
Recruiting In Vitro Transcribed mRNA against Cancer Immunotherapy: A Contemporary Appraisal of the Current Landscape

 , ,
, ,  and
and
Abstract
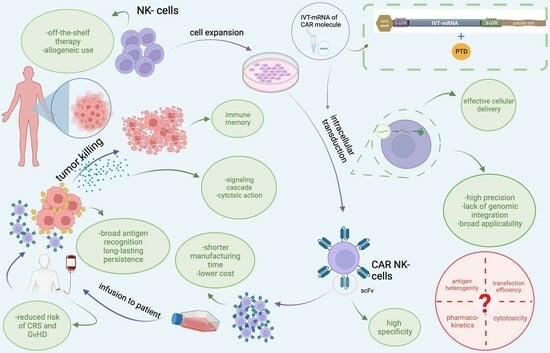
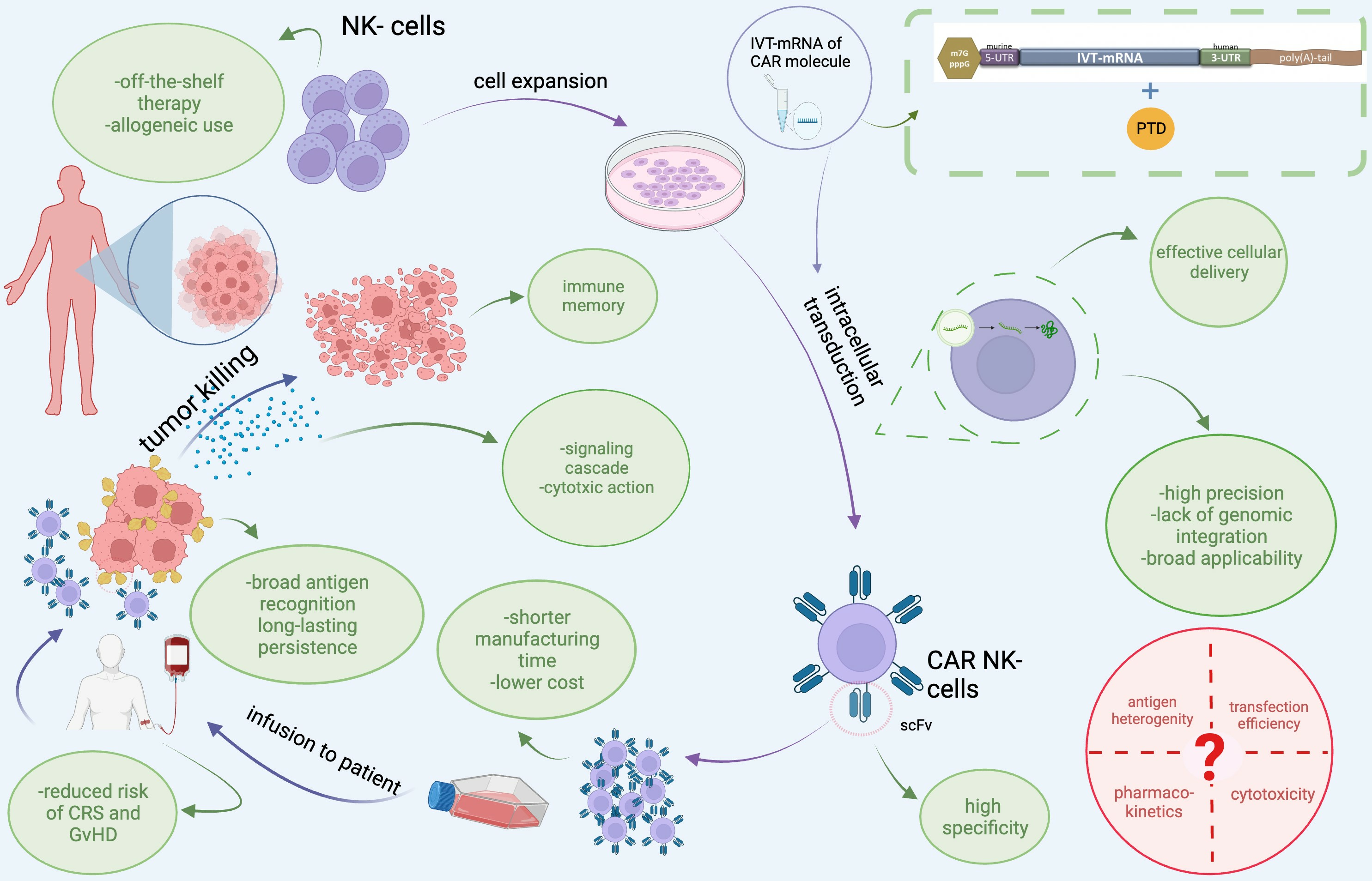
:
1. IVT-mRNA: A Comprehensive Exploration of the Technology
2. IVT-mRNA Innovations against Cancer: A Leap in Cancer Immunotherapy
3. Empowering the Immune System: The IVT-mRNA Vaccines and Neoantigens in Cancer Immunotherapy
4. Exploration of Cancer Immunotherapy: From mAbs to Adoptive Cell Therapy
5. Chimeric Antigen Receptor (CAR) T-Cells: Clinical Applications and Ongoing Trials in Cancer Immunotherapy
5.1. Clinical Implementation of CAR T-Cell Therapy
5.2. Next-Generation and Alternative TAA-Targeting CAR T Therapies in Clinical Studies

{kind=link}
| Code of CAR T Therapy | Construct | Malignancy | Transduction Method | Clinical Study-Result |
|---|---|---|---|---|
| Hematological Malignancies | ||||
| NCT01853631 [160] | CD19-CD3ζ-CD28-4-1BB (3rd generation) | non-Hodgkin’s lymphoma | retrovirus | Higher expansion and persistence of the 3rd generation CAR |
| NCT04381741 [168] | CD19-CD8- 4-1BB-CD3ζ-IL-7-CCL19 (TRUCK) | Large B cell lymphoma | lentivirus | ORR: 5/7 patients |
| NCT04557436 [169] | CD19 (Universal) | Pediatric, refractory B- ALL | lentivirus | Expansion of engineered CAR T-cells, but with serious side effects (Phase I) |
| NCT03016377 [164] | CD19-CD3ζ-4-1ΒΒ, Inducible caspase 9 (switchable) | Adult B-ALL | virus-induced | Improvement in CAR T therapy-induced side effects (Phase I) |
| NCT03233854 [170] | CD19vH-CD22vL-hinge- CD22vH-CD19vL-4-1BB-CD3ζ (Biphasic) | Adults B-ALL, Large B cell lymphoma | lentivirus | 100% response with 88% CR (B-ALL) (Phase I) |
| Solid Tumors | ||||
| NCT03980288 [167] | GPC3-4-1BB-CD3ζ-Runt-related transcription factor 3 (RUNX3) | Hepatocellular carcinoma | lentivirus | Safety evaluation-Phase I |
| NCT02209376 [171] | EGFRvIII | Glioblastoma | lentivirus | Case report-prolongation in life expectancy in |
| NCT03740256 | HER2 CAR T therapy and oncolytic virus | Breast Cancer | lentivirus | Recruiting |
| NCT05681650 | HypoSti.CAR-HER2 T-cell therapy (Switchable) | Breast and other HER2+ Cancers | retrovirus | Not yet recruiting |
6. Exploring the Synergy of CAR T-Cells and mRNA in Cancer Immunotherapy
7. Recruitment of Our Novel, Patented Delivery Method of IVT-mRNAs via PTD Technology to Transduce CAR into NK-92 Cells
8. Enhancing CAR NK Cell Potency: A Spotlight on Bioenergetics
9. Challenges and Limitations of IVT-mRNA Cancer Immunotherapy Strategies
10. Future Directions in Cancer Immunotherapy: Paving the Way for Advancements
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADCC | Antibody-Dependent Cellular Cytotoxicity |
| ALL | Acute Lymphocytic Leukemia |
| APCs | Antigen Presenting Cells |
| ARCAs | Anti-Reverse Cap Analogs |
| BCMA | B-cell Maturation Antigen |
| CAR | Chimeric Antigen Receptor |
| CDC | Complement-Dependent Cytotoxicity |
| CPPs | Cell Penetrating Peptides |
| CRS | Cytokine Release Syndrome |
| CTL | cytotoxic T-cell |
| DCs | Dendritic Cells |
| GvHD | Graft-versus-Host Disease |
| EGF | Epidermal Growth Factor |
| ErbB | Epidermal growth factor receptor |
| HER2 | Human Epidermal growth factor receptor 2 |
| HLA | Human Leukocyte Antigen |
| GMP | Good Manufacturing Practice |
| IFN | Interferon |
| iPSCs | induced Pluripotent Stem Cells |
| IVT-mRNAs | in vitro transcribed (IVT)-mRNAs |
| LNPs | Lipid Nanoparticles |
| mAbs | monoclonal antibodies |
| MHC | Major Histocompatibility Complex |
| NK cells | Natural Killer cells |
| OV | Oncolytic Viruses |
| PD1 | Programmed Death 1 |
| PTD | Protein Transduction Domain |
| r/r | refractory and/or relapsed |
| scFv | single chain Fragment variant |
| TAAs | Tumor-Associated Antigens |
| TCR | T-Cell Receptor |
| TILs | Tumor Infiltrating Lymphocytes |
| TNFα | Tumor Necrosis Factor α |
| TSAs | Tumor-Specific Antigens |
| UTRs | untranslated regions |
| Ψ | pseudouridine |
References
- Kim, Y.K. RNA therapy: Rich history, various applications and unlimited future prospects. Exp. Mol. Med. 2022, 54, 455–465. [Google Scholar] [CrossRef] [PubMed]
- RNA Based Therapeutics Market Overview 2030. Available online: https://www.alliedmarketresearch.com/rna-based-therapeutics-market (accessed on 15 September 2023).
- Gomez-Aguado, I.; Rodriguez-Castejon, J.; Beraza-Millor, M.; Rodriguez-Gascon, A.; Del Pozo-Rodriguez, A.; Solinis, M.A. mRNA delivery technologies: Toward clinical translation. Int. Rev. Cell Mol. Biol. 2022, 372, 207–293. [Google Scholar] [CrossRef]
- Wei, H.; Zheng, L.; Wang, Z. mRNA therapeutics: New vaccination and beyond. Fundam. Res. 2023, 3, 749–759. [Google Scholar] [CrossRef]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Foltopoulou, P.F.; Ingendoh-Tsakmakidis, A.; Tsiftsoglou, A.S.; Vizirianakis, I.S.; Pappas, I.S.; Papadopoulou, L.C. Protein Transduction Domain-Mediated Delivery of Recombinant Proteins and In Vitro Transcribed mRNAs for Protein Replacement Therapy of Human Severe Genetic Mitochondrial Disorders: The Case of Sco2 Deficiency. Pharmaceutics 2023, 15, 286. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. In Vitro-Transcribed (IVT)-mRNA CAR Therapy Development. Methods Mol. Biol. 2020, 2086, 87–117. [Google Scholar] [CrossRef]
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Gros, F.; Hiatt, H.; Gilbert, W.; Kurland, C.G.; Risebrough, R.W.; Watson, J.D. Unstable ribonucleic acid revealed by pulse labelling of Escherichia coli. Nature 1961, 190, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Dolgin, E. The tangled history of mRNA vaccines. Nature 2021, 597, 318–324. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro rats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. The messenger’s great message for vaccination. Expert. Rev. Vaccines 2015, 14, 153–156. [Google Scholar] [CrossRef]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nanotoday 2019, 28, 100766. [Google Scholar] [CrossRef]
- Rohner, E.; Yang, R.; Foo, K.S.; Goedel, A.; Chien, K.R. Unlocking the promise of mRNA therapeutics. Nat. Biotechnol. 2022, 40, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Offord, C.; Cohen, J. Award honors pair for mRNA work key to COVID-19 vaccines. Science 2023, 382, 22. [Google Scholar] [CrossRef]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Mir, F.F.; Jhunjhunwala, S.; Kaczmarek, J.C.; Hurtado, J.E.; Yang, J.H.; Webber, M.J.; Kowalski, P.S.; Heartlein, M.W.; DeRosa, F.; et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials 2016, 109, 78–87. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Kariko, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef]
- Kocmik, I.; Piecyk, K.; Rudzinska, M.; Niedzwiecka, A.; Darzynkiewicz, E.; Grzela, R.; Jankowska-Anyszka, M. Modified ARCA analogs providing enhanced translational properties of capped mRNAs. Cell Cycle 2018, 17, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Miliotou, A.N.; Pappas, I.S.; Vizirianakis, I.S.; Papadopoulou, L.C. In Vitro-Transcribed mRNAs as a New Generation of Therapeutics in the Dawn of Twenty-First Century: Exploitation of Peptides as Carriers for Their Intracellular Delivery. In Messenger RNA Therapeutics; RNA, Technologies; Jurga, S., Barciszewski, J., Eds.; Springer: Cham, Switzerland, 2022; Volume 133. [Google Scholar]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Jiang, H.; et al. Algorithm for optimized mRNA design improves stability and immunogenicity. Nature 2023, 621, 396–403. [Google Scholar] [CrossRef]
- Rosa, S.S.; Nunes, D.; Antunes, L.; Prazeres, D.M.F.; Marques, M.P.C.; Azevedo, A.M. Maximizing mRNA vaccine production with Bayesian optimization. Biotechnol. Bioeng. 2022, 119, 3127–3139. [Google Scholar] [CrossRef]
- Sunita; Sajid, A.; Singh, Y.; Shukla, P. Computational tools for modern vaccine development. Hum. Vaccin. Immunother. 2020, 16, 723–735. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, N.; Wang, Z.; Xu, X. Structure-based design of oligomeric receptor-binding domain (RBD) recombinant proteins as potent vaccine candidates against SARS-CoV-2. Hum. Vaccin. Immunother. 2023, 19, 2174755. [Google Scholar] [CrossRef]
- Rappaport, A.R.; Hong, S.J.; Scallan, C.D.; Gitlin, L.; Akoopie, A.; Boucher, G.R.; Egorova, M.; Espinosa, J.A.; Fidanza, M.; Kachura, M.A.; et al. Low-dose self-amplifying mRNA COVID-19 vaccine drives strong protective immunity in non-human primates against SARS-CoV-2 infection. Nat. Commun. 2022, 13, 3289. [Google Scholar] [CrossRef]
- Deo, S.; Desai, K.; Patare, A.; Wadapurkar, R.; Rade, S.; Mahudkar, S.; Sathe, M.; Srivastava, S.; Prasanna, P.; Singh, A. Evaluation of self-amplifying mRNA platform for protein expression and genetic stability: Implication for mRNA therapies. Biochem. Biophys. Res. Commun. 2023, 680, 108–118. [Google Scholar] [CrossRef]
- Li, W.; Huang, Z.; MacKay, J.A.; Grube, S.; Szoka, F.C., Jr. Low-pH-sensitive poly(ethylene glycol) (PEG)-stabilized plasmid nanolipoparticles: Effects of PEG chain length, lipid composition and assembly conditions on gene delivery. J. Gene Med. 2005, 7, 67–79. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. Liposomes in Drug Delivery: How It All Happened. Pharmaceutics 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.N.; Lee, S.Y.; Lee, S.; Youn, H.; Im, H.J. Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics 2022, 12, 7509–7531. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Beasock, D.; Fessler, A.; Szebeni, J.; Ljubimova, J.Y.; Afonin, K.A.; Dobrovolskaia, M.A. To PEGylate or not to PEGylate: Immunological properties of nanomedicine’s most popular component, polyethylene glycol and its alternatives. Adv. Drug Deliv. Rev. 2022, 180, 114079. [Google Scholar] [CrossRef] [PubMed]
- Hillary, V.E.; Ceasar, S.A. An update on COVID-19: SARS-CoV-2 variants, antiviral drugs, and vaccines. Heliyon 2023, 9, e13952. [Google Scholar] [CrossRef]
- Bormann, M.; van de Sand, L.; Witzke, O.; Krawczyk, A. Recent Antiviral Treatment and Vaccination Strategies Against SARS-CoV-2. Klin. Monbl. Augenheilkd. 2021, 238, 569–578. [Google Scholar] [CrossRef]
- Ye, Z.; Harmon, J.; Ni, W.; Li, Y.; Wich, D.; Xu, Q. The mRNA Vaccine Revolution: COVID-19 Has Launched the Future of Vaccinology. ACS Nano 2023, 17, 15231–15253. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Gong, Y.; Lin, X.; Zhao, Y.; Zhi, D.; Zhou, Q.; Zhang, S. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. 2018, 7, 473–479. [Google Scholar] [CrossRef]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.M. Cationic lipids activate intracellular signaling pathways. Adv. Drug Deliv. Rev. 2012, 64, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Higgins, J.; Woods, A.; Levy, B.; Xia, Y.; Hsiao, C.J.; Acosta, E.; Almarsson, O.; Moore, M.J.; Brito, L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control Release 2021, 335, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, D.; Kiselev, M.A. Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application. Pharmaceutics 2022, 14, 543. [Google Scholar] [CrossRef] [PubMed]
- Delehedde, C.; Even, L.; Midoux, P.; Pichon, C.; Perche, F. Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles. Pharmaceutics 2021, 13, 945. [Google Scholar] [CrossRef]
- Yang, L.; Gong, L.; Wang, P.; Zhao, X.; Zhao, F.; Zhang, Z.; Li, Y.; Huang, W. Recent Advances in Lipid Nanoparticles for Delivery of mRNA. Pharmaceutics 2022, 14, 2682. [Google Scholar] [CrossRef]
- Shahzad, M.M.; Mangala, L.S.; Han, H.D.; Lu, C.; Bottsford-Miller, J.; Nishimura, M.; Mora, E.M.; Lee, J.W.; Stone, R.L.; Pecot, C.V.; et al. Targeted delivery of small interfering RNA using reconstituted high-density lipoprotein nanoparticles. Neoplasia 2011, 13, 309–319. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Pappas, I.S.; Spyroulias, G.; Vlachaki, E.; Tsiftsoglou, A.S.; Vizirianakis, I.S.; Papadopoulou, L.C. Development of a novel PTD-mediated IVT-mRNA delivery platform for potential protein replacement therapy of metabolic/genetic disorders. Mol. Ther. Nucleic Acids 2021, 26, 694–710. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Miliotou, A.N.; Ntenti, C.; Pappas, I.S.; Papadopoulou, L.C. An Innovative PTD-IVT-mRNA Delivery Platform for CAR Immunotherapy of ErbB(+) Solid Tumor Neoplastic Cells. Biomedicines 2022, 10, 2885. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Sahu, I.; Haque, A.; Weidensee, B.; Weinmann, P.; Kormann, M.S.D. Recent Developments in mRNA-Based Protein Supplementation Therapy to Target Lung Diseases. Mol. Ther. 2019, 27, 803–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Tran, D.M.; Cavedon, A.; Cai, X.; Rajendran, R.; Lyle, M.J.; Martini, P.G.V.; Miao, C.H. Treatment of Hemophilia A Using Factor VIII Messenger RNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2020, 20, 534–544. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S. From COVID-19 to Cancer mRNA Vaccines: Moving From Bench to Clinic in the Vaccine Landscape. Front. Immunol. 2021, 12, 679344. [Google Scholar] [CrossRef]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert. Rev. Vaccines 2015, 14, 161–176. [Google Scholar] [CrossRef]
- Arya, S.; Lin, Q.; Zhou, N.; Gao, X.; Huang, J.D. Strong Immune Responses Induced by Direct Local Injections of Modified mRNA-Lipid Nanocomplexes. Mol. Ther. Nucleic Acids 2020, 19, 1098–1109. [Google Scholar] [CrossRef]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer 2018, 6, 125. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, C.L.; Haanen, J.B.; Met, O.; Svane, I.M. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef] [PubMed]
- Breda, L.; Papp, T.E.; Triebwasser, M.P.; Yadegari, A.; Fedorky, M.T.; Tanaka, N.; Abdulmalik, O.; Pavani, G.; Wang, Y.; Grupp, S.A.; et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science 2023, 381, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, K. George Klein: 1925-2016. Proc. Natl. Acad. Sci. USA 2017, 114, 3275–3277. [Google Scholar] [CrossRef]
- Zhang, R.; Billingsley, M.M.; Mitchell, M.J. Biomaterials for vaccine-based cancer immunotherapy. J. Control Release 2018, 292, 256–276. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef]
- Esprit, A.; de Mey, W.; Bahadur Shahi, R.; Thielemans, K.; Franceschini, L.; Breckpot, K. Neo-Antigen mRNA Vaccines. Vaccines 2020, 8, 776. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Bok, R.A. Treatment of prostate cancer: Therapeutic potential of targeted immunotherapy with APC8015. Ther. Clin. Risk Manag. 2008, 4, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, F.; Chang, Y.; Zhao, Q.; He, X. Advances of mRNA vaccine in tumor: A maze of opportunities and challenges. Biomark. Res. 2023, 11, 6. [Google Scholar] [CrossRef]
- Zahm, C.D.; Moseman, J.E.; Delmastro, L.E.; Mcneel, D.G. PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy. Oncoimmunology 2021, 10, 1912892. [Google Scholar] [CrossRef] [PubMed]
- Dorrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells-An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M. The use of RNA-based treatments in the field of cancer immunotherapy. Mol. Cancer 2023, 22, 106. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Ma, X.L.; Yang, L.; Wei, Y.Q.; Peng, Y.; Wei, X.W. Progress in Neoantigen Targeted Cancer Immunotherapies. Front. Cell Dev. Biol. 2020, 8, 728. [Google Scholar] [CrossRef]
- Vizirianakis, I.S.; Mystridis, G.A.; Avgoustakis, K.; Fatouros, D.G.; Spanakis, M. Enabling personalized cancer medicine decisions: The challenging pharmacological approach of PBPK models for nanomedicine and pharmacogenomics (Review). Oncol. Rep. 2016, 35, 1891–1904. [Google Scholar] [CrossRef]
- Coley, W.B. The Treatment of Malignant Tumors by Repeated Inoculations of Erysipelas: With a Report of Ten Original Cases. Am. J. Med. Sci. 1991, 105, 487. [Google Scholar] [CrossRef]
- Burnet, M. Cancer; a biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M. The role of innate immune signals in antitumor immunity. Oncoimmunology 2012, 1, 189–194. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-alpha and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef]
- Raeber, M.E.; Sahin, D.; Karakus, U.; Boyman, O. A systematic review of interleukin-2-based immunotherapies in clinical trials for cancer and autoimmune diseases. EBioMedicine 2023, 90, 104539. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Redelman-Sidi, G. BCG in Bladder Cancer Immunotherapy. Cancers 2022, 14, 3073. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Parray, H.A.; Shukla, S.; Samal, S.; Shrivastava, T.; Ahmed, S.; Sharma, C.; Kumar, R. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol. 2020, 85, 106639. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.-M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) Anti-CD20 Monoclonal Antibody Therapy in Patients With Relapsed Low-Grade Non-Hodgkin’s Lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Callejo, D.; González-Rincón, J.; Sánchez, A.; Provencio, M.; Sánchez-Beato, M. Action and resistance of monoclonal CD20 antibodies therapy in B-cell Non-Hodgkin Lymphomas. Cancer Treat. Rev. 2015, 41, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, R.; Newman, R.A.; Hanna, N.; Anderson, D.R. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [CrossRef]
- Andemariam, B.; Leonard, J.P. Radioimmunotherapy with tositumomab and iodine-131 tositumomab for non-Hodgkin’s lymphoma. Biol. Targets Ther. 2007, 1, 113–120. [Google Scholar]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Geens, M.; Stappers, S.; Konings, H.; De Winter, B.Y.; Specenier, P.; Van Meerbeeck, J.P.; Verpooten, G.A.; Abrams, S.; Janssens, A.; Peeters, M.; et al. Epidermal growth factor as a potential prognostic and predictive biomarker of response to platinum-based chemotherapy. PLoS ONE 2021, 16, e0252646. [Google Scholar] [CrossRef]
- Peng, D.; Fan, Z.; Lu, Y.; DeBlasio, T.; Scher, H.; Mendelsohn, J. Anti-epidermal growth factor receptor monoclonal antibody 225 up-regulates p27KIP1 and induces G1 arrest in prostatic cancer cell line DU145. Cancer Res. 1996, 56, 3666–3669. [Google Scholar]
- Wu, X.; Fan, Z.; Masui, H.; Rosen, N.; Mendelsohn, J. Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J. Clin. Investig. 1995, 95, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2(+) breast cancer: A review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Hodeib, M.; Serna-Gallegos, T.; Tewari, K.S. A review of HER2-targeted therapy in breast and ovarian cancer: Lessons from antiquity—CLEOPATRA and PENELOPE. Future Oncol. 2015, 11, 3113–3131. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.H.; Kim, M.R.; Jang, J.H.; Na, H.J.; Lee, S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Qiao, M.; Jiang, T.; Zhou, C. Shining light on advanced NSCLC in 2017: Combining immune checkpoint inhibitors. J. Thorac. Dis. 2018, 10 (Suppl. 13), S1534–S1546. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102–124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Stevanovic, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.K.; Fernandes, R.A.; Seymour, L.W. Oncolytic viruses and antibodies: Are they more successful when delivered separately or when engineered as a single agent? J. Immunother. Cancer 2023, 11, e006518. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J. Immunother. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- Finck, A.V.; Blanchard, T.; Roselle, C.P.; Golinelli, G.; June, C.H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 2022, 28, 678–689. [Google Scholar] [CrossRef]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22–28. [Google Scholar] [CrossRef]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Letourneur, F.; Klausner, R.D. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Varela-Rohena, A.; Carpenito, C.; Perez, E.E.; Richardson, M.; Parry, R.V.; Milone, M.; Scholler, J.; Hao, X.; Mexas, A.; Carroll, R.G.; et al. Genetic engineering of T cells for adoptive immunotherapy. Immunol. Res. 2008, 42, 166–181. [Google Scholar] [CrossRef]
- Awasthi, R.; Maier, H.J.; Zhang, J.; Lim, S. Kymriah(R) (tisagenlecleucel)—An overview of the clinical development journey of the first approved CAR-T therapy. Hum. Vaccin. Immunother. 2023, 19, 2210046–2210054. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.C.; Hu, Z.H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef]
- Landsburg, D.J.; Frigault, M.; Heim, M.; Foley, S.R.; Hill, B.T.; Ho, C.M.; Jacobson, C.A.; Jaglowski, S.; Locke, F.L.; Ram, R.; et al. Real-World Outcomes for Patients with Relapsed or Refractory (R/R) Aggressive B-Cell Non-Hodgkin’s Lymphoma (aBNHL) Treated with Commercial Tisagenlecleucel: Subgroup Analyses from the Center for International Blood and Marrow Transplant Research (CIBMTR) Registry. Blood 2022, 140 (Suppl. 1), 1584–1587. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T therapy beyond cancer: The evolution of a living drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Farooq, U.; Ghobadi, A. Axicabtagene Ciloleucel, an Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Relapsed or Refractory Large B-Cell Lymphoma: Practical Implications for the Community Oncologist. Oncologist 2020, 25, e138–e146. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009, 32, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chavda, V.P.; Bezbaruah, R.; Dhamne, H.; Yang, D.H.; Zhao, H.B. CAR T-Cell therapy for the management of mantle cell lymphoma. Mol. Cancer 2023, 22, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef]
- Sanoyan, D.A.; Seipel, K.; Bacher, U.; Kronig, M.N.; Porret, N.; Wiedemann, G.; Daskalakis, M.; Pabst, T. Real-life experiences with CAR T-cell therapy with idecabtagene vicleucel (ide-cel) for triple-class exposed relapsed/refractory multiple myeloma patients. BMC Cancer 2023, 23, 345. [Google Scholar] [CrossRef]
- Ramsborg, C.G.; Guptill, P.; Weber, C.; Christin, B.; Larson, R.P.; Lewis, K.; Mallaney, M.; Bowen, M.; Higham, E.; Albertson, T. JCAR017 Is a Defined Composition CAR T Cell Product with Product and Process Controls That Deliver Precise Doses of CD4 and CD8 CAR T Cell to Patients with NHL. Blood 2017, 130, 4471. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; June, C.H. Predicting Dangerous Rides in CAR T Cells: Bridging the Gap between Mice and Humans. Mol. Ther. 2018, 26, 1401–1403. [Google Scholar] [CrossRef]
- Bouchkouj, N.; Lin, X.; Wang, X.; Przepiorka, D.; Xu, Z.; Purohit-Sheth, T.; Theoret, M. FDA Approval Summary: Brexucabtagene Autoleucel for Treatment of Adults With Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Oncologist 2022, 27, 892–899. [Google Scholar] [CrossRef]
- Totzeck, M.; Michel, L.; Lin, Y.; Herrmann, J.; Rassaf, T. Cardiotoxicity from chimeric antigen receptor-T cell therapy for advanced malignancies. Eur. Heart J. 2022, 43, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, J.; Jasinski, M.; Basak, G.W. Next generations of CAR-T cells—New therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Jan, M.; Scarfo, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Slabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295. [Google Scholar] [CrossRef]
- Kvorjak, M.; Ruffo, E.; Tivon, Y.; So, V.; Parikh, A.B.; Deiters, A.; Lohmueller, J. Conditional control of universal CAR T cells by cleavable OFF-switch adaptors. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Savoldo, B.; Lau, W.; Rubinos, C.; Grover, N.; Armistead, P.; Coghill, J.; Hagan, R.S.; Morrison, K.; Buchanan, F.B.; et al. Utility of a safety switch to abrogate CD19.CAR T-cell-associated neurotoxicity. Blood 2021, 137, 3306–3309. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, S.; Qi, D.; Ma, P.; Fang, Y.; Jiang, N.; Wu, E.; Li, N. Clinical Investigations of CAR-T Cell Therapy for Solid Tumors. Front. Immunol. 2022, 13, 896685. [Google Scholar] [CrossRef]
- Fu, Q.; Zheng, Y.; Fang, W.; Zhao, Q.; Zhao, P.; Liu, L.; Zhai, Y.; Tong, Z.; Zhang, H.; Lin, M.; et al. RUNX-3-expressing CAR T cells targeting glypican-3 in patients with heavily pretreated advanced hepatocellular carcinoma: A phase I trial. EClinicalMedicine 2023, 63, 102175. [Google Scholar] [CrossRef]
- Qian, W.; Zhao, A.; Liu, H.; Lei, W.; Liang, Y.; Yuan, X. Safety and Efficacy of CD19 CAR-T Cells Co-Expressing IL-7 and CCL19 in Combination with Anti-PD-1 Antibody for Refractory/Relapsed DLBCL: Preliminary Data from the Phase Ⅰb Trial (NCT04381741). Blood 2021, 138, 3843. [Google Scholar] [CrossRef]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; Nishimura, M.I. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar] [CrossRef]
- Foster, J.B.; Barrett, D.M.; Kariko, K. The Emerging Role of In Vitro-Transcribed mRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ponzo, M.; Nicolette, C.A.; Tcherepanova, I.Y.; Biondi, A.; Magnani, C.F. The Past, Present, and Future of Non-Viral CAR T Cells. Front. Immunol. 2022, 13, 867013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.; Cohen, C.J.; Gattinoni, L.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 2006, 13, 151–159. [Google Scholar] [CrossRef]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Van De Parre, T.J.; Martinet, W.; Schrijvers, D.M.; Herman, A.G.; De Meyer, G.R. mRNA but not plasmid DNA is efficiently transfected in murine J774A.1 macrophages. Biochem. Biophys. Res. Commun. 2005, 327, 356–360. [Google Scholar] [CrossRef]
- Van den Bosch, G.A.; Ponsaerts, P.; Nijs, G.; Lenjou, M.; Vanham, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F. Ex vivo induction of viral antigen-specific CD8 T cell responses using mRNA-electroporated CD40-activated B cells. Clin. Exp. Immunol. 2005, 139, 458–467. [Google Scholar] [CrossRef]
- Smits, E.; Ponsaerts, P.; Lenjou, M.; Nijs, G.; Van Bockstaele, D.R.; Berneman, Z.N.; Van Tendeloo, V.F. RNA-based gene transfer for adult stem cells and T cells. Leukemia 2004, 18, 1898–1902. [Google Scholar] [CrossRef]
- Birkholz, K.; Hombach, A.; Krug, C.; Reuter, S.; Kershaw, M.; Kampgen, E.; Schuler, G.; Abken, H.; Schaft, N.; Dorrie, J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009, 16, 596–604. [Google Scholar] [CrossRef]
- Yoon, S.H.; Lee, J.M.; Cho, H.I.; Kim, E.K.; Kim, H.S.; Park, M.Y.; Kim, T.G. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009, 16, 489–497. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [PubMed]
- Lehner, M.; Gotz, G.; Proff, J.; Schaft, N.; Dorrie, J.; Full, F.; Ensser, A.; Muller, Y.A.; Cerwenka, A.; Abken, H.; et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE 2012, 7, e31210. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Woll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Yokoyama, W.M. Natural killer cell immune responses. Immunol. Res. 2005, 32, 317–325. [Google Scholar] [CrossRef]
- Leong, J.W.; Fehniger, T.A. Human NK cells: SET to kill. Blood 2011, 117, 2297–2298. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [CrossRef]
- Sungur, C.M.; Murphy, W.J. Positive and negative regulation by NK cells in cancer. Crit. Rev. Oncog. 2014, 19, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-The-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. The NK-92 cell line-30 years later: Its impact on natural killer cell research and treatment of cancer. Cytotherapy 2023, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Karvouni, M.; Vidal-Manrique, M.; Lundqvist, A.; Alici, E. Engineered NK Cells Against Cancer and Their Potential Applications Beyond. Front. Immunol. 2022, 13, 825979. [Google Scholar] [CrossRef]
- Biederstadt, A.; Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 2021, 114, 554–571. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar] [PubMed]
- Burger, M.C.; Forster, M.T.; Romanski, A.; Strassheimer, F.; Macas, J.; Zeiner, P.S.; Steidl, E.; Herkt, S.; Weber, K.J.; Schupp, J.; et al. Intracranial injection of NK cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro-Oncology 2023, 25, 2058–2071. [Google Scholar] [CrossRef] [PubMed]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef]
- Brasseur, R.; Divita, G. Happy birthday cell penetrating peptides: Already 20 years. Biochim. Biophys. Acta 2010, 1798, 2177–2181. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papagiannopoulou, D.; Vlachaki, E.; Samiotaki, M.; Laspa, D.; Theodoridou, S.; Tsiftsoglou, A.S.; Papadopoulou, L.C. PTD-mediated delivery of alpha-globin chain into Kappa-562 erythroleukemia cells and alpha-thalassemic (HBH) patients’ RBCs ex vivo in the frame of Protein Replacement Therapy. J. Biol. Res. 2021, 28, 16. [Google Scholar] [CrossRef]
- Kaiafas, G.C.; Papagiannopoulou, D.; Miliotou Alpha, N.; Tsingotjidou, A.S.; Chalkidou, P.C.; Tsika, A.C.; Spyroulias, G.A.; Tsiftsoglou, A.S.; Papadopoulou, L.C. In vivo biodistribution study of TAT-L-Sco2 fusion protein, developed as protein therapeutic for mitochondrial disorders attributed to SCO2 mutations. Mol. Genet. Metab. Rep. 2020, 25, 100683. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Ingendoh-Tsakmakidis, A.; Mpoutoureli, C.N.; Tzikalou, L.D.; Spyridou, E.D.; Gavriilidis, G.I.; Kaiafas, G.C.; Ntaska, A.T.; Vlachaki, E.; Panayotou, G.; et al. Production and Transduction of a Human Recombinant beta-Globin Chain into Proerythroid K-562 Cells To Replace Missing Endogenous beta-Globin. Mol. Pharm. 2018, 15, 5665–5677. [Google Scholar] [CrossRef]
- Foltopoulou, P.F.; Tsiftsoglou, A.S.; Bonovolias, I.D.; Ingendoh, A.T.; Papadopoulou, L.C. Intracellular delivery of full length recombinant human mitochondrial L-Sco2 protein into the mitochondria of permanent cell lines and SCO2 deficient patient’s primary cells. Biochim. Biophys. Acta 2010, 1802, 497–508. [Google Scholar] [CrossRef]
- Lai, H.C.; Chang, C.J.; Yang, C.H.; Hsu, Y.J.; Chen, C.C.; Lin, C.S.; Tsai, Y.H.; Huang, T.T.; Ojcius, D.M.; Tsai, Y.H.; et al. Activation of NK cell cytotoxicity by the natural compound 2,3-butanediol. J. Leukoc. Biol. 2012, 92, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Minamino, Y.; Kakudo, K.; Nozaki, M. Resistance of oral squamous cell carcinoma cells to cetuximab is associated with EGFR insensitivity and enhanced stem cell-like potency. Oncol. Rep. 2014, 32, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.M.; Foster, J.; Van Der Stegen, S.J.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L.; et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203 e198. [Google Scholar] [CrossRef]
- Assmann, N.; Finlay, D.K. Metabolic regulation of immune responses: Therapeutic opportunities. J. Clin. Investig. 2016, 126, 2031–2039. [Google Scholar] [CrossRef]
- Beezhold, K.; Byersdorfer, C.A. Targeting immuno-metabolism to improve anti-cancer therapies. Cancer Lett. 2018, 414, 127–135. [Google Scholar] [CrossRef]
- Sohn, H.; Cooper, M.A. Metabolic regulation of NK cell function: Implications for immunotherapy. Immunometabolism 2023, 5, e00020. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Bialkowski, L.; Van der Jeught, K.; Renmans, D.; van Weijnen, A.; Heirman, C.; Keyaerts, M.; Breckpot, K.; Thielemans, K. Adjuvant-Enhanced mRNA Vaccines. Methods Mol. Biol. 2017, 1499, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Posteraro, B.; Pastorino, R.; Di Giannantonio, P.; Ianuale, C.; Amore, R.; Ricciardi, W.; Boccia, S. The link between genetic variation and variability in vaccine responses: Systematic review and meta-analyses. Vaccine 2014, 32, 1661–1669. [Google Scholar] [CrossRef]
- Heymans, S.; Cooper, L.T. Myocarditis after COVID-19 mRNA vaccination: Clinical observations and potential mechanisms. Nat. Rev. Cardiol. 2022, 19, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Monge, S.; Pastor-Barriuso, R.; Hernan, M.A. The imprinting effect of covid-19 vaccines: An expected selection bias in observational studies. BMJ 2023, 381, e074404. [Google Scholar] [CrossRef] [PubMed]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Lower, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Le Moignic, A.; Malard, V.; Benvegnu, T.; Lemiegre, L.; Berchel, M.; Jaffres, P.A.; Baillou, C.; Delost, M.; Macedo, R.; Rochefort, J.; et al. Preclinical evaluation of mRNA trimannosylated lipopolyplexes as therapeutic cancer vaccines targeting dendritic cells. J. Control Release 2018, 278, 110–121. [Google Scholar] [CrossRef]
- Guo, M.; Duan, X.; Peng, X.; Jin, Z.; Huang, H.; Xiao, W.; Zheng, Q.; Deng, Y.; Fan, N.; Chen, K.; et al. A lipid-based LMP2-mRNA vaccine to treat nasopharyngeal carcinoma. Nano Res. 2023, 16, 5357–5367. [Google Scholar] [CrossRef]
- Segel, M.; Lash, B.; Song, J.; Ladha, A.; Liu, C.C.; Jin, X.; Mekhedov, S.L.; Macrae, R.K.; Koonin, E.V.; Zhang, F. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 2021, 373, 882–889. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamsi, H.O.; Alhazzani, W.; Wolff, R.A. Extended RAS testing in metastatic colorectal cancer-Refining the predictive molecular biomarkers. J. Gastrointest. Oncol. 2015, 6, 314–321. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Wang, G.; Yang, W.; Xu, Y. mRNA-Based Therapeutics in Cancer Treatment. Pharmaceutics 2023, 15, 622. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.; Cerullo, V. Cancer immunotherapies: A hope for the uncurable? Front. Mol. Med. Sec. Gene Virother. 2023, 3, 1140977. [Google Scholar] [CrossRef]
- Zhang, L.; More, K.R.; Ojha, A.; Jackson, C.B.; Quinlan, B.D.; Li, H.; He, W.; Farzan, M.; Pardi, N.; Choe, H. Effect of mRNA-LNP components of two globally-marketed COVID-19 vaccines on efficacy and stability. NPJ Vaccines 2023, 8, 156. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Lang, F.; Schrors, B.; Lower, M.; Tureci, O.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Tureci, O.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Larcombe-Young, D.; Papa, S.; Maher, J. PanErbB-targeted CAR T-cell immunotherapy of head and neck cancer. Expert. Opin. Biol. Ther. 2020, 20, 965–970. [Google Scholar] [CrossRef]
| mRNA COVID-19 Vaccines | mRNA Vaccines for Cancer | |
|---|---|---|
| Purpose | To trigger an immune response against a virus. | To stimulate the immune system to target and destroy cancer cells. |
| Antigen target | Spike protein of the SARS-CoV-2 virus. | Target tumor-specific/tumor-associated antigens unique to cancer cells or overexpressed in cancer antigens. Would be patient-specific or common among certain cancer types. |
| Immuneresponse | To generate neutralizing antibodies and activate the immune system to recognize and attack the SARS-CoV-2 virus. | To stimulate a robust T-cell-mediated immune response to target and eliminate cancer cells. Focus is on cytotoxic T cells. |
| Personalization | Not personalized. The same for everyone receiving the vaccine. | Could be designed to be personalized based on the patient’s specific tumor antigens (neoantigens), making them unique to each patient. |
| Immunogenicity | Spike protein is highly immunogenic vaccines induce a strong and rapid immune response. | Tumor antigens might not be very immunogenic, Additional strategies might be required to enhance their immunogenicity. |
| Clinical development | Been developed in a remarkably short timeframe due to the urgency of the COVID-19 pandemic. Enrollment of thousands healthy individuals in clinical trials. | Still in various stages of clinical development and face a longer and more complex path to approval, Clinical trials with a limited patient pool. |
| Manufacturing and distribution | Manufactured and distributed globally on a large scale to address the pandemic, They require distribution chains. | Manufacturing and distribution would be patient-specific or limited to specific cancer types, A different logistical challenge. |
| Clinical outcome | Significant efficacy in preventing COVID-19 infection and severe disease in large clinical trials. | Efficacy and clinical outcome vary by the type of cancer, patient-specific factors, and the stage of development. |
| CAR T-Cells | CAR NK-Cells | |
|---|---|---|
| Cell Origin | - Usually derived from the patient’s own T-cells (autologous) - Universal T-cells via genetic engineering (in clinical trials) | - Derived from various sources, including patient, healthy donors, induced pluripotent stem cells (iPSCs), and cell lines - They are often allogeneic, making them an off-the-shelf treatment option |
| Target antigens | - Specific antigens expressed on the surface of cancer cells - Target antigen is predetermined - Usually, a protein associated with the cancer (TAAs/TSAs) | - NK cells have the inherent ability to recognize a broad spectrum of antigens on target cells - This makes them potentially suitable for a wider range of cancer types and other diseases |
| Specificity | - Highly specific to the chosen target antigen - They may not have the same natural ability to recognize and kill cancer cells as NK cells | - They combine the specificity of CARs with the natural cytotoxicity of NK cells, allowing them to target and kill cancer cells both specifically and through their innate mechanisms |
| Graft-vs-Host Disease (GvHD) | - Risk of GvHD when using allogeneic CAR T-cells, as they are derived from a donor and can recognize normal host tissues as foreign | -Less likely to cause GvHD due to their natural ability to distinguish between healthy and abnormal cells |
| Manufacturing Complexity | - Manufacturing could be complex and time-consuming, often requiring genetic modification, expansion, as well as selection of T-cells, cryopreservation, and transport facilities | - Manufacturing is generally simpler and faster, making them more accessible for patients |
| Cytokine Release Syndrome (CRS) | - CAR T-cell therapy is associated with a higher risk of CRS, a potentially severe immune reaction -Specialized personnel in hospital is needed to counteract the CRS | - CAR NK cells have a significant lower risk of causing CRS, which is a major advantage in terms of safety |
| Persistence | - CAR T-cells tend to persist in the body | - CAR NK cells may exhibit exhaustion - Trials to enhance their persistence via 3rd generation CAR NK cells and adjustment of their bioenergetics needs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miliotou, A.N.; Georgiou-Siafis, S.K.; Ntenti, C.; Pappas, I.S.; Papadopoulou, L.C. Recruiting In Vitro Transcribed mRNA against Cancer Immunotherapy: A Contemporary Appraisal of the Current Landscape. Curr. Issues Mol. Biol. 2023, 45, 9181-9214. https://doi.org/10.3390/cimb45110576
Miliotou AN, Georgiou-Siafis SK, Ntenti C, Pappas IS, Papadopoulou LC. Recruiting In Vitro Transcribed mRNA against Cancer Immunotherapy: A Contemporary Appraisal of the Current Landscape. Current Issues in Molecular Biology. 2023; 45(11):9181-9214. https://doi.org/10.3390/cimb45110576
Chicago/Turabian StyleMiliotou, Androulla N., Sofia K. Georgiou-Siafis, Charikleia Ntenti, Ioannis S. Pappas, and Lefkothea C. Papadopoulou. 2023. "Recruiting In Vitro Transcribed mRNA against Cancer Immunotherapy: A Contemporary Appraisal of the Current Landscape" Current Issues in Molecular Biology 45, no. 11: 9181-9214. https://doi.org/10.3390/cimb45110576