


Design of β-Keto Esters with Antibacterial Activity: Synthesis, In Vitro Evaluation, and Theoretical Assessment of Their Reactivity and Quorum-Sensing Inhibition Capacity

 , , , and
, , , and
Abstract
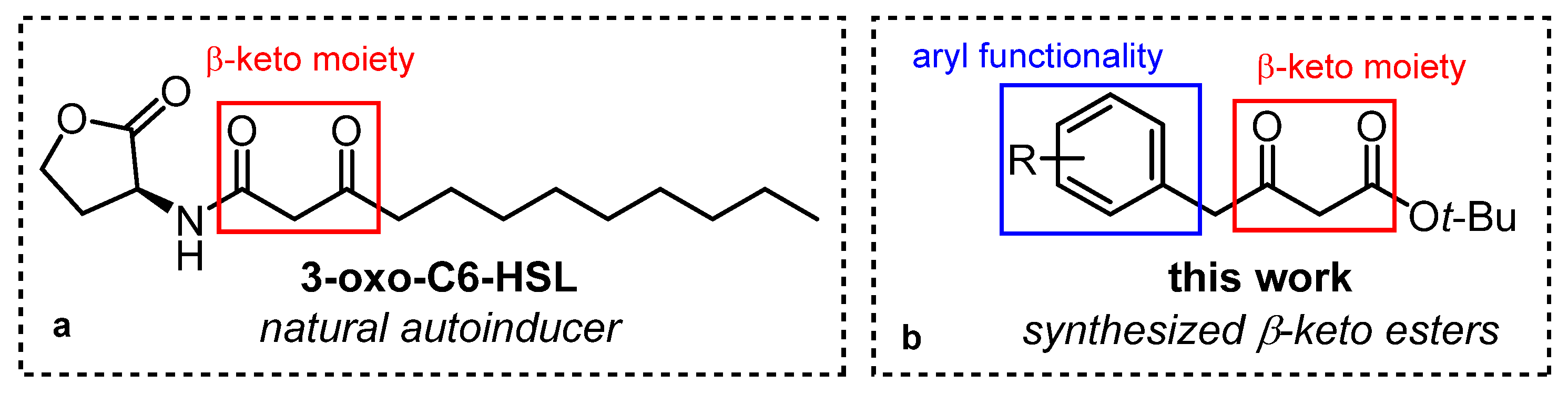
:1. Introduction
2. Results and Discussion
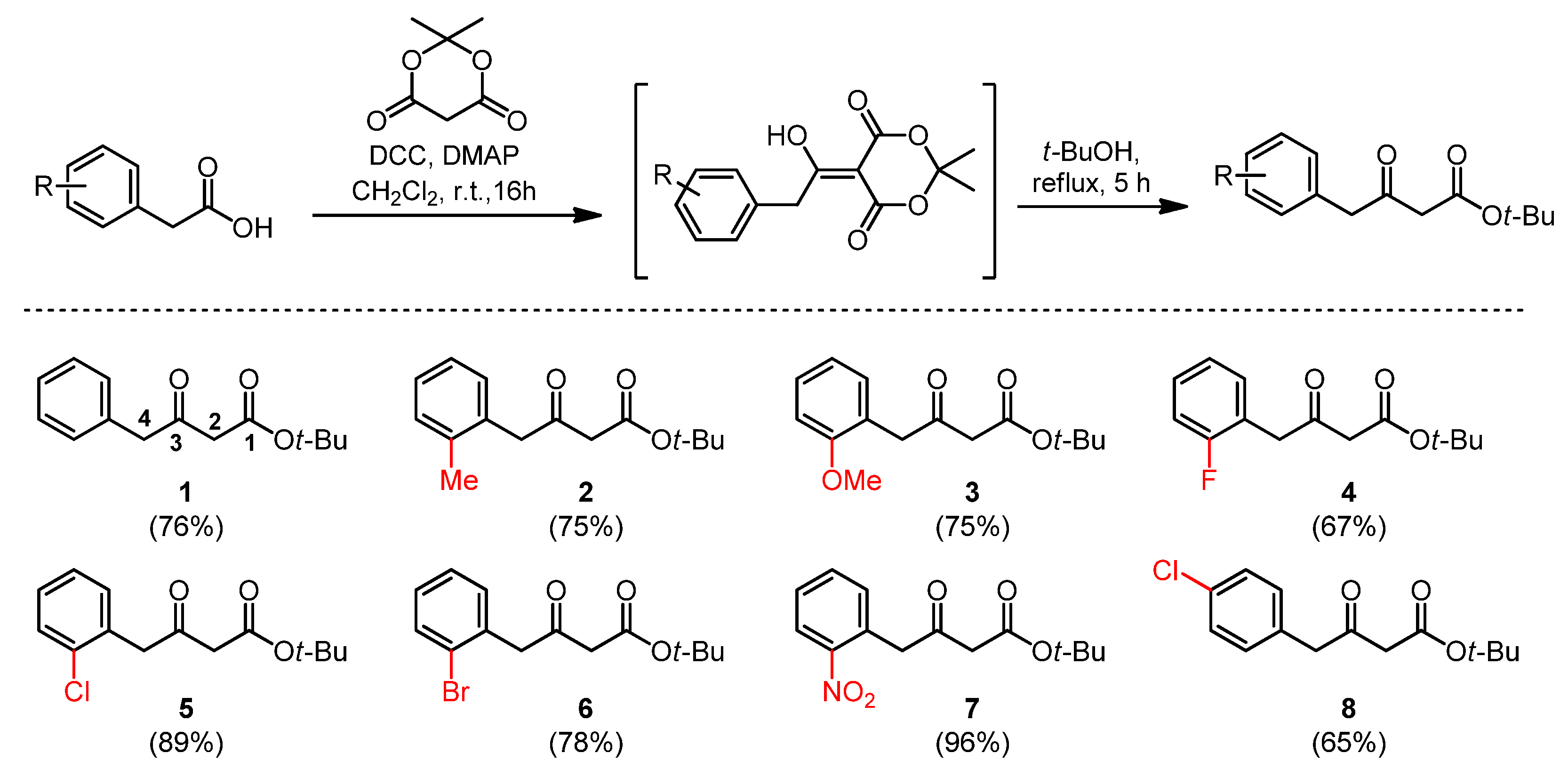
2.1. Synthesis and Spectroscopic Characterisation of β-Keto Esters
2.2. Computational Analysis of the Reactivity and ADME Properties
2.2.1. ADME Properties of the β-Keto Esters
2.2.2. Reactivity Indices Based on Electronic Structure
2.3. In Silico Analysis of Quorum-Sensing Activity
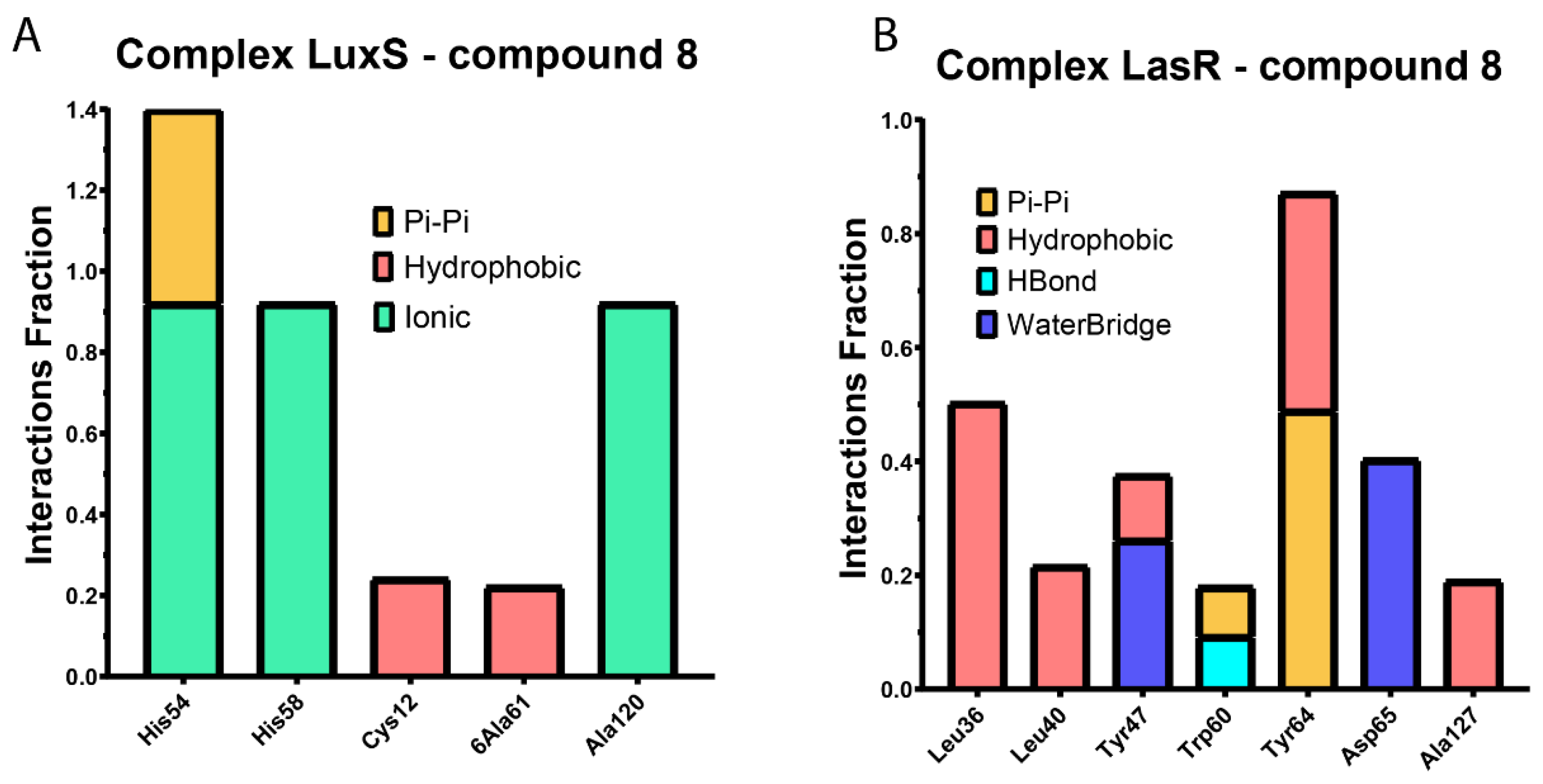
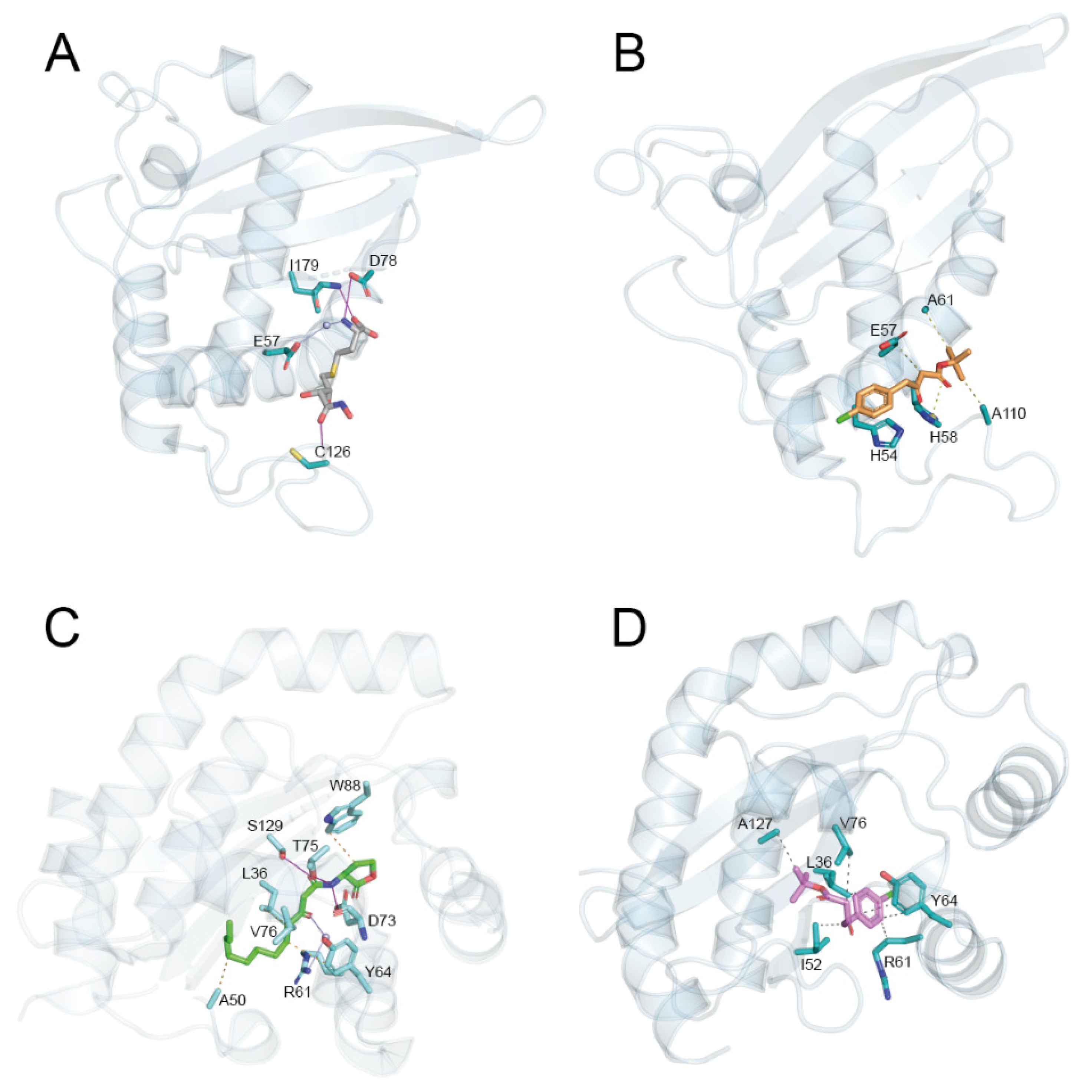
2.3.1. Molecular Docking
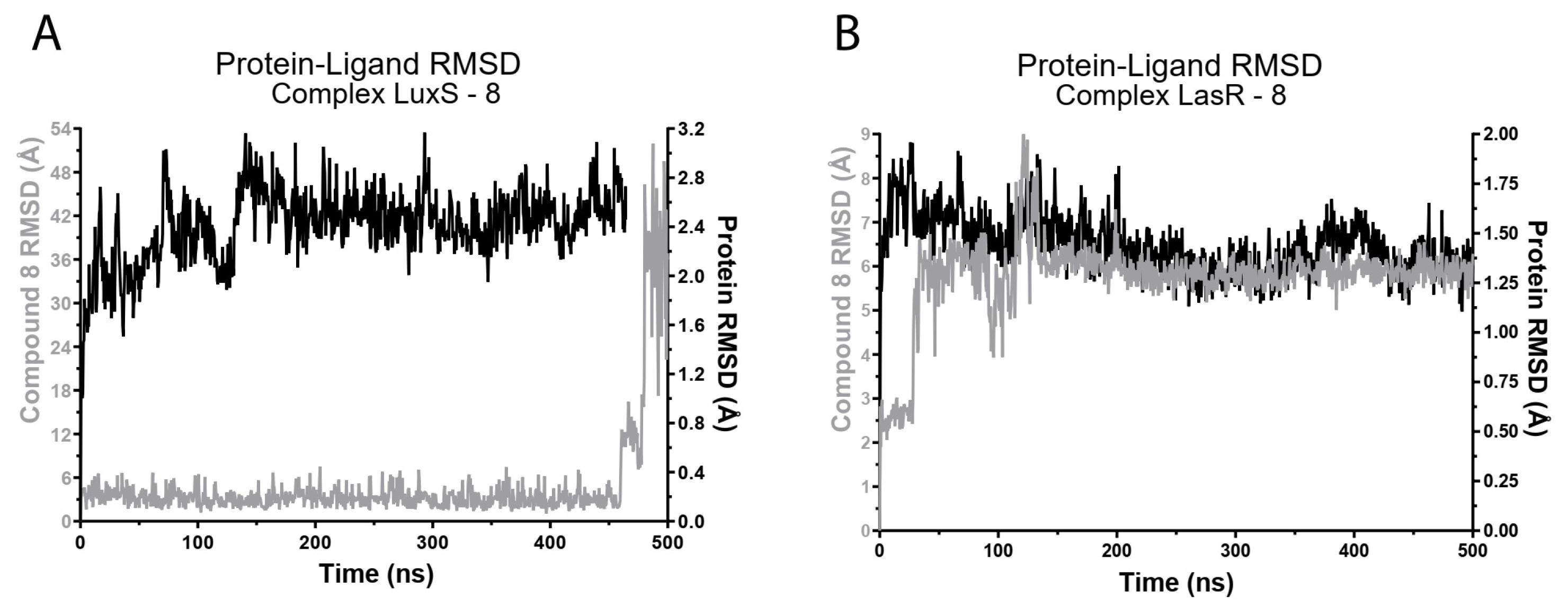
2.3.2. Molecular Dynamics
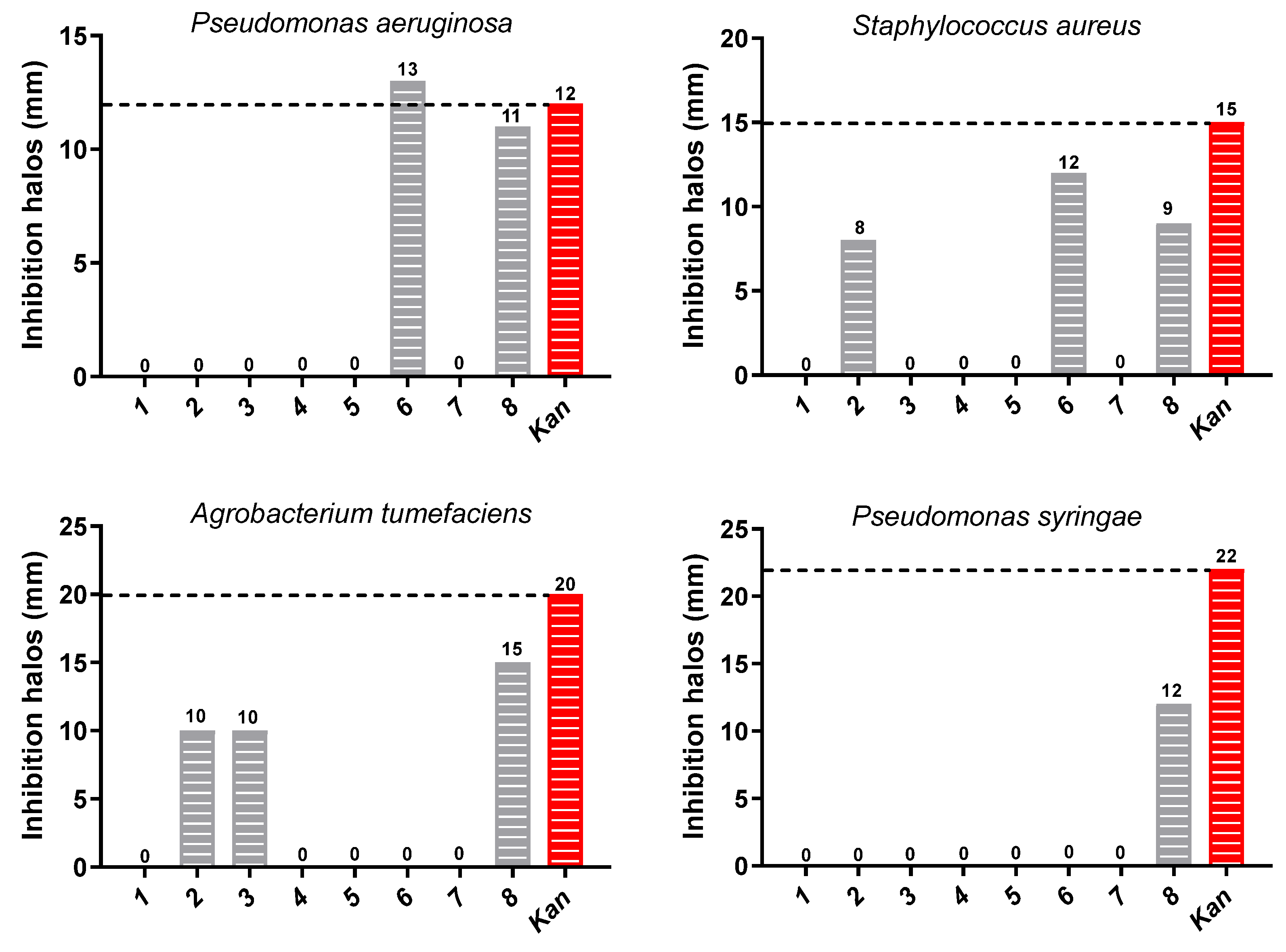
2.4. Antibacterial Activity
3. Materials and Methods
3.1. Synthesis of β-Keto Esters
3.2. ADME Properties’ Evaluation
3.3. Quantum Chemical Calculation
3.4. Docking and DM Calculations
3.5. Antibacterial Activity
3.5.1. Strain and Growth Conditions
3.5.2. Paper-Disk Diffusion Method
3.5.3. Minimum Inhibitory Concentration (MIC)
3.5.4. Minimum Bactericidal Concentration (MCB)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antimicrobial Resistance Division; WHO. Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014; p. 256. [Google Scholar]
- Neu, H.C. The crisis in antibiotic-resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Huigens, R.W.; Richards, J.J.; Parise, G.; Ballard, T.E.; Zeng, W.; Deora, R.; Melander, C. Inhibition of Pseudomonas aeruginosa biofilm formation with bromoageliferin analogues. J. Am. Chem. Soc. 2007, 129, 6966–6967. [Google Scholar] [CrossRef] [PubMed]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The Formation of Biofilms by Pseudomonas aeruginosa: A Review of the Natural and Synthetic Compounds Interfering with Control Mechanisms. Biomed. Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef]
- Engebrecht, J.; Nealson, K.; Silverman, M. Bacterial Bioluminescence—Isolation and Genetic-Analysis of Functions from Vibrio fischeri. Cell 1983, 32, 773–781. [Google Scholar] [CrossRef]
- Geske, G.D.; Oneill, J.C.; Miller, D.M.; Wezeman, R.J.; Mattmann, M.E.; Lin, Q.; Blackwell, H.E. Comparative analyses of N-acylated homoserine Lactones reveal unique structural features that dictate their ability to activate or inhibit quorum sensing. Chembiochem 2008, 9, 389–400. [Google Scholar] [CrossRef]
- Wei, Z.Y.; Li, T.; Gu, Y.; Zhang, Q.; Wang, E.H.; Li, W.B.; Wang, X.; Li, Y.; Li, H.Y. Design, Synthesis, and Biological Evaluation of N-Acyl-Homoserine Lactone Analogs of Quorum Sensing in Pseudomonas aeruginosa. Front. Chem. 2022, 10, 948687. [Google Scholar] [CrossRef]
- Qiang, Z.; Li, S.Z.; Queneau, Y.; Soulere, L. Synthesis of new 1,4-and 1,5-disubstituted N-ethyl acetate and N-alpha-butyro-gamma-lactone alkylimidazole derivatives as N-acylhomoserine lactone analogs. J. Heterocycl. Chem. 2021, 58, 2298–2303. [Google Scholar] [CrossRef]
- Frezza, M.; Soulere, L.; Reverchon, S.; Guiliani, N.; Jerez, C.; Queneau, Y.; Doutheau, A. Synthetic homoserine lactone-derived sulfonylureas as inhibitors of Vibrio fischeri quorum sensing regulator. Bioorg. Med. Chem. 2008, 16, 3550–3556. [Google Scholar] [CrossRef] [PubMed]
- Manson, D.E.; O’Reilly, M.C.; Nyffeler, K.E.; Blackwell, H.E. Design, Synthesis, and Biochemical Characterization of Non-Native Antagonists of the Pseudomonas aeruginosa Quorum Sensing Receptor LasR with Nanomolar IC50 Values. ACS Infect. Dis. 2020, 6, 649–661. [Google Scholar] [CrossRef]
- Marsden, D.M.; Nicholson, R.L.; Skindersoe, M.E.; Galloway, W.; Sore, H.F.; Givskov, M.; Salmond, G.P.C.; Ladlow, M.; Welchd, M.; Spring, D.R. Discovery of a quorum sensing modulator pharmacophore by 3D small-molecule microarray screening. Org. Biomol. Chem. 2010, 8, 5313–5323. [Google Scholar] [CrossRef] [PubMed]
- Boursier, M.E.; Combs, J.B.; Blackwell, H.E. N-Acyl L-Homocysteine Thiolactones Are Potent and Stable Synthetic Modulators of the RhlR Quorum Sensing Receptor in Pseudomonas aeruginosa. ACS Chem. Biol. 2019, 14, 186–191. [Google Scholar] [CrossRef]
- Guo, N.; Bai, X.; Shen, Y.; Zhang, T.H. Target-based screening for natural products against Staphylococcus aureus biofilms. Crit. Rev. Food Sci. Nutr. 2023, 63, 2216–2230. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.B.; Grenier, D.; Yi, L. Regulatory Mechanisms of the LuxS/AI-2 System and Bacterial Resistance. Antimicrob. Agents Chemother. 2019, 63, 12. [Google Scholar] [CrossRef]
- Xavier, K.B.; Bassler, B.L. LuxS quorum sensing: More than just a numbers game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef]
- Zhang, B.Z.; Ku, X.G.; Zhang, X.Q.; Zhang, Y.; Chen, G.; Chen, F.Z.; Zeng, W.; Li, J.; Zhu, L.; He, Q.G. The AI-2/luxS Quorum Sensing System Affects the Growth Characteristics, Biofilm Formation, and Virulence of Haemophilus parasuis. Front. Cell. Infect. Microbiol. 2019, 9, 15. [Google Scholar] [CrossRef]
- Murray, E.J.; Crowley, R.C.; Truman, A.; Clarke, S.R.; Cottam, J.A.; Jadhav, G.P.; Steele, V.R.; O’Shea, P.; Lindholm, C.; Cockayne, A.; et al. Targeting Staphylococcus aureus Quorum Sensing with Nonpeptidic Small Molecule Inhibitors. J. Med. Chem. 2014, 57, 2813–2819. [Google Scholar] [CrossRef]
- Forschner-Dancause, S.; Poulin, E.; Meschwitz, S. Quorum Sensing Inhibition and Structure-Activity Relationships of beta-Keto Esters. Molecules 2016, 21, 971. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Fujita, T. P-σ-π Analysis: A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Issa, N.T.; Wathieu, H.; Ojo, A.; Byers, S.W.; Dakshanamurthy, S. Drug Metabolism in Preclinical Drug Development: A Survey of the Discovery Process, Toxicology, and Computational Tools. Curr. Drug Metab. 2017, 18, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, R.; Subramanian, V.; Roy, D.R.; Chattaraj, P.K. Electrophillicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Group philicity and electrophilicity as possible descriptors for modeling ecotoxicity applied to chlorophenols. Chem. Res. Toxicol. 2006, 19, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Patra, S.G.; Chattaraj, P.K. Quantitative Structure-Toxicity Relationship in Bioactive Molecules from a Conceptual DFT Perspective. Pharmaceuticals 2022, 15, 1383. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Geohagen, B.C.; Nordstroem, L.U. Mechanisms of soft and hard electrophile toxicities. Toxicology 2019, 418, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Gacitua, M.; Carreno, A.; Morales-Guevara, R.; Paez-Hernandez, D.; Martinez-Araya, J.I.; Araya, E.; Preite, M.; Otero, C.; Rivera-Zaldivar, M.M.; Silva, A.; et al. Physicochemical and Theoretical Characterization of a New Small Non-Metal Schiff Base with a Differential Antimicrobial Effect against Gram-Positive Bacteria. Int. J. Mol. Sci. 2022, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Zamora, P.P.; Bieger, K.; Cuchillo, A.; Tello, A.; Muena, J.P. Theoretical determination of a reaction intermediate: Fukui function analysis, dual reactivity descriptor and activation energy. J. Mol. Struct. 2021, 1227, 129369. [Google Scholar] [CrossRef]
- Hennessy, M.C.; O’Sullivan, T.P. Recent advances in the transesterification of beta-keto esters. RSC Adv. 2021, 11, 22859–22920. [Google Scholar] [CrossRef]
- Rao, G.B.D.; Anjaneyulu, B.; Kaushik, M.P. Greener and expeditious one-pot synthesis of dihydropyrimidinone derivatives using non-commercial beta-ketoesters via the Biginelli reaction. RSC Adv. 2014, 4, 43321–43325. [Google Scholar] [CrossRef]
- Bacha, K.; Tariku, Y.; Gebreyesus, F.; Zerihun, S.; Mohammed, A.; Weiland-Brauer, N.; Schmitz, R.A.; Mulat, M. Antimicrobial and anti-Quorum Sensing activities of selected medicinal plants of Ethiopia: Implication for development of potent antimicrobial agents. BMC Microbiol. 2016, 16, 139. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.T.; Huang, X.Q.; Yang, H.X.; Niu, X.Q.; Li, D.X.; Yang, C.; Li, L.; Zou, L.T.; Qiu, Z.W.; Wu, S.H.; et al. Antibacterial Activity and Anti-Quorum Sensing Mediated Phenotype in Response to Essential Oil from Melaleuca bracteata Leaves. Int. J. Mol. Sci. 2019, 20, 5696. [Google Scholar] [CrossRef] [PubMed]
- Simirgiotis, M.J.; Burton, D.; Parra, F.; López, J.; Muñoz, P.; Escobar, H.; Parra, C. Antioxidant and Antibacterial Capacities of Origanum vulgare L. Essential Oil from the Arid Andean Region of Chile and its Chemical Characterization by GC-MS. Metabolites 2020, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, B.; Parra, C.; Bonjoch, J. Organocatalyzed Asymmetric Synthesis of Morphans. Org. Lett. 2013, 15, 2458–2461. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zou, Y.; Nair, S.K. LasR-OC12 HSL Complex. PDB ID: 3IX3 2009. Available online: https://www.wwpdb.org/pdb?id=pdb_00003ix3 (accessed on 14 September 2023).
- Shen, G.; Rajan, R.; Zhu, J.G.; Bell, C.E.; Pei, D.H. Design and synthesis of substrate and intermediate analogue inhibitors of S-ribosylhomocysteinase. J. Med. Chem. 2006, 49, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins-Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: Manhattan, NY, USA, 2006; p. 43. [Google Scholar]
- Lu, C.; Wu, C.J.; Ghoreishi, D.; Chen, W.; Wang, L.L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Physicochemical Properties | Lipophilicity | Water Solubility | Pharmacokinetics | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MW 1 | Rot. Bond 2 | HB-A 3 | HB-D 4 | TPSA 5 | Consensus Log Po/w 6 | Solubility (mol/L) | GI Abs 7 | BBB 8 | log Kp (cm/s) 9 | |
| 1 | 234.29 | 6 | 3 | 0 | 43.37 | 2.6 | 8.67 × 10−5 | High | Yes | −5.92 |
| 2 | 248.32 | 6 | 3 | 0 | 43.37 | 2.97 | 3.56 × 10−5 | High | Yes | −5.75 |
| 3 | 264.32 | 7 | 4 | 0 | 52.6 | 2.63 | 6.53 × 10−5 | High | Yes | −6.12 |
| 4 | 252.28 | 6 | 4 | 0 | 43.37 | 2.96 | 4.58 × 10−5 | High | Yes | −5.96 |
| 5 | 268.74 | 6 | 3 | 0 | 43.37 | 3.18 | 2.13 × 10−5 | High | Yes | −5.68 |
| 6 | 313.19 | 6 | 3 | 0 | 43.37 | 3.24 | 1.29 × 10−5 | High | Yes | −5.91 |
| 7 | 279.29 | 7 | 5 | 0 | 89.19 | 1.93 | 3.69 × 10−4 | High | No | −6.31 |
| 8 | 268.74 | 6 | 3 | 0 | 43.37 | 3.19 | 2.13 × 10−5 | High | Yes | −5.68 |
| ID | Lipinski # Violations 1 | Ghose # Violations 2 | Veber # Violations 3 | Egan # Violations 4 | Muegge # Violations 5 |
|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0 | 0 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 | 0 | 0 |
| 6 | 0 | 0 | 0 | 0 | 0 |
| 7 | 0 | 0 | 0 | 0 | 0 |
| 8 | 0 | 0 | 0 | 0 | 0 |
| Compound | IPv | EAv | η | μ | ω |
|---|---|---|---|---|---|
| 1 | 8.98 | −0.77 | 9.75 | −4.11 | 0.86 |
| 2 | 8.76 | −0.79 | 9.55 | −3.98 | 0.83 |
| 3 | 8.27 | −0.89 | 9.17 | −3.69 | 0.74 |
| 4 | 9.12 | −0.76 | 9.88 | −4.18 | 0.88 |
| 5 | 9.05 | −0.73 | 9.78 | −4.16 | 0.88 |
| 6 | 8.99 | −0.72 | 9.72 | −4.14 | 0.88 |
| 7 | 9.58 | 0.63 | 8.95 | −5.10 | 1.46 |
| 8 | 8.91 | −0.62 | 9.53 | −4.15 | 0.90 |
| Compound | Atom | fk+ | fk− | fk2 | |
|---|---|---|---|---|---|
| 1 | C1 | 0.53 | −0.18 | 0.71 | 5.56 |
| C3 | 0.84 | 0.21 | 0.63 | 4.90 | |
| 2 | C1 | 0.61 | −0.17 | 0.78 | 6.33 |
| C3 | 0.25 | 0.16 | 0.09 | 0.77 | |
| 3 | C1 | 0.44 | −0.16 | 0.60 | 5.25 |
| C3 | −0.15 | 0.11 | −0.25 | −2.23 | |
| 4 | C1 | 0.34 | −0.18 | 0.51 | 3.90 |
| C3 | −0.11 | 0.25 | −0.36 | −2.71 | |
| 5 | C1 | 0.17 | −0.16 | 0.33 | 2.54 |
| C3 | 0.16 | 0.16 | 0.00 | 0.03 | |
| 6 | C1 | 0.07 | −0.17 | 0.24 | 1.86 |
| C3 | 0.35 | 0.09 | 0.26 | 2.02 | |
| 7 | C1 | −0.14 | −0.28 | 0.14 | 1.30 |
| C3 | 0.26 | 0.83 | −0.57 | −5.27 | |
| 8 | C1 | 0.33 | −0.16 | 0.49 | 4.01 |
| C3 | 0.76 | 0.16 | 0.60 | 4.89 |
| Compound | LuxS | LasR | ||
|---|---|---|---|---|
| Docking Score (kcal/mol) | MMGBSA ΔG Bind (kcal/mol) | Docking Score (kcal/mol) | MMGBSA ΔG Bind (kcal/mol) | |
| 1 | −3.781 | −22.82 | −6.405 | −69.71 |
| 2 | −3.475 | −29.17 | −2.742 | −75.95 |
| 3 | −4.052 | −28.77 | −5.253 | −71.19 |
| 4 | −4.265 | −28.34 | −7.291 | −73.54 |
| 5 | −3.981 | −28.55 | −7.439 | −80.07 |
| 6 | −4.011 | −28.35 | −4.335 | −77.37 |
| 7 | −1.188 | −31.69 | −3.555 | −73.08 |
| 8 | −4.085 | −31.38 | −4.649 | −77.67 |
| Bacteria | MIC 1 (mg/mL) | Kan 3 (µg/mL) | MBC 2 (mg/mL) | Kan 3 (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 6 | 8 | 2 | 3 | 6 | 8 | |||
| Pathogenic | ||||||||||
| Pseudomonas aeruginosa (ATCC 19429) | ND | ND | 0.32 | 0.63 | 5.00 | ND | ND | 2.50 | 5.00 | 10.00 |
| Staphylococcus aureus (ATCC 29737) | 0.63 | ND | 0.63 | 0.32 | 2.50 | 5.00 | ND | 5.00 | 2.50 | 10.00 |
| Phytopathogenic | ||||||||||
| Pseudomonas syringae (MF547632) | ND | ND | ND | 1.25 | 1.25 | ND | ND | ND | 5.00 | 2.50 |
| Agrobacterium tumefaciens (ATCC 19358) | 0.16 | 0.16 | ND | 0.08 | 1.25 | 2.50 | 2.50 | ND | 1.25 | 5.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Soto-Tapia, E.; Linares-Pipón, C.; Bradshaw, B.; Valenzuela-Hormazabal, P.; Ramírez, D.; Muñoz-Torres, P.; Parra, C. Design of β-Keto Esters with Antibacterial Activity: Synthesis, In Vitro Evaluation, and Theoretical Assessment of Their Reactivity and Quorum-Sensing Inhibition Capacity. Pharmaceuticals 2023, 16, 1339. https://doi.org/10.3390/ph16101339
Martínez-Cifuentes M, Soto-Tapia E, Linares-Pipón C, Bradshaw B, Valenzuela-Hormazabal P, Ramírez D, Muñoz-Torres P, Parra C. Design of β-Keto Esters with Antibacterial Activity: Synthesis, In Vitro Evaluation, and Theoretical Assessment of Their Reactivity and Quorum-Sensing Inhibition Capacity. Pharmaceuticals. 2023; 16(10):1339. https://doi.org/10.3390/ph16101339
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Emmanuel Soto-Tapia, Camila Linares-Pipón, Ben Bradshaw, Paulina Valenzuela-Hormazabal, David Ramírez, Patricio Muñoz-Torres, and Claudio Parra. 2023. "Design of β-Keto Esters with Antibacterial Activity: Synthesis, In Vitro Evaluation, and Theoretical Assessment of Their Reactivity and Quorum-Sensing Inhibition Capacity" Pharmaceuticals 16, no. 10: 1339. https://doi.org/10.3390/ph16101339