polymerase II transcription by Sp1. Trends Biochem. Sci. 1986, 11, 20–23. [Google Scholar] [CrossRef]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Khachigian, L. Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell. Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef]
- Suzuki, T.; Kimura, A.; Nagai, R.; Horikoshi, M. Regulation of interaction of the acetyltransferase region of p300 and the DNA-binding domain of Sp1 on and through DNA binding. Genes Cells 2000, 5, 29–41. [Google Scholar] [CrossRef]
- Jackson, S.P.; Tjian, R. O-glycosylation of eukaryotic transcription factors: Implications for mechanisms of transcriptional regulation. Cell 1988, 55, 125–133. [Google Scholar] [CrossRef]
- Stielow, B.; Sapetschnig, A.; Wink, C.; Kruger, I.; Suske, G. SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep. 2008, 9, 899–906. [Google Scholar] [CrossRef]
- Wei, S.; Chuang, H.C.; Tsai, W.C.; Yang, H.C.; Ho, S.R.; Paterson, A.J.; Kulp, S.K.; Chen, C.S. Thiazolidinediones mimic glucose starvation in facilitating Sp1 degradation through the up-regulation of beta-transducin repeat-containing protein. Mol. Pharmacol. 2009, 76, 47–57. [Google Scholar] [CrossRef]
- Marin, M.; Karis, A.; Visser, P.; Grosveld, F.; Philipsen, S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell 1997, 89, 619–628. [Google Scholar] [CrossRef]
- Gilmour, J.; Assi, S.A.; Jaegle, U.; Kulu, D.; van de Werken, H.; Clarke, D.; Westhead, D.R.; Philipsen, S.; Bonifer, C. A crucial role for the ubiquitously expressed transcription factor Sp1 at early stages of hematopoietic specification. Development 2014, 141, 2391–2401. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Davie, J.R. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat.-Anat. Anz. 2010, 192, 275–283. [Google Scholar] [CrossRef]
- Hjaltelin, J.X.; Izarzugaza, J.M.G.; Jensen, L.J.; Russo, F.; Westergaard, D.; Brunak, S. Identification of hyper-rewired genomic stress non-oncogene addiction genes across 15 cancer types. NPJ Syst. Biol. Appl. 2019, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Brahimi-Horn, M.C.; Berta, M.A.; Benizri, E.; Bilton, R.L.; Dayan, F.; Ginouvès, A.; Berra, E.; Pouysségur, J. HIF-1: Master and commander of the hypoxic world. A pharmacological approach to its regulation by siRNAs. Biochem. Pharmacol. 2004, 68, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors--similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Archer, M.C. Role of sp transcription factors in the regulation of cancer cell metabolism. Genes Cancer 2011, 2, 712–719. [Google Scholar] [CrossRef]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sun, Y.; Luo, J.; Wu, M.; Li, J.; Cao, Y. Specificity protein 1 regulates gene expression related to fatty acid metabolism in goat mammary epithelial cells. Int. J. Mol. Sci. 2015, 16, 1806–1820. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate is a metabolic checkpoint of anti- tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.-C. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef]
- Yang, L.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef]
- Böttcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology 2018, 7, e1445454. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA- associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor- repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Doetzlhofer, A.; Kroboth, K.; Wintersberger, E.; Seiser, C. Alternate activation of two divergently transcribed mouse genes from a bidirectional promoter is linked to changes in histone modification. J. Biol. Chem. 2003, 278, 1784–1793. [Google Scholar] [CrossRef]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef]
- Kim, S.J.; Glick, A.; Sporn, M.B.; Roberts, A.B. Characterization of the promoter region of the human transforming growth factor-beta 1 gene. J. Biol. Chem. 1989, 264, 402–408. [Google Scholar] [CrossRef]
- Qureshi, H.Y.; Sylvester, J.; El Mabrouk, M.; Zafarullah, M. TGF-beta-induced expression of tissue inhibitor of metalloproteinases-3 gene in chondrocytes is mediated by extracellular signal-regulated kinase pathway and Sp1 transcription factor. J. Cell. Physiol. 2005, 203, 345–352. [Google Scholar] [CrossRef]
- Wang, J.P.; Wen, M.H.; Chen, Y.T.; Lee, H.H.; Chiang, E.R.; Lee, Y.T.; Liu, C.L.; Chen, T.H.; Hung, S.C. Trichostatin A inhibits TGF-β1 induced in vitro chondrogenesis of hMSCs through Sp1 suppression. Differentiation 2011, 81, 119–126. [Google Scholar] [CrossRef]
- Datta, P.K.; Blake, M.C.; Moses, H.L. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-beta-induced physical and functional interactions between smads and Sp1. J. Biol. Chem. 2000, 275, 40014–40019. [Google Scholar] [CrossRef]
- Fajardo, O.A.; Thompson, K.; Parapuram, S.K.; Liu, S.; Leask, A. Mithramycin reduces expression of fibro-proliferative mRNAs in human gingival fibroblasts. Cell Prolif. 2011, 44, 166–173. [Google Scholar] [CrossRef]
- Su, F.; Geng, J.; Li, X.; Qiao, C.; Luo, L.; Feng, J.; Dong, X.; Lv, M. SP1 promotes tumor angiogenesis and invasion by activating VEGF expression in an acquired trastuzumab-resistant ovarian cancer model. Oncol. Rep. 2017, 38, 2677–2684. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Shu, H.K. EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 2007, 67, 6121–6129. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Hirche, C.; Wissmann, C.; Ebert, M.P.; Höcker, M. Transcription of the vascular endothelial growth factor receptor-3 (VEGFR3) gene is regulated by the zinc finger proteins Sp1 and Sp3 and is under epigenetic control: Transcription of vascular endothelial growth factor receptor 3. Cell Oncol. 2014, 37, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef]
- Nam, E.H.; Lee, Y.; Zhao, X.F.; Park, Y.K.; Lee, J.W.; Kim, S. ZEB2-Sp1 cooperation induces invasion by upregulating cadherin-11 and integrin a5 expression. Carcinogenesis 2014, 35, 302–314. [Google Scholar] [CrossRef]
- Kolesnikoff, N.; Attema, J.L.; Roslan, S.; Bert, A.G.; Schwarz, Q.P.; Gregory, P.A.; Goodall, G.J. Specificity protein 1 (Sp1) maintains basal epithelial expression of the miR-200 family: Implications for epithelial-mesenchymal transition. J. Biol. Chem. 2014, 289, 11194–11205. [Google Scholar] [CrossRef]
- Qian, Y.; Yao, W.; Yang, T.; Yang, Y.; Liu, Y.; Shen, Q.; Zhang, J.; Qi, W.; Wang, J. aPKC-i/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017, 66, 1165–1182. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Gupta, V.K.; McGinn, O.; Nomura, A.; Sharma, N.S.; Arora, N.; Giri, B.; Dudeja, V.; Saluja, A.K.; Banerjee, S. Inhibition of Sp1 prevents ER homeostasis and causes cell death by lysosomal membrane permeabilization in pancreatic cancer. Sci. Rep. 2017, 7, 1564. [Google Scholar] [CrossRef] [PubMed]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 28. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: A complex problem. Trends Cell Biol. 2023, 33, 235–246. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Cencioni, C.; Scagnoli, F.; Spallotta, F.; Nasi, S.; Illi, B. The “Superoncogene” Myc at the Crossroad between Metabolism and Gene Expression in Glioblastoma Multiforme. Int. J. Mol. Sci. 2023, 24, 4217. [Google Scholar] [CrossRef]
- Pirity, M.; Blanck, J.K.; Schreiber-Agus, N. Lessons learned from Myc/Max/Mad knockout mice. Curr. Top. Microbiol. Immunol. 2006, 302, 205–234. [Google Scholar] [CrossRef]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef]
- See, Y.X.; Chen, K.; Fullwood, M.J. MYC overexpression leads to increased chromatin interactions at super-enhancers and MYC binding sites. Genome Res. 2022, 32, 629–642. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Karadkhelkar, N.M.; Lin, M.; Eubanks, L.M.; Janda, K.D. Demystifying the Druggability of the MYC Family of Oncogenes. J. Am. Chem. Soc. 2023, 145, 3259–3269. [Google Scholar] [CrossRef]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lin, J.; Jiang, M.; He, X.; Wang, K.; Wang, W.; Hu, C.; Shen, Z.; He, Z.; Lin, H.; et al. HIF-1α Promotes the Metastasis of Esophageal Squamous Cell Carcinoma by Targeting SP1. J. Cancer 2020, 11, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, M.; Noé, V.; Ciudad, C.J. Transcriptional regulation of the human Sp1 gene promoter by the specificity protein (Sp) family members nuclear factor Y (NF-Y) and E2F. Biochem. J. 2003, 371 Pt 2, 265–275. [Google Scholar] [CrossRef]
- Nicolás, M.; Noé, V.; Jensen, K.B.; Ciudad, C.J. Cloning and characterization of the 5′-flanking region of the human transcription factor Sp1 gene. J. Biol. Chem. 2001, 276, 22126–22132. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Leung, S.W.; Semenza, G.L. The human hypoxia-inducible factor 1alpha gene: HIF1A structure and evolutionary conservation. Genomics 1998, 52, 159–165. [Google Scholar] [CrossRef]
- Kato, A.; Ng, S.; Thangasamy, A.; Han, H.; Zhou, W.; Raeppel, S.; Fallon, M.; Guha, S.; Ammanamanchi, S. A potential signaling axis between RON kinase receptor and hypoxia-inducible factor-1 alpha in pancreatic cancer. Mol. Carcinog. 2021, 60, 734–745. [Google Scholar] [CrossRef]
- Liu, J.; Levens, D. Making myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32. [Google Scholar] [CrossRef]
- Geltinger, C.; Hörtnagel, K.; Polack, A. TATA box and Sp1 sites mediate the activation of c-myc promoter P1 by immunoglobulin kappa enhancers. Gene Expr. 1996, 6, 113–127. [Google Scholar]
- Wierstra, I.; Alves, J. The c-myc promoter: Still MysterY and challenge. Adv. Cancer Res. 2008, 99, 113–333. [Google Scholar] [CrossRef]
- Biswas, S.; Mukherjee, R.; Tapryal, N.; Singh, A.K.; Mukhopadhyay, C.K. Insulin regulates hypoxia-inducible factor-1α transcription by reactive oxygen species sensitive activation of Sp1 in 3T3-L1 preadipocyte. PLoS ONE 2013, 8, e62128. [Google Scholar] [CrossRef] [PubMed]
- Lafleur, V.N.; Richard, S.; Richard, D.E. Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the protein arginine methyltransferase PRMT1. Mol. Biol. Cell 2014, 25, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, Z.; Huang, W.; Lin, M.; Liu, W.; Liu, K.; Li, C. microRNA-124-3p attenuates myocardial injury in sepsis via modulating SP1/HDAC4/HIF-1α axis. Cell Death Discov. 2022, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Wang, X.; Li, C.X.; Liu, X.Y.; Guo, X.J.; Li, Y.; Chen, Y.L.; Ye, H.X.; Chen, H.S. SP1 Promotes HDAC4 Expression and Inhibits HMGB1 Expression to Reduce Intestinal Barrier Dysfunction, Oxidative Stress, and Inflammatory Response after Sepsis. J. Innate Immun. 2022, 14, 366–379. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Baddela, V.S.; Sharma, A.; Michaelis, M.; Vanselow, J. HIF1 driven transcriptional activity regulates steroidogenesis and proliferation of bovine granulosa cells. Sci. Rep. 2020, 10, 3906. [Google Scholar] [CrossRef]
- Jeong, J.K.; Park, S.Y. Transcriptional regulation of specific protein 1 (SP1) by hypoxia-inducible factor 1 alpha (HIF-1α) leads to PRNP expression and neuroprotection from toxic prion peptide. Biochem. Biophys. Res. Commun. 2012, 429, 93–98. [Google Scholar] [CrossRef]
- Culver, C.; Melvin, A.; Mudie, S.; Rocha, S. HIF-1α depletion results in SP1-mediated cell cycle disruption and alters the cellular response to chemotherapeutic drugs. Cell Cycle 2011, 10, 1249–1260. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.X.; Qian, D.Z.; Dai, M.S. Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8, 590576. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; and Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Ricci, M.S.; Scata, K.A.; Arsham, A.M.; Simon, M.C.; Dicker, D.T.; El-Deiry, W.S. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol. Ther. 2005, 4, 1285–1294. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Koshiji, M.; To, K.K.; Hammer, S.; Kumamoto, K.; Harris, A.L.; Modrich, P.; Huang, L.E. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell. 2005, 17, 793–803. [Google Scholar] [CrossRef]
- To, K.K.; Sedelnikova, O.A.; Samons, M.; Bonner, W.M.; Huang, L.E. The phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repression. EMBO J. 2006, 25, 4784–4794. [Google Scholar] [CrossRef]
- Seira, N.; Yamagata, K.; Fukushima, K.; Araki, Y.; Kurata, N.; Yanagisawa, N.; Mashimo, M.; Nakamura, H.; Regan, J.W.; Murayama, T.; et al. Cellular density-dependent increases in HIF-1alpha compete with c-Myc to down-regulate human EP4 receptor promoter activity through Sp-1-binding region. Pharmacol. Res. Perspect. 2018, 6, e00441. [Google Scholar] [CrossRef]
- Wong, W.J.; Qiu, B.; Nakazawa, M.S.; Qing, G.; Simon, M.C. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol. Cell. Biol. 2013, 33, 3494–3504. [Google Scholar] [CrossRef]
- Li, Q.; Kluz, T.; Sun, H.; Costa, M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PLoS ONE 2009, 4, e8531. [Google Scholar] [CrossRef] [PubMed]
- Zarrabi, A.J.; Kao, D.; Nguyen, D.T.; Loscalzo, J.; Handy, D.E. Hypoxia-induced suppression of c-Myc by HIF-2alpha in human pulmonary endothelial cells attenuates TFAM expression. Cell. Signal. 2017, 38, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Florczyk, U.; Czauderna, S.; Stachurska, A.; Tertil, M.; Nowak, W.; Kozakowska, M.; Poellinger, L.; Jozkowicz, A.; Loboda, A.; Dulak, J. Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic. Biol. Med. 2011, 51, 1882–1892. [Google Scholar] [CrossRef]
- Thaysen, J.; Boisen, A.; Hansen, O.; Bouwstra, S. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef]
- Hoefflin, R.; Harlander, S.; Schäfer, S.; Metzger, P.; Kuo, F.; Schönenberger, D.; Adlesic, M.; Peighambari, A.; Seidel, P.; Chen, C.-Y.; et al. HIF-1alpha and HIF-2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 2020, 11, 4111. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Yan, H.-L.; Zhang, Y.; Hao, L.-Q.; Zhu, X.-T.; Mei, Q.; Sun, S.-H. c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene 2015, 34, 1393–1406. [Google Scholar] [CrossRef]
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.-A.; et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha dependent pathway in multiple myeloma. Cancer Res. 2009, 69, 5082–5090. [Google Scholar] [CrossRef]
- Chen, C.; Cai, S.; Wang, G.; Cao, X.; Yang, X.; Luo, X.; Feng, Y.; Hu, J. c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1alpha. Biochem. Biophys. Res. Commun. 2013, 430, 505–511. [Google Scholar] [CrossRef]
- Weili, Z.; Zhikun, L.; Jianmin, W.; Qingbao, T. Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-Myc/hypoxia inducible factor-1 alpha pathway. J. Cell. Biochem. 2019, 120, 201–212. [Google Scholar] [CrossRef]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef]
- Fu, R.; Chen, Y.; Wang, X.-P.; An, T.; Tao, L.; Zhou, Y.-X.; Huang, Y.-J.; Chen, B.-A.; Li, Z.-Y.; You, Q.-D.; et al. Wogonin inhibits multiple myeloma-stimulated angiogenesis via c-Myc/VHL/HIF-1alpha signaling axis. Oncotarget 2016, 7, 5715–5727. [Google Scholar] [CrossRef]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647 e637. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell. 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2α Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef]
- Soleymani Abyaneh, H.; Gupta, N.; Alshareef, A.; Gopal, K.; Lavasanifar, A.; Lai, R. Hypoxia Induces the Acquisition of Cancer Stem-like Phenotype Via Upregulation and Activation of Signal Transducer and Activator of Transcription-3 (STAT3) in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancer Microenviron. 2018, 11, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Parisi, F.; Wirapati, P.; Naef, F. Identifying synergistic regulation involving c-Myc and sp1 in human tissues. Nucleic Acids Res. 2007, 35, 1098–1107. [Google Scholar] [CrossRef]
- Su, T.; Liu, P.; Ti, X.; Wu, S.; Xue, X.; Wang, Z.; Dioum, E.; Zhang, Q. HΙF1α, EGR1 and SP1 co-regulate the erythropoietin receptor expression under hypoxia: An essential role in the growth of non-small cell lung cancer cells. Cell Commun. Signal. 2019, 17, 152. [Google Scholar] [CrossRef] [PubMed]
- Miki, N.; Ikuta, M.; Matsui, T. Hypoxia-induced activation of the retinoic acid receptor-related orphan receptor alpha4 gene by an interaction between hypoxia-inducible factor-1 and Sp1. J. Biol. Chem. 2004, 279, 15025–15031. [Google Scholar] [CrossRef]
- Koizume, S.; Ito, S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Sakuma, Y.; Osaka, H.; Takano, Y.; Ruf, W.; Miyagi, Y. HIF2α-Sp1 interaction mediates a deacetylation-dependent FVII-gene activation under hypoxic conditions in ovarian cancer cells. Nucleic Acids Res. 2012, 40, 5389–5401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.-X.; Zhou, S.-Y.; Wang, S.-X.; Zheng, K.; Xu, D.-D.; Liu, Y.-T.; Wang, X.-Y.; Yan, H.-Z.; Zhang, L.; et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol. Cancer 2015, 14, 56. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, M.; Luo, X.; Zhang, L.; Niu, Z.; Peng, C.; Ma, J.; Peng, S.; Zhou, H.; Xiang, B.; et al. Transcriptional regulation of BRD7 expression by Sp1 and c-Myc. BMC Mol. Biol. 2008, 9, 111. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Taira, T.; Kanaya, T.; Itoh, H.; Yutsudo, M.; Ariga, H.; Inoue, M. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000, 28, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Liu, G.H.; Zhang, H.; Xing, S.; Hu, L.J.; Zhao, W.F.; Xie, B.; Li, M.Z.; Zeng, B.H.; Li, Y.; et al. Sp1 and c-Myc regulate transcription of BMI1 in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2929–2944. [Google Scholar] [CrossRef]
- Gopisetty, G.; Xu, J.; Sampath, D.; Colman, H.; Puduvalli, V.K. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene 2013, 32, 3119–3129. [Google Scholar] [CrossRef]
- Kong, L.M.; Liao, C.G.; Zhang, Y.; Xu, J.; Li, Y.; Huang, W.; Zhang, Y.; Bian, H.; Chen, Z.N. A regulatory loop involving miR-22, Sp1, and c-Myc modulates CD147 expression in breast cancer invasion and metastasis. Cancer Res. 2014, 74, 3764–3778. [Google Scholar] [CrossRef]
- Nishi, H.; Nakada, T.; Kyo, S.; Inoue, M.; Shay, J.W.; Isaka, K. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol. Cell. Biol. 2004, 24, 6076–6083. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Xia, L.; Ning, X.; Liu, L.; Sun, W.; Huang, C.; Wang, H.; Sun, S. Hypoxia-induced Bmi1 promotes renal tubular epithelial cell-mesenchymal transition and renal fibrosis via PI3K/Akt signal. Mol. Biol. Cell 2014, 25, 2650–2659. [Google Scholar] [CrossRef]
- Ke, X.; Fei, F.; Chen, Y.; Xu, L.; Zhang, Z.; Huang, Q.; Zhang, H.; Yang, H.; Chen, Z.; Xing, J. Hypoxia upregulates CD147 through a combined effect of HIF-1α and Sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis 2012, 33, 1598–1607. [Google Scholar] [CrossRef]
- Ohnishi, S.; Maehara, O.; Nakagawa, K.; Kameya, A.; Otaki, K.; Fujita, H.; Higashi, R.; Takagi, K.; Asaka, M.; Sakamoto, N.; et al. Hypoxia-inducible factors activate CD133 promoter through ETS family transcription factors. PLoS ONE 2013, 8, e66255. [Google Scholar] [CrossRef]
- Barbato, L.; Bocchetti, M.; Di Biase, A.; Regad, T. Cancer Stem Cells and Targeting Strategies. Cells 2019, 8, 926. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008, 99, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhao, Y.; Wang, S.; Jia, W.; Kang, J.; Zhu, J. Human Telomerase Reverse Transcriptase (hTERT) Transcription Requires Sp1/Sp3 Binding to the Promoter and a Permissive Chromatin Environment. J. Biol. Chem. 2015, 290, 30193–30203. [Google Scholar] [CrossRef]
- Kong, L.M.; Liao, C.G.; Fei, F.; Guo, X.; Xing, J.L.; Chen, Z.N. Transcription factor Sp1 regulates expression of cancer-associated molecule CD147 in human lung cancer. Cancer Sci. 2010, 101, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Luo, Z.; Zhou, G.; Song, C.; Yu, F.; Xiang, J.; Li, G. Cis-regulatory functions of overlapping HIF-1alpha/E-box/AP-1-like sequences of CD164. BMC Mol. Biol. 2011, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, N.; Ulibarri, I.F.; Kauppila, A.; Luosujärvi, H.; Rivinoja, A.; Pospiech, H.; Kellokumpu, I.; Kellokumpu, S. Hypoxia upregulates carcinoembryonic antigen expression in cancer cells. Int. J. Cancer 2007, 121, 2443–2450. [Google Scholar] [CrossRef]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef]
- Khattar, E.; Tergaonkar, V. Transcriptional Regulation of Telomerase Reverse Transcriptase (TERT) by MYC. Front. Cell Dev. Biol. 2017, 5, 1. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Vaishnave, S. BMI1 and PTEN are key determinants of breast cancer therapy: A plausible therapeutic target in breast cancer. Gene 2018, 678, 302–311. [Google Scholar] [CrossRef]
- Hardenbol, P.; Van Dyke, M.W. In vitro inhibition of c-myc transcription by mithramycin. Biochem. Biophys. Res. Commun. 1992, 185, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Holstege, C.P. Mithramycin. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 347–348. ISBN 9780123864550. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Jauset, T.; Massó-Vallés, D.; Martínez-Martín, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; Serrano Del Pozo, E.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, eaar5012. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, T.; Zhao, X.; Huang, D.; Huang, J.; Huang, Y.; Huang, Q.; Liang, Z.; Chen, C.; Chen, M.; et al. HIF1α-SP1 interaction disrupts the circ-0001875/miR-31-5p/SP1 regulatory loop under a hypoxic microenvironment and promotes non-small cell lung cancer progression. J. Exp. Clin. Cancer Res. 2022, 41, 156. [Google Scholar] [CrossRef] [PubMed]
- Gnabre, J.N.; Brady, J.N.; Clanton, D.J.; Ito, Y.; Dittmer, J.; Bates, R.B.; Huang, R.C. Inhibition of human immunodeficiency virus type 1 transcription and replication by DNA sequence-selective plant lignans. Proc. Natl. Acad. Sci. USA 1995, 92, 11239–11243. [Google Scholar] [CrossRef]
- Gnabre, J.N.; Ito, Y.; Ma, Y.; Huang, R.C. Isolation and anti-HIV-1 lignans from Larreatridentata by counter-current chromatography. J. Chromatogr. A 1996, 719, 353–364. [Google Scholar] [CrossRef]
- Gnabre, J.N.; Huang, R.C.; Bates, R.B.; Burns, J.J.; Caldera, S.; Malcomson, M.E.; McClure, K.J. Characterization of Anti-HIV lignans from Larreatridentata. Tetrahedron 1995, 51, 12203–12210. [Google Scholar] [CrossRef]
- Hwu, J.R.; Tseng, W.N.; Gnabre, J.; Giza, P.; Huang, R.C.C. Antiviral activities of methylated nordihydroguaiaretic acids. 1. Synthesis, structure identification, and inhibition of Tat regulated HIV transactivation. J. Med. Chem. 1998, 41, 2994–3000. [Google Scholar] [CrossRef]
- Chen, H.; Teng, L.; Li, J.N.; Park, R.; Mold, D.E.; Gnabre, J.; Hwu, J.R.; Tseng, W.N.; Huang, R.C. Antiviral activities of methylated nordihydroguaiaretic acids. 2. Targeting herpes simplex virus replication by the mutation insensitive transcription inhibitor tetra-O-methyl-NDGA. J. Med. Chem. 1998, 41, 3001–3007. [Google Scholar] [CrossRef]
- Heller, J.D.; Kuo, J.; Wu, T.C.; Kast, W.M.; Huang, R.C. Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res. 2001, 61, 5499–5504. [Google Scholar]
- Park, R.; Chang, C.C.; Liang, Y.C.; Chung, Y.; Henry, R.A.; Lin, E.; Mold, D.E.; Huang, R.C. Systemic treatment with tetra-O-methyl nordihydroguaiaretic acid suppresses the growth of human xenograft tumors. Clin. Cancer Res. 2005, 11, 4601–4609. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Huang, R.C. Tetra-O-Methyl Nordihydroguaiaretic Acid Broadly Suppresses Cancer Metabolism and Synergistically Induces Strong Anticancer Activity in Combination with Etoposide, Rapamycin and UCN-01. PLoS ONE 2016, 11, e0148685. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.C.; Kimura, K. Methods for Inhibition of BNIP3 and Prevention and Treatment of Ischemia Reperfusion Injury by Tetra-O-Methyl Nordihydroguaiaretic Acid. U.S. 9456995B2, 18 July 2013. [Google Scholar]
- Huang, R.C.; Kimura, K. Suppression of Cancer Growth and Metastasis Using Nordihydroguaiaretic Acid Derivatives with Metabolic Modulators. U.S. 2011/0014192 A1, 20 January 2011. [Google Scholar]
- Huang, R.C.; Kimura, K. Suppression of Cancer Growth and Metastasis Using Nordihydroguaiaretic Acid Derivatives with 7-hydroxystaurosporine. U.S. 2013/0053335 A1, 28 February 2013. [Google Scholar]
- Huang, R.C.; Mold, D.; Ruland, C.; Liang, Y.C.; Chun, J.H. Compositions Comprising NDGA Derivatives and Sorafenib and Their Use in Treatment of Cancer. PCT/US13/24595, 4 June 2013. U.S. 2015/0018302 A1, 15 January 2015. [Google Scholar]
- Chang, C.C.; Liang, Y.C.; Klutz, A.; Hsu, C.I.; Lin, C.F.; Mold, D.E.; Chou, T.C.; Lee, Y.C.; Huang, R.C.C. Reversal of multidrug resistance by two nordihydroguaiaretic acid derivatives, M4N and maltose-M3N, and their use in combination with doxorubicin or paclitaxel. Cancer Chemother. Pharmacol. 2006, 58, 640–653. [Google Scholar] [CrossRef]
- Kimura, K.; Chun, J.H.; Lin, Y.; Liang, Y.; Jackson, T.L.B.; Huang, R.C.C. Tetra-O-methyl-nordihydroguaiaretic acid inhibits energy metabolism and synergistically induces anticancer effects with temozolomide on LN229 glioblastoma tumors implanted in mice while preventing obesity in normal mice that consume high-fat diets. PLoS ONE 2023, 18, e0285536. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Heller, J.D.; Kuo, J.; Huang, R.C. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc. Natl. Acad. Sci. USA 2004, 101, 13239–13244. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Peereboom, D.; Rosenfeld, M.R.; Mikkelsen, T.; Supko, J.G.; Desideri, S.; Adult Brain Tumor Consortium. Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro-Oncology 2012, 14, 511–517. [Google Scholar] [CrossRef]
- Nair, M.K.; Pandey, M.; Huang, R.C.C. A Single Group Study into the Effect of Intralesional Tetra-O-Methyl Nordihydroguaiaretic Acid (M4N) in Oral Squamous Cell Carcinoma. World J. Med. Res. 2019, 8, 1–10. [Google Scholar]
- Huang, R.C.; Chun, J.; Lin, Y.; Liang, Y.; Liao, K.W.; Jackson, T.; Lai, C.; Mold, D. Formulations of Terameprocol and Temozolomide and Their Use in Stimulation of Humoral Immunity in Tumors. PCT/US2020/062288, 25 November 2020. WO/2021/108601, 3 June 2021. [Google Scholar]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Tornin, J.; Martinez-Cruzado, L.; Santos, L.; Rodriguez, A.; Núñez, L.E.; Oro, P.; Hermosilla, M.A.; Allonca, E.; Fernández-García, M.T.; Astudillo, A.; et al. Inhibition of SP1 by the mithramycin analog EC-8042 efficiently targets tumor initiating cells in sarcoma. Oncotarget 2016, 7, 30935–30950. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, K.; Jackson, T.L.B.; Huang, R.C.C. Interaction and Collaboration of SP1, HIF-1, and MYC in Regulating the Expression of Cancer-Related Genes to Further Enhance Anticancer Drug Development. Curr. Issues Mol. Biol. 2023, 45, 9262-9283. https://doi.org/10.3390/cimb45110580
Kimura K, Jackson TLB, Huang RCC. Interaction and Collaboration of SP1, HIF-1, and MYC in Regulating the Expression of Cancer-Related Genes to Further Enhance Anticancer Drug Development. Current Issues in Molecular Biology. 2023; 45(11):9262-9283. https://doi.org/10.3390/cimb45110580
Chicago/Turabian StyleKimura, Kotohiko, Tiffany L. B. Jackson, and Ru Chih C. Huang. 2023. "Interaction and Collaboration of SP1, HIF-1, and MYC in Regulating the Expression of Cancer-Related Genes to Further Enhance Anticancer Drug Development" Current Issues in Molecular Biology 45, no. 11: 9262-9283. https://doi.org/10.3390/cimb45110580