




An Innovative Approach to Address Neurodegenerative Diseases through Kinase-Targeted Therapies: Potential for Designing Covalent Inhibitors
Abstract
:
1. Introduction
1.1. Protein Kinases in Drug Discovery
1.1.1. Protein Kinases in Neurodegenerative Diseases
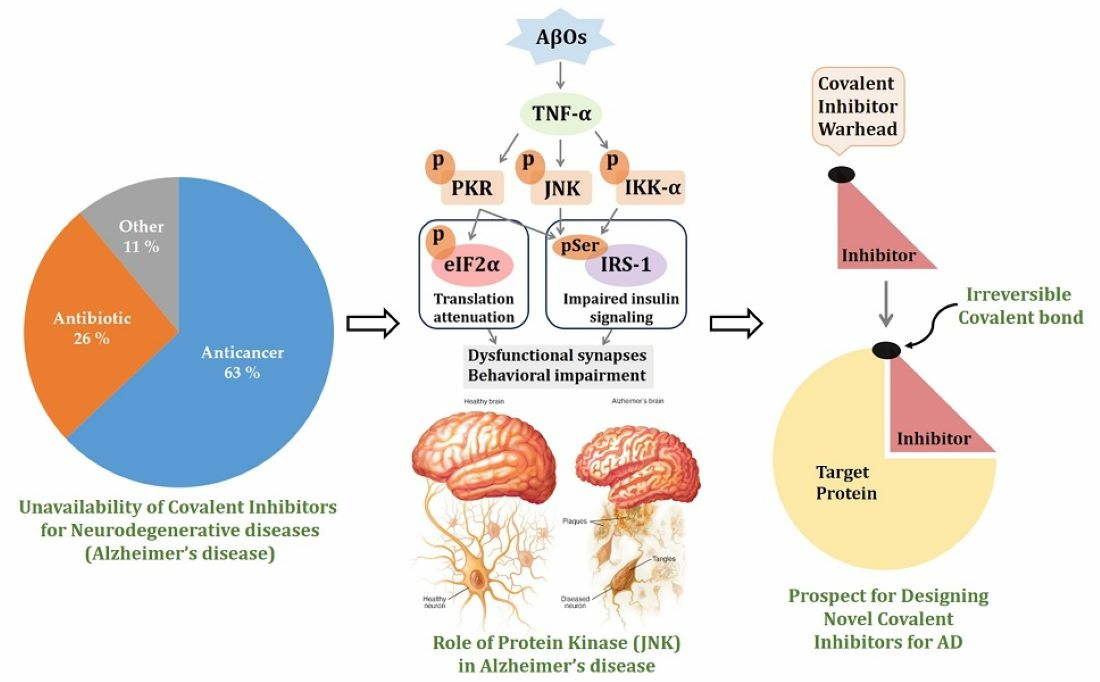
1.1.2. JNK3 in Neurodegenerative Diseases
1.2. Recent Studies on Neurodegenerative Diseases
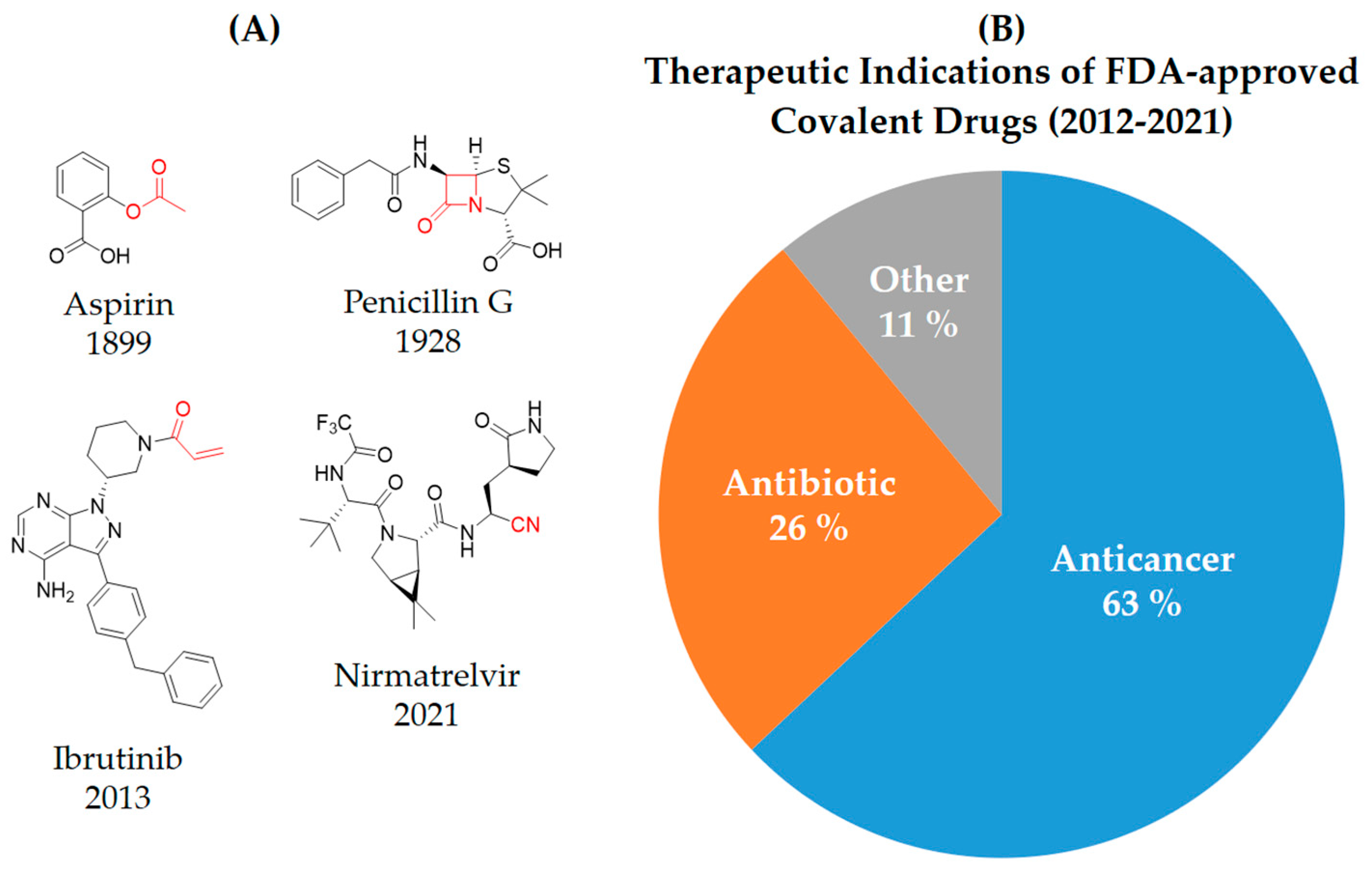
2. Covalent Inhibitors
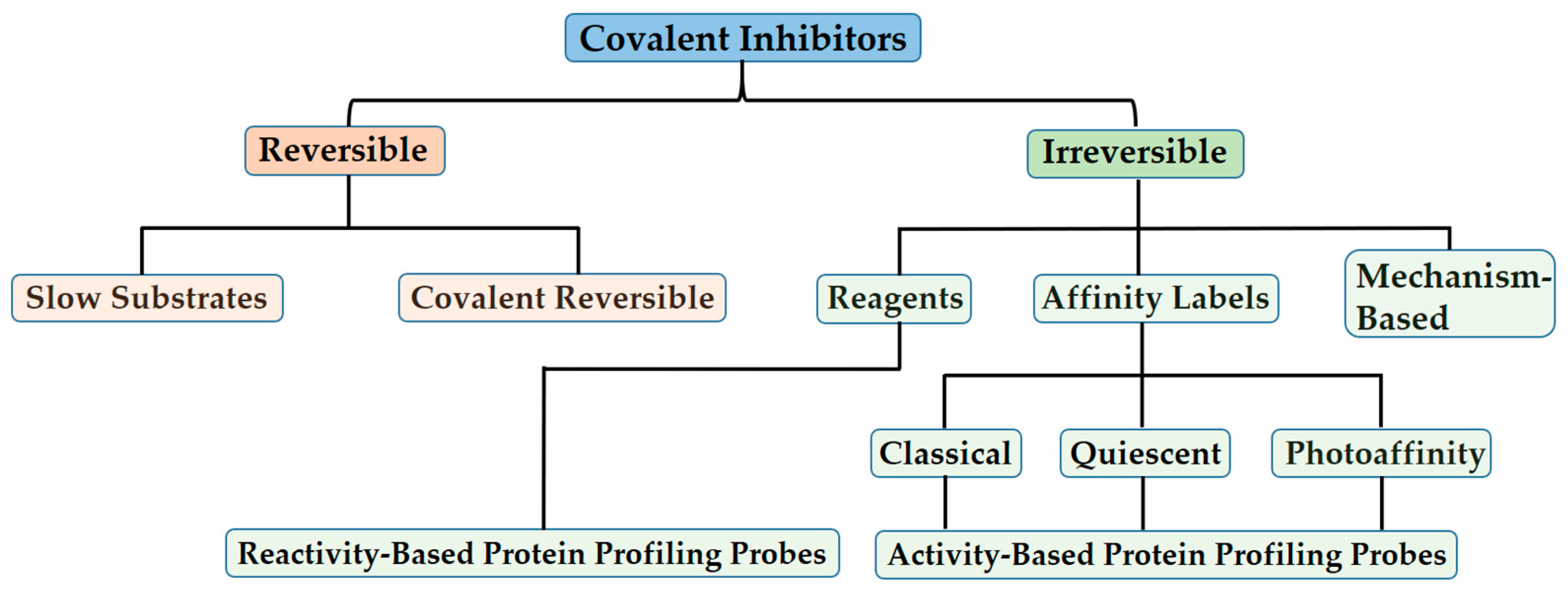
3. Types of Covalent Inhibitors
3.1. Covalent Reversible Inhibitors
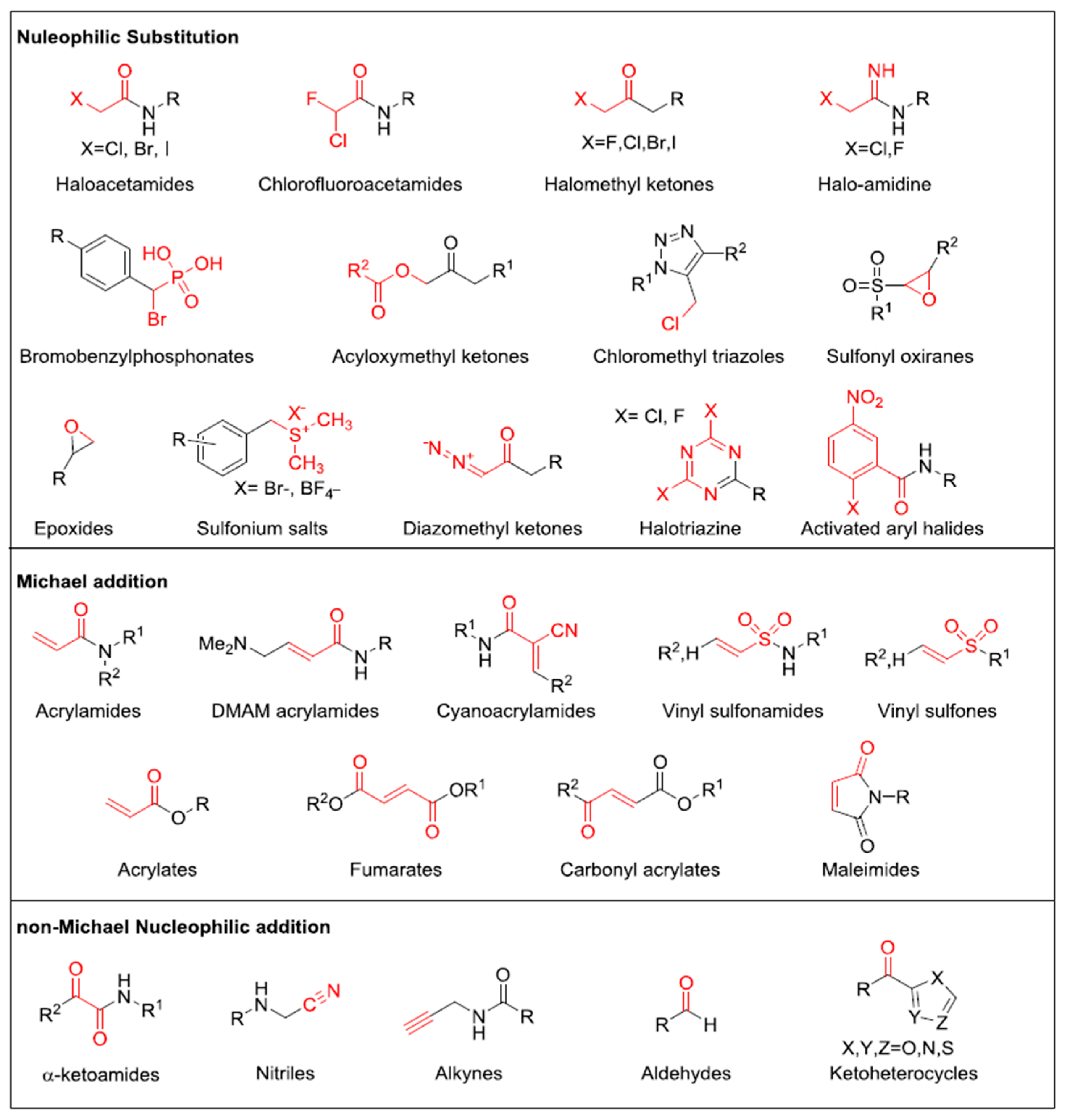
3.2. Covalent Irreversible Inhibitors
3.2.1. Residue-Specific Reagents
3.2.2. Affinity Labels
3.2.3. Covalent Mechanism-Based Enzyme Inactivators
4. Advantages and Disadvantages of Covalent Inhibitors
4.1. Potency
4.2. Pharmacodynamics (PD)
4.3. Drug Dosing
4.4. Drug Resistance
4.5. Target Scope
5. Disadvantages
6. Need for Covalent Kinase Inhibitors
6.1. Importance of Cysteine Residue
6.2. Opportunities for Covalent Kinase Inhibitors (CKIs) by Targeting Kinases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Cao, J.; Maddox, S.M. Opportunities and challenges for the development of covalent chemical immunomodulators. Bioorg. Med. Chem. 2019, 27, 3421–3439. [Google Scholar]
- Cheng, H.; Nair, S.K.; Murray, B.W. Recent progress on third generation covalent EGFR inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1861–1868. [Google Scholar] [CrossRef]
- Fan, S.; Yue, L.; Wan, W.; Zhang, Y.; Zhang, B.; Otomo, C.; Li, Q.; Lin, T.; Hu, J.; Xu, P.; et al. Inhibition of autophagy by a small molecule through covalent modification of the LC3 protein. Angew. Chem. Int. Ed. 2021, 60, 26105–26114. [Google Scholar] [CrossRef]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative diseases: Regenerative mechanisms and novel therapeutic approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- David, M.A.; Tayebi, M.J. Detection of protein aggregates in brain and cerebrospinal fluid derived from multiple sclerosis patients. Front. Neurol. 2014, 5, 251. [Google Scholar] [CrossRef]
- Altman, J. Are new neurons formed in the brains of adult mammals? Science 1962, 135, 1127–1128. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Dickinson-Anson, H.; Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996, 16, 2027–2033. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking traumatic brain injury to chronic traumatic encephalopathy: Identification of potential mechanisms leading to neurofibrillary tangle development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef]
- Hatters, D.M. Protein misfolding inside cells: The case of huntingtin and Huntington’s disease. IUBMB Life 2008, 60, 724–728. [Google Scholar] [CrossRef]
- Woolley, J.D.; Khan, B.K.; Murthy, N.K.; Miller, B.L.; Rankin, K.P. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: Rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J. Clin. Psychiatry 2011, 72, 4437. [Google Scholar] [CrossRef]
- Vadakkan, K.I. Neurodegenerative disorders share common features of “loss of function” states of a proposed mechanism of nervous system functions. BioMedicine 2016, 83, 412–430. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimer Dement. 2012, 8, 131–168. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.B. Extended Applications of Small-Molecule Covalent Inhibitors toward Novel Therapeutic Targets. Pharmaceuticals 2022, 15, 1478. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.P.; Hah, J.-M. An Intriguing Purview on the Design of Macrocyclic Inhibitors for Unexplored Protein Kinases through Their Binding Site Comparison. Pharmaceuticals 2023, 16, 1009. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.; Yang, S.; Lee, J.; Moon, H.; Kim, J.; Jung, H.; Im, D.; Oh, Y.; Jang, M.; Cho, H.J. Discovery of novel imidazole chemotypes as isoform-selective JNK3 inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2023, 245, 114894. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.; Moon, H.; Yang, S.; Lee, J.; Baek, J.; Kim, H.; Cho, H.; Hwang, K.; Ahn, S.; Kim, Y.J.; et al. Carbamate JNK3 Inhibitors Show Promise as Effective Treatments for Alzheimer’s Disease: In Vivo Studies on Mouse Models. J. Med. Chem. 2023, 66, 6372–6390. [Google Scholar] [CrossRef]
- Chen, Y.; He, H.; Lin, B.; Chen, Y.; Deng, X.; Jiang, W.; Zhou, R. RRx-001 ameliorates inflammatory diseases by acting as a potent covalent NLRP3 inhibitor. Cell. Mol. Immunol. 2021, 18, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Bratkowski, M.; Burdett, T.C.; Danao, J.; Wang, X.; Mathur, P.; Gu, W.; Beckstead, J.A.; Talreja, S.; Yang, Y.-S.; Danko, G.; et al. Uncompetitive, adduct-forming SARM1 inhibitors are neuroprotective in preclinical models of nerve injury and disease. Neuron 2022, 110, 3711–3726.e3716. [Google Scholar] [CrossRef]
- Huang, J.; Chen, L.; Wu, J.; Ai, D.; Zhang, J.-Q.; Chen, T.-G.; Wang, L. Targeting the PI3K/AKT/mTOR signaling pathway in the treatment of human diseases: Current status, trends, and solutions. J. Med. Chem. 2022, 65, 16033–16061. [Google Scholar] [CrossRef] [PubMed]
- Tuley, A.; Fast, W. The taxonomy of covalent inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A.J. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Mukherjee, H.; Grimster, N.P. Beyond cysteine: Recent developments in the area of targeted covalent inhibition. Curr. Opin. Chem. Biol. 2018, 44, 30–38. [Google Scholar] [CrossRef]
- Singh, J. The ascension of targeted covalent inhibitors. J. Med. Chem. 2022, 65, 5886–5901. [Google Scholar] [CrossRef]
- Chen, P.; Sun, J.; Zhu, C.; Tang, G.; Wang, W.; Xu, M.; Xiang, M.; Zhang, C.J.; Zhang, Z.M.; Gao, L.; et al. Cell-Active, Reversible, and Irreversible Covalent Inhibitors That Selectively Target the Catalytic Lysine of BCR-ABL Kinase. Angew. Chem. Int. Ed. 2022, 61, e202203878. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, J.P.; Le, X.; Vijayan, R.; Hicks, J.K.; Heeke, S.; Elamin, Y.Y.; Lin, H.Y.; Udagawa, H.; Skoulidis, F.; Tran, H.; et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 2021, 597, 732–737. [Google Scholar]
- Copeland, R. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide to Chemists and Pharmacologists; Wiley-Interscience: Hoboken, NJ, USA, 2005. [Google Scholar]
- Holdgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132. [Google Scholar]
- Silverman, R. Introduction in “Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology”; CRC Press: Boca Raton, FL, USA, 1988; Volume 1. [Google Scholar]
- So, W.H.; Zhang, Y.; Kang, W.; Wong, C.T.; Sun, H.; Xia, J. Site-selective covalent reactions on proteinogenic amino acids. Curr. Opin. Biotechnol. 2017, 48, 220–227. [Google Scholar] [PubMed]
- Fernhoff, N.B.; Derbyshire, E.R.; Marletta, M.A. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc. Natl. Acad. Sci. USA 2009, 106, 21602–21607. [Google Scholar] [CrossRef]
- Plapp, B.V. Application of affinity labeling for studying structure and function of enzymes. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 87, pp. 469–499. [Google Scholar]
- Silverman, R.B. Mechanism-based enzyme inactivators. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1995; Volume 249, pp. 240–283. [Google Scholar]
- Abdeldayem, A.; Raouf, Y.S.; Constantinescu, S.N.; Moriggl, R.; Gunning, P.T. Advances in covalent kinase inhibitors. Chem. Soc. Rev. 2020, 49, 2617–2687. [Google Scholar] [PubMed]
- Ward, R.A.; Colclough, N.; Challinor, M.; Debreczeni, J.E.; Eckersley, K.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J. Med. Chem. 2015, 58, 4790–4801. [Google Scholar]
- Smith, A.J.; Zhang, X.; Leach, A.G.; Houk, K. Beyond picomolar affinities: Quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar]
- Swaika, A.; Hammond, W.A.; Joseph, R.W. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol. Immunol. 2015, 67, 4–17. [Google Scholar]
- Van Kasteren, S.I.; Neefjes, J.; Ovaa, H. Creating molecules that modulate immune responses. Nat. Rev. Chem. 2018, 2, 184–193. [Google Scholar]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar]
- Hildeman, D.A.; Mitchell, T.; Kappler, J.; Marrack, P. T cell apoptosis and reactive oxygen species. J. Clin. Investig. 2003, 111, 575–581. [Google Scholar] [PubMed]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goettl, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. The Journal of the American Society of Hematology. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood J. Am. Soc. Hematol. 2012, 120, 4621–4634. [Google Scholar]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar]
- Smith, G.A.; Uchida, K.; Weiss, A.; Taunton, J. Essential biphasic role for JAK3 catalytic activity in IL-2 receptor signaling. Nat. Chem. Biol. 2016, 12, 373–379. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Structure | Targets | Therapeutic Indication | Warhead | Ref (Approval Date) |
|---|---|---|---|---|
 Wortmannin | Kinase (PI3K) | N/A | Furan | [24] |
 A5 | Kinase (BCR-ABL) | Anticancer (chronic myeloid leukemia) | Aldehyde | [28] |
 Acalabrutinib | Kinase (BTK) | Anticancer (mantle cell lymphoma | 2-butyneamide | 31 October 2017 |
 Zanubrutinib | Kinase (BTK) | Anticancer (mantle cell lymphoma | Acrylamide | 14 November 2019 |
 Afatinib | Kinase (EGFR T790M and pan-HER) | Anticancer (NSCLC) | Acrylamide | 12 July 2013 |
 Neratinib | Kinase (pan-HER) | Anticancer (breast cancer) | Acrylamide | 17 July 2017 |
 Dacomitinib | Kinase (pan-HER) | Anticancer (NSCLC) | Acrylamide | 27 September 2018 |
 Mobocertinib | Kinase (EGFR ex20ins) | Anticancer (NSCLC) | Acrylamide | 15 September 2021 |
 Lazertinib | Kinase (EGFR) | Anticancer (NSCLC) | Acrylamide | Accelerated approval, 21 May 2021, combination with amivantamab |
 Nazartinib | Kinase (EGFR) | Anticancer (NSCLC) | Acrylamide | [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhujbal, S.P.; Hah, J.-M. An Innovative Approach to Address Neurodegenerative Diseases through Kinase-Targeted Therapies: Potential for Designing Covalent Inhibitors. Pharmaceuticals 2023, 16, 1295. https://doi.org/10.3390/ph16091295
Bhujbal SP, Hah J-M. An Innovative Approach to Address Neurodegenerative Diseases through Kinase-Targeted Therapies: Potential for Designing Covalent Inhibitors. Pharmaceuticals. 2023; 16(9):1295. https://doi.org/10.3390/ph16091295
Chicago/Turabian StyleBhujbal, Swapnil P., and Jung-Mi Hah. 2023. "An Innovative Approach to Address Neurodegenerative Diseases through Kinase-Targeted Therapies: Potential for Designing Covalent Inhibitors" Pharmaceuticals 16, no. 9: 1295. https://doi.org/10.3390/ph16091295