


Molecular Dynamics Simulations of Drug-Conjugated Cell-Penetrating Peptides


Abstract
:1. Introduction

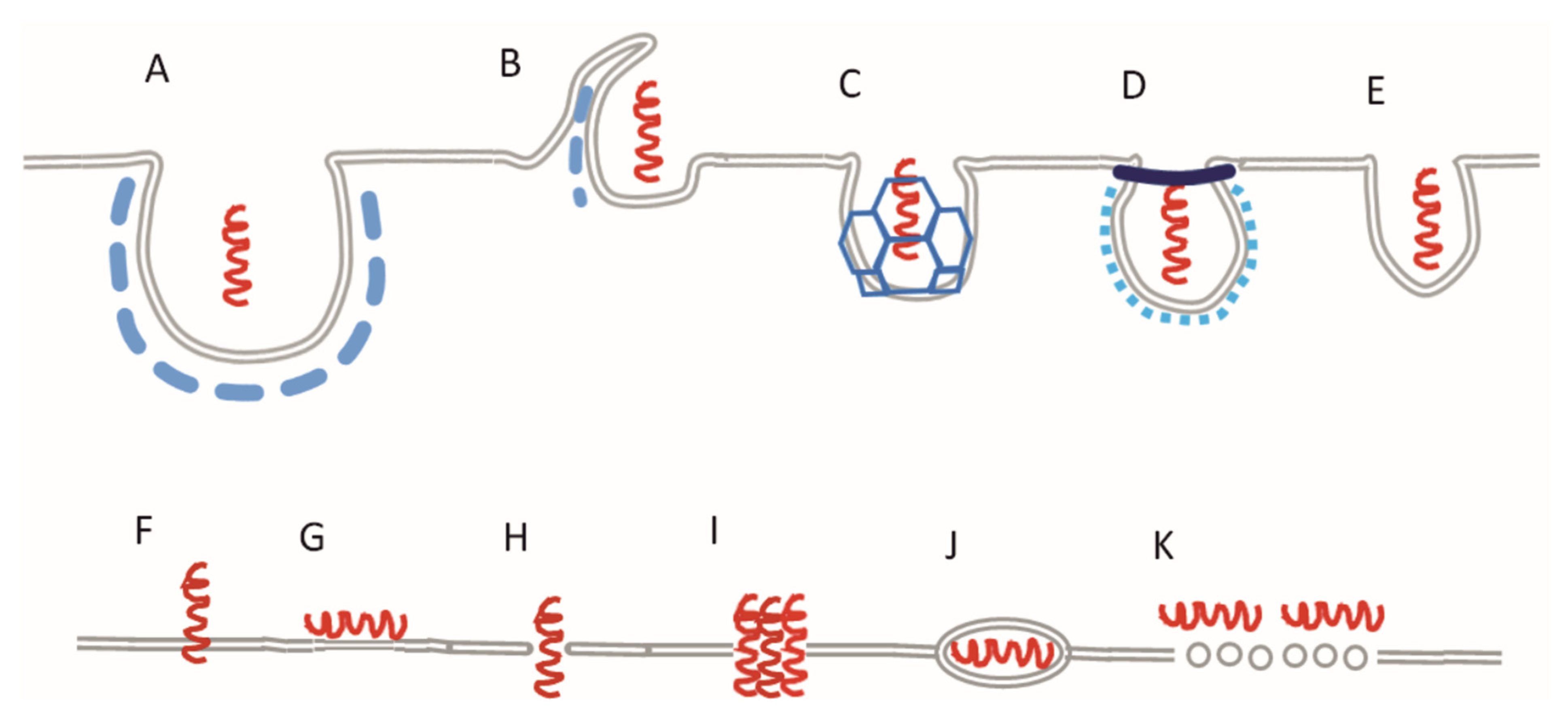{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Molar Weight (g/mol) | Experimental logP |
|---|---|---|
| Doxorubicin | 543.52 | 0.32 [59] |
| Zidovudine | 267.24 | 0.04 [60] |
| Rasagiline | 171.24 | 2.462 [61] |
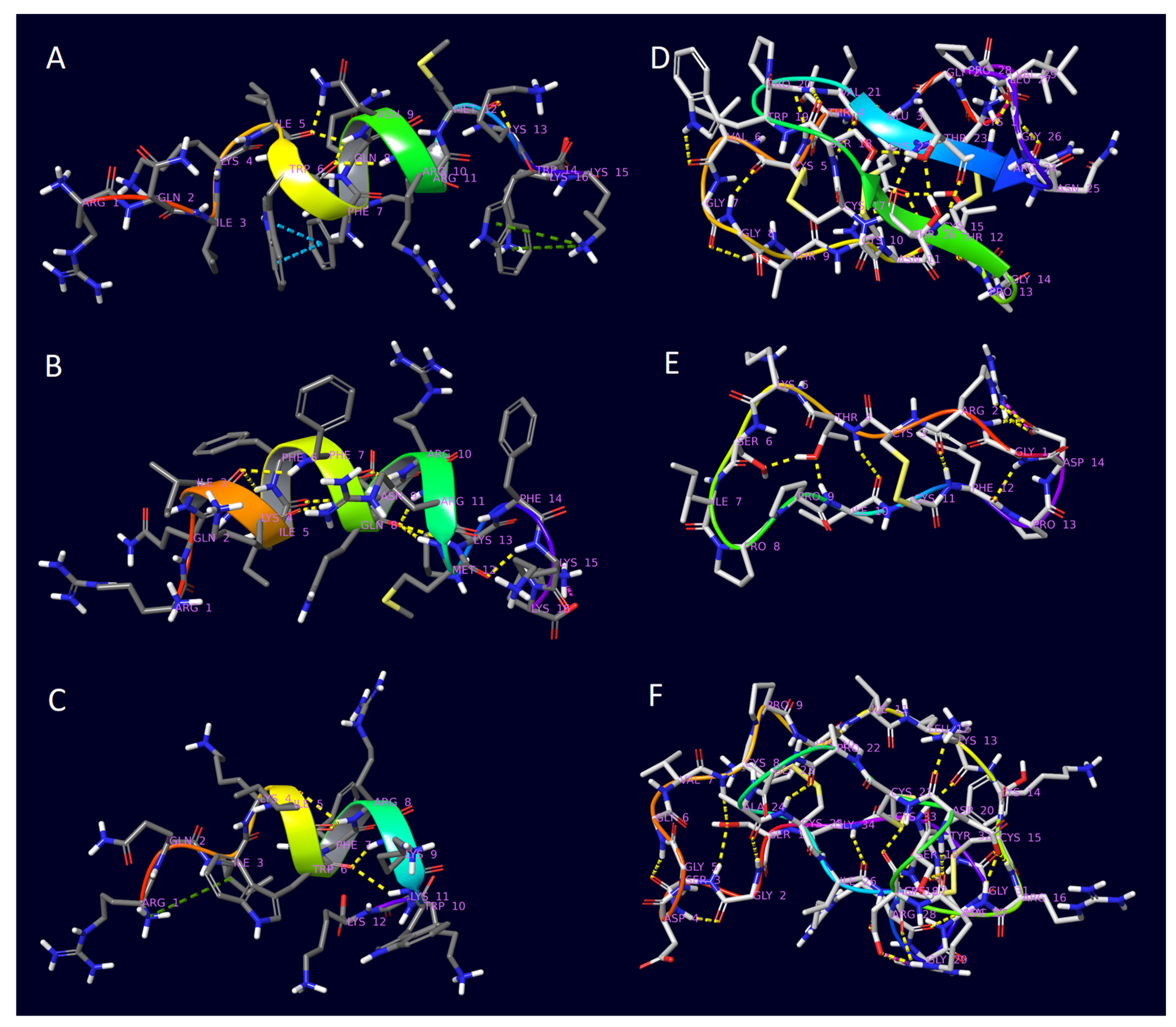
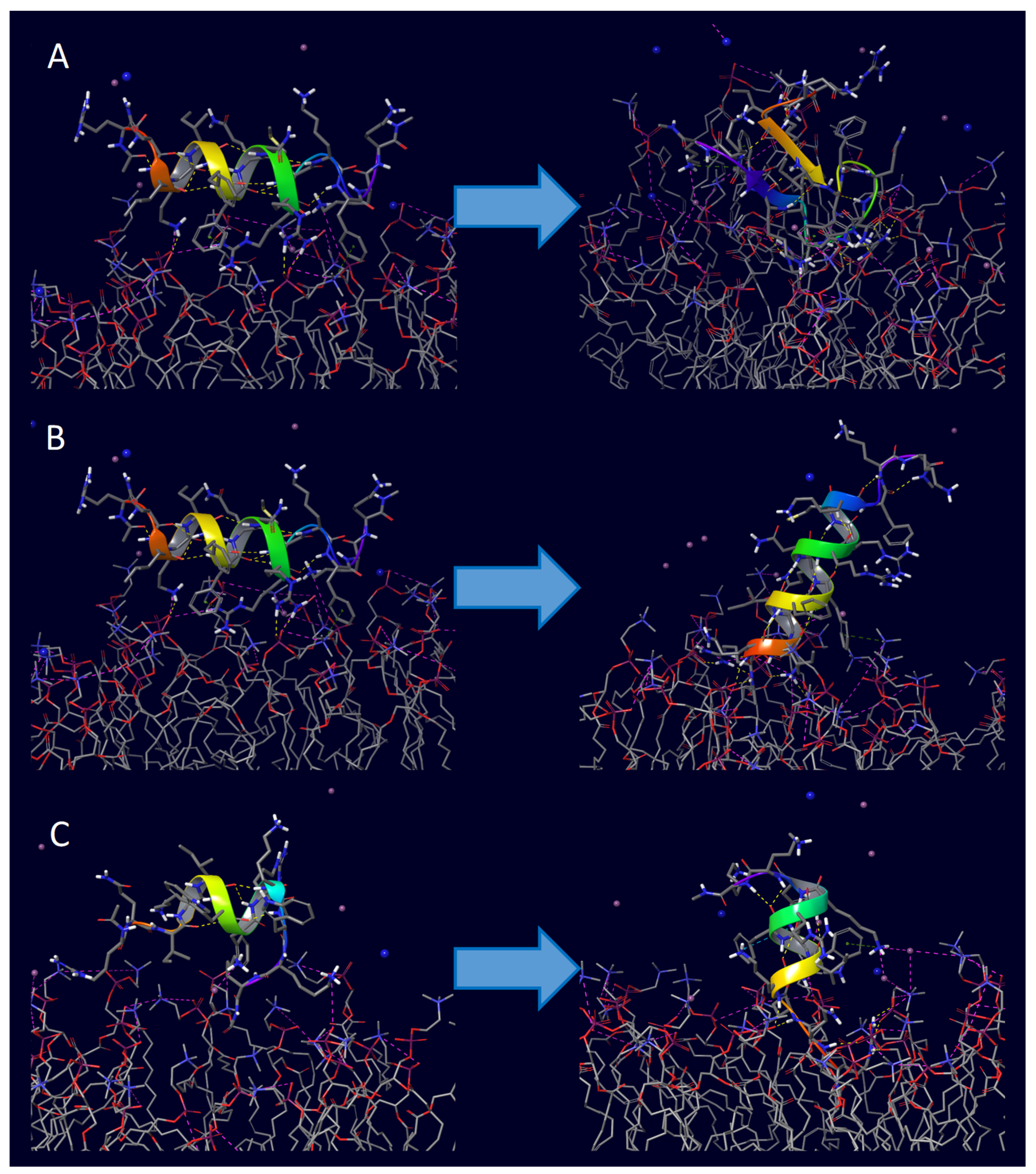
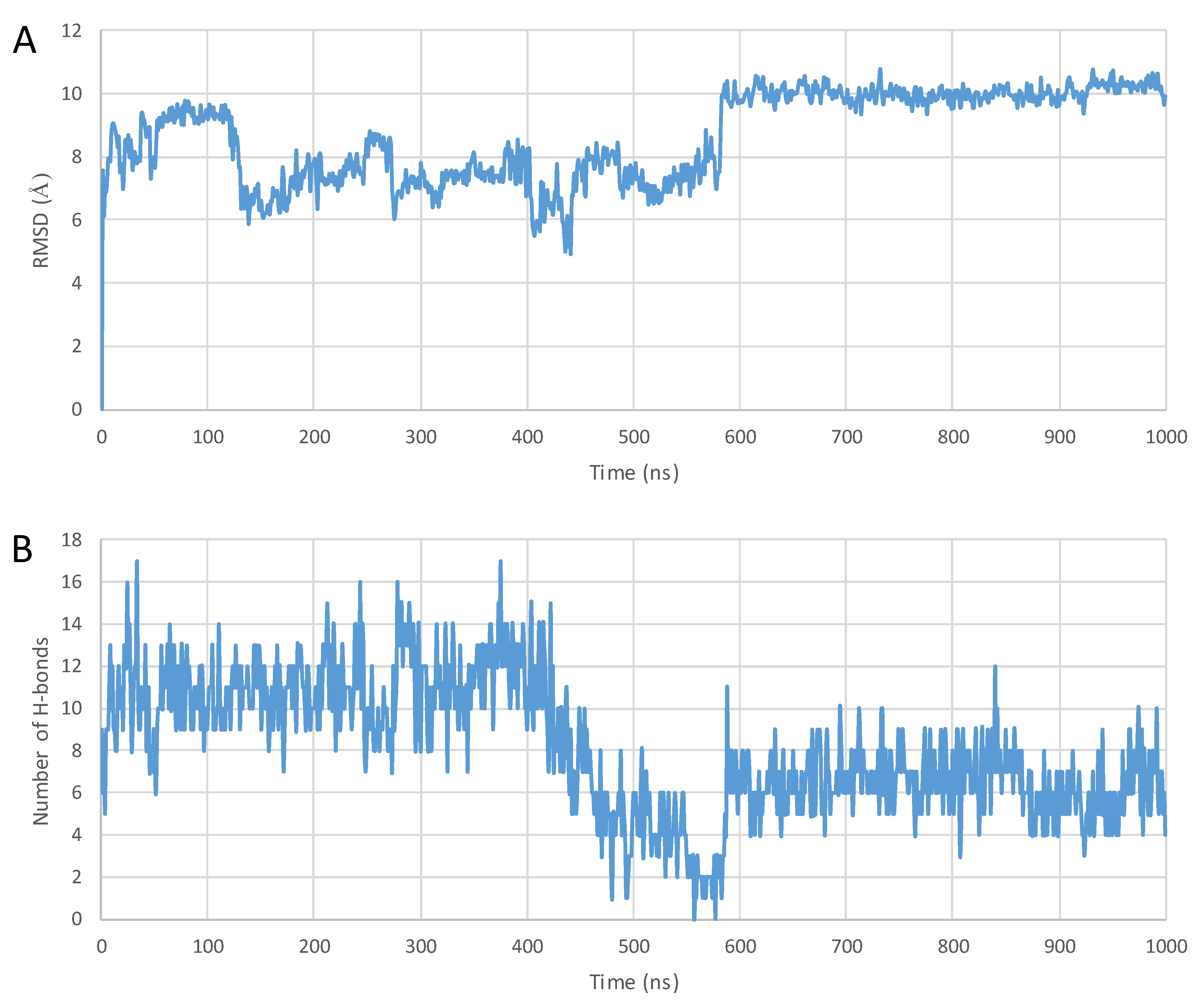
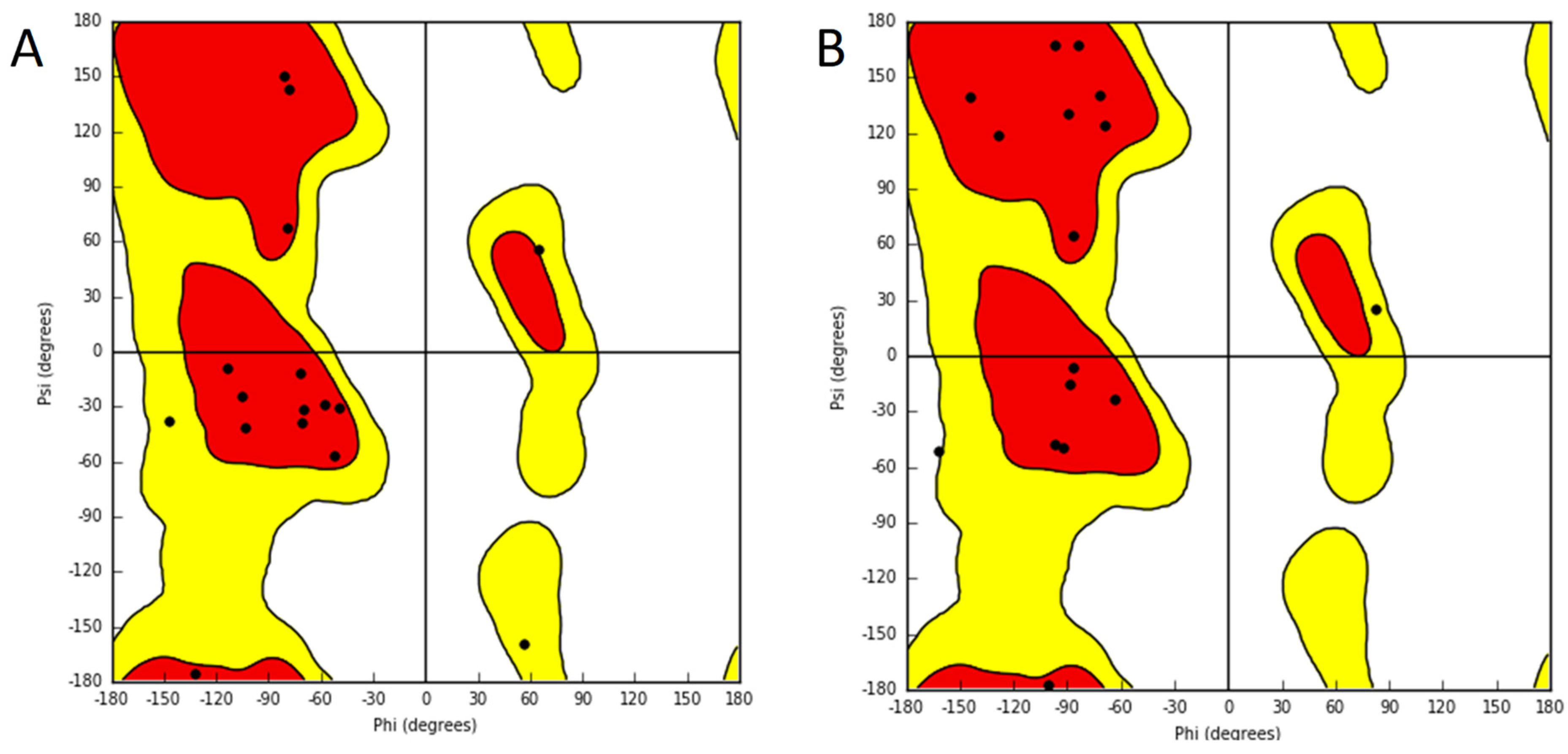
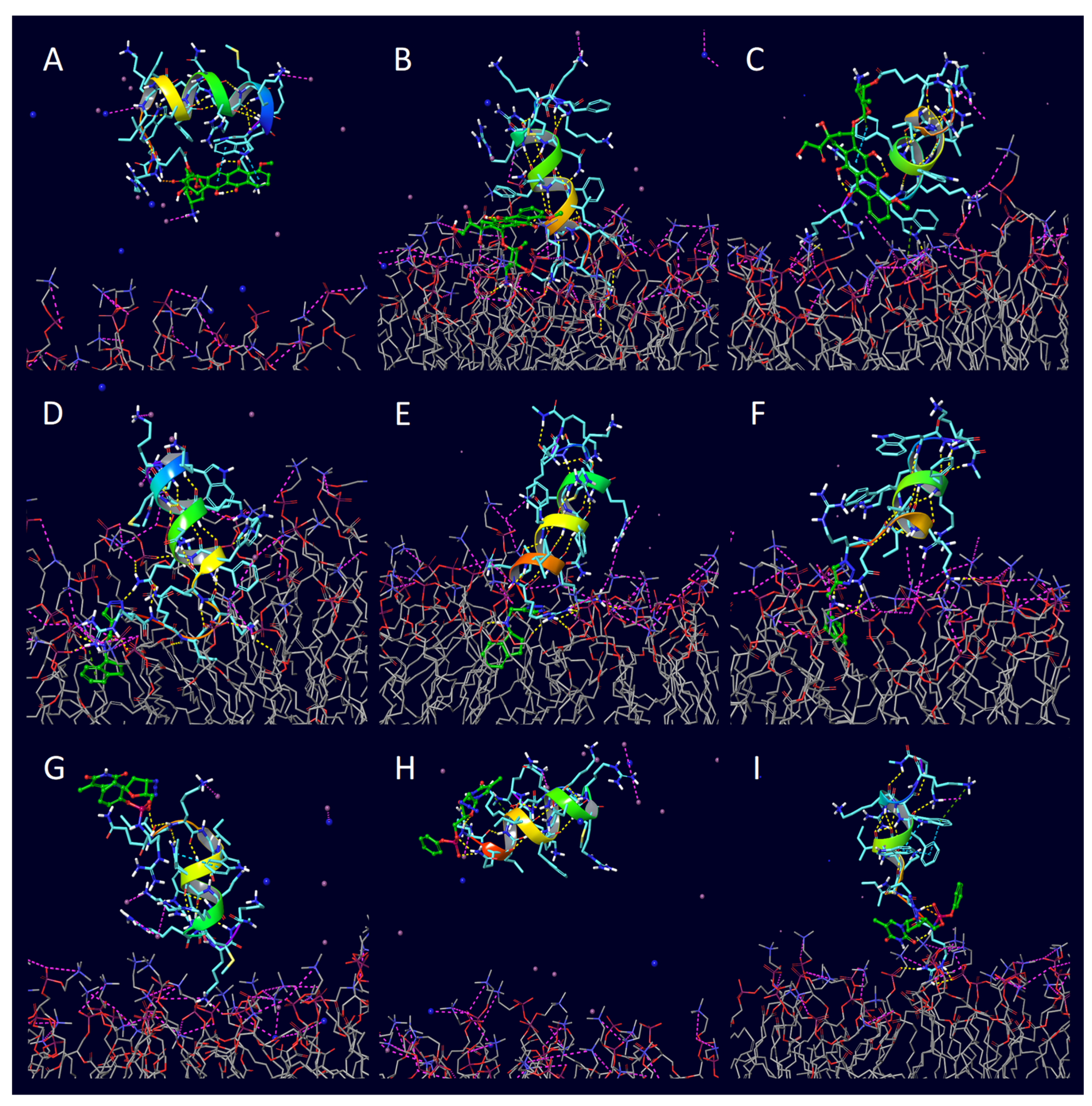
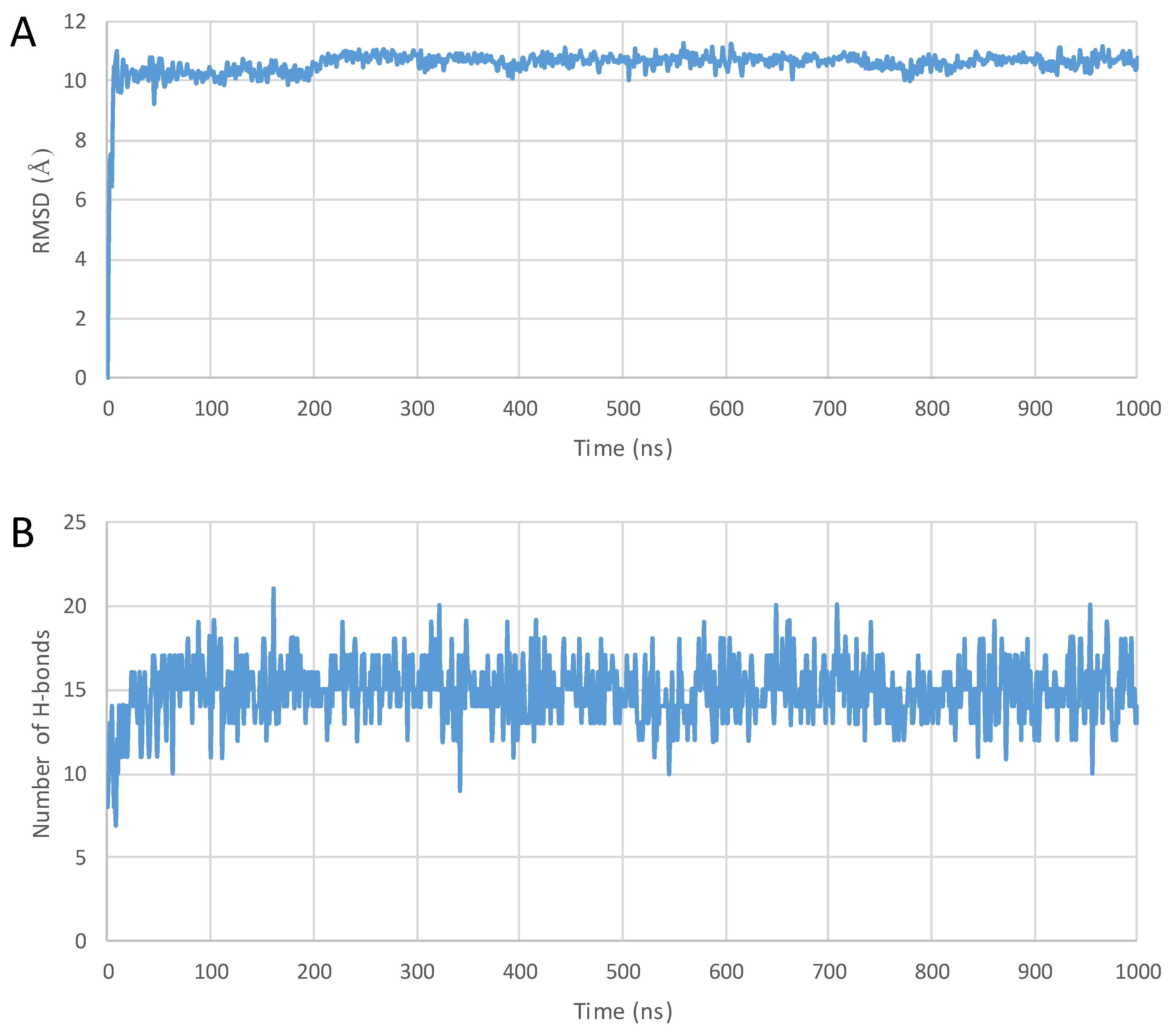
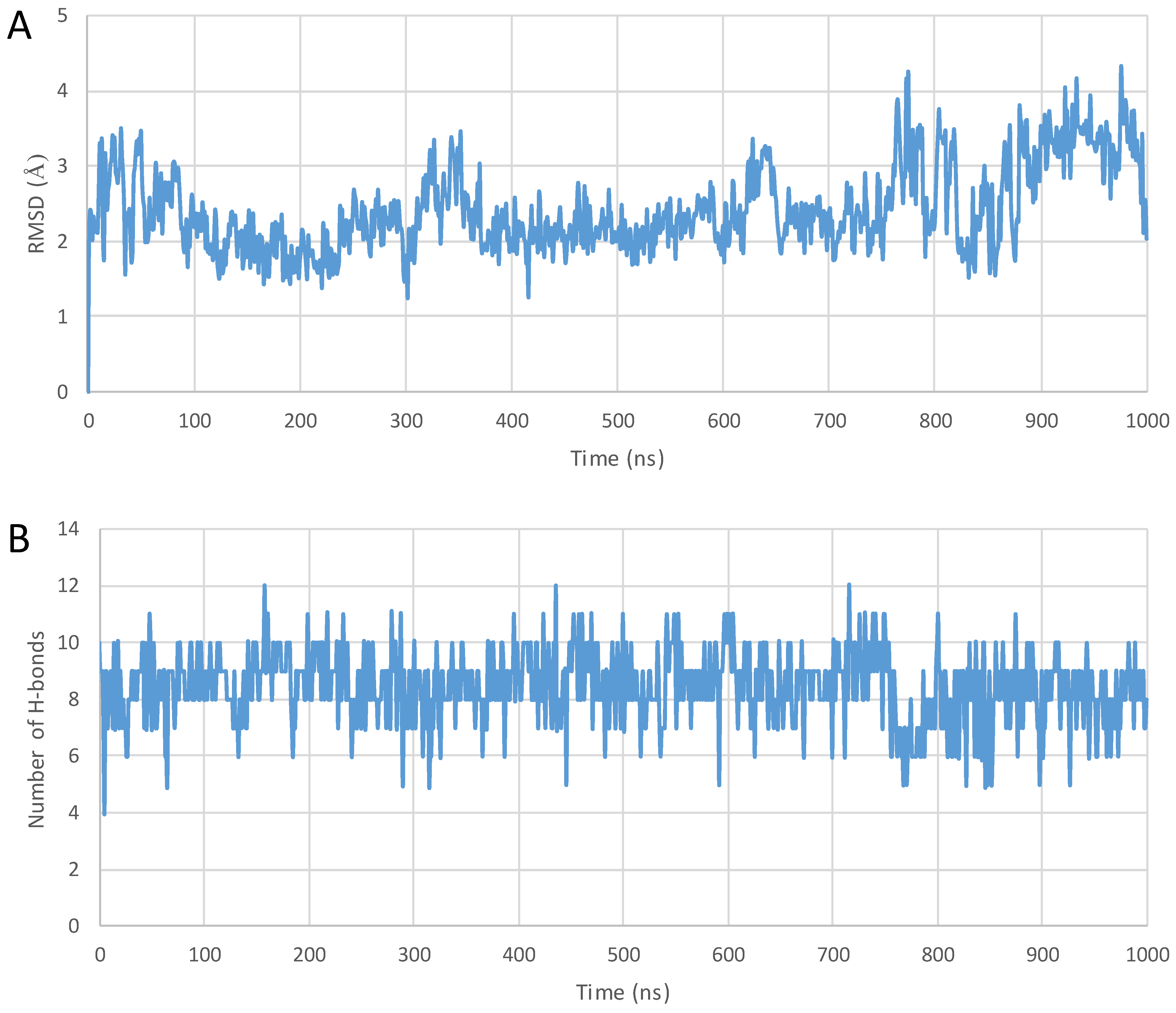
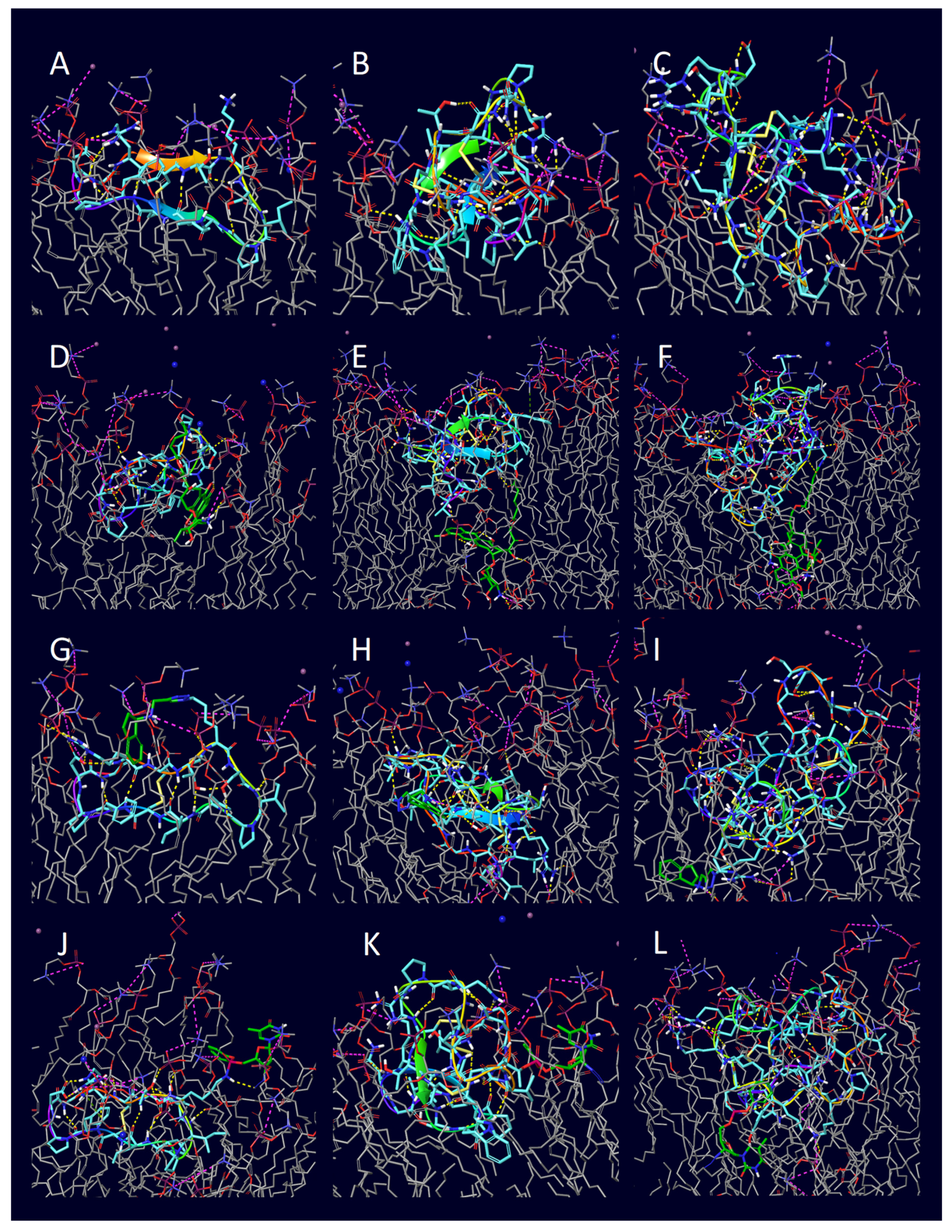
2. Results
3. Discussion
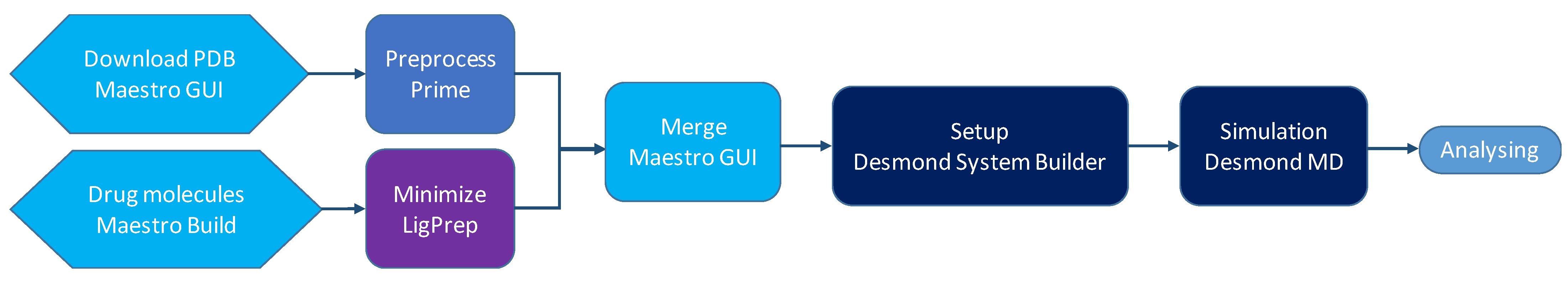
4. Methods
4.1. Preparation of Peptides and Conjugates
4.2. Setup and Building of the Systems
4.3. MD Simulations
4.4. Analyzing the Structures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Jafarzade, B.S.; Mardani, G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides 2017, 87, 50–63. [Google Scholar] [CrossRef]
- Gautam, A.; Singh, H.; Tyagi, A.; Chaudhary, K.; Kumar, R.; Kapoor, P.; Raghava, G.P. CPPsite: A curated database of cell penetrating peptides. Database 2012, 2012, bas015. [Google Scholar] [CrossRef] [PubMed]
- Bashyal, S.; Noh, G.; Keum, T.; Choi, Y.W.; Lee, S. Cell penetrating peptides as an innovative approach for drug delivery; then, present and the future. J. Pharm. Investig. 2016, 46, 205–220. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef]
- Wang, S.; Zhelev, N.Z.; Duff, S.; Fischer, P.M. Synthesis and biological activity of conjugates between paclitaxel and the cell delivery vector penetratin. Bioorg. Med. Chem. Lett. 2006, 16, 2628–2631. [Google Scholar] [CrossRef]
- Diedrichsen, R.G.; Harloff-Helleberg, S.; Werner, U.; Besenius, M.; Leberer, E.; Kristensen, M.; Nielsen, H.M. Revealing the importance of carrier-cargo association in delivery of insulin and lipidated insulin. J. Control. Release 2021, 338, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Pari, E.; Horvati, K.; Bosze, S.; Biri-Kovacs, B.; Szeder, B.; Zsila, F.; Kiss, E. Drug conjugation induced modulation of structural and membrane interaction features of cationic cell-permeable peptides. Int. J. Mol. Sci. 2020, 21, 2197. [Google Scholar] [CrossRef]
- Balogh, B.; Ivanczi, M.; Nizami, B.; Beke-Somfai, T.; Mandity, I.M. ConjuPepDB: A database of peptide-drug conjugates. Nucleic Acids Res. 2021, 49, D1102–D1112. [Google Scholar] [CrossRef]
- Liotard, J.F.; Mehiri, M.; Di Giorgio, A.; Boggetto, N.; Reboud-Ravaux, M.; Aubertin, A.M.; Condom, R.; Patino, N. AZT and AZT-monophosphate prodrugs incorporating HIV-protease substrate fragment: Synthesis and evaluation as specific drug delivery systems. Antivir. Chem. Chemother. 2006, 17, 193–213. [Google Scholar] [CrossRef]
- Nasrolahi Shirazi, A.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Alves, C.; Sharma, V.; Lazaro, D.F.; Silva, S.; Gomes, P.; Outeiro, T.F. A new MAP-Rasagiline conjugate reduces alpha-synuclein inclusion formation in a cell model. Pharmacol. Rep. 2020, 72, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J.; Buchoux, S.; Toupe, J.; Sani, M.A.; Jean-Francois, F.; Khemtemourian, L.; Grelard, A.; Loudet-Courreges, C.; Laguerre, M.; Elezgaray, J.; et al. Membrane interacting peptides: From killers to helpers. Curr. Protein Pept. Sci. 2012, 13, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Asrorov, A.M.; Wang, H.; Zhang, M.; Wang, Y.; He, Y.; Sharipov, M.; Yili, A.; Huang, Y. Cell penetrating peptides: Highlighting points in cancer therapy. Drug Dev. Res. 2023; early view. [Google Scholar] [CrossRef]
- Mansur, A.A.P.; Carvalho, S.M.; Lobato, Z.I.P.; Leite, M.F.; Cunha, A.D.S., Jr.; Mansur, H.S. Design and Development of Polysaccharide-Doxorubicin-Peptide Bioconjugates for Dual Synergistic Effects of Integrin-Targeted and Cell-Penetrating Peptides for Cancer Chemotherapy. Bioconj. Chem. 2018, 29, 1973–2000. [Google Scholar] [CrossRef]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Cell penetrating peptides: How do they do it? J. Biol. Phys. 2007, 33, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Durzynska, J.; Przysiecka, L.; Nawrot, R.; Barylski, J.; Nowicki, G.; Warowicka, A.; Musidlak, O.; Gondzicka-Jozefiak, A. Viral and other cell-penetrating peptides as vectors of therapeutic agents in medicine. J. Pharmacol. Exp. Ther. 2015, 354, 32–42. [Google Scholar] [CrossRef]
- Strandberg, E.; Wadhwani, P.; Burck, J.; Anders, P.; Mink, C.; van den Berg, J.; Ciriello, R.A.M.; Melo, M.N.; Castanho, M.; Bardaji, E.; et al. Temperature-Dependent Re-alignment of the Short Multifunctional Peptide BP100 in Membranes Revealed by Solid-State NMR Spectroscopy and Molecular Dynamics Simulations. ChemBioChem 2023, 24, e202200602. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.C.L.; da Costa, K.S.; Taube, P.S.; Lima, A.H.; Junior, C.d.S.d.S. Biological Membrane-Penetrating Peptides: Computational Prediction and Applications. Front. Cell. Infect. Microbiol. 2022, 12, 838259. [Google Scholar] [CrossRef]
- Kabelka, I.; Brozek, R.; Vacha, R. Selecting Collective Variables and Free-Energy Methods for Peptide Translocation across Membranes. J. Chem. Inf. Model. 2021, 61, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Jobin, M.-L.; Vamparys, L.; Deniau, R.; Grelard, A.; Mackereth, C.D.; Fuchs, P.F.J.; Alves, I.D. Biophysical insight on the membrane insertion of an arginine-rich cell-penetrating peptide. Int. J. Mol. Sci. 2019, 20, 4441. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Garcia, A.E. Free energy of translocating an arginine-rich cell-penetrating peptide across a lipid bilayer suggests pore formation. Biophys. J. 2013, 104, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Vogel, A.; Correia, I.; Lequin, O.; Olausson, B.E.; Desbat, B.; Sagan, S.; Alves, I.D. Membrane interactions of two arginine-rich peptides with different cell internalization capacities. Biochim. Biophys. Acta 2012, 1818, 1755–1763. [Google Scholar] [CrossRef]
- Reid, L.M.; Verma, C.S.; Essex, J.W. The role of molecular simulations in understanding the mechanisms of cell-penetrating peptides. Drug Discov. Today 2019, 24, 1821–1835. [Google Scholar] [CrossRef]
- Gao, X.; Hong, S.; Liu, Z.; Yue, T.; Dobnikar, J.; Zhang, X. Membrane potential drives direct translocation of cell-penetrating peptides. Nanoscale 2019, 11, 1949–1958. [Google Scholar] [CrossRef]
- Trofimenko, E.; Grasso, G.; Heulot, M.; Chevalier, N.; Deriu, M.A.; Dubuis, G.; Arribat, Y.; Serulla, M.; Michel, S.; Vantomme, G.; et al. Genetic, cellular, and structural characterization of the membrane potential-dependent cell-penetrating peptide translocation pore. eLife 2021, 10, e69832. [Google Scholar] [CrossRef] [PubMed]
- Blumer, M.; Harris, S.; Li, M.; Martinez, L.; Untereiner, M.; Saeta, P.N.; Carpenter, T.S.; Ingolfsson, H.I.; Bennett, W.F.D. Simulations of Asymmetric Membranes Illustrate Cooperative Leaflet Coupling and Lipid Adaptability. Front. Cell Dev. Biol. 2020, 8, 575. [Google Scholar] [CrossRef]
- Lattig-Tunnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Christiaens, B.; Vandekerckhove, J.; Prochiantz, A.; Rosseneu, M. Penetratin-membrane association: W48/R52/W56 shield the peptide from the aqueous phase. Biophys. J. 2005, 88, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef] [PubMed]
- Akabori, K.; Huang, K.; Treece, B.W.; Jablin, M.S.; Maranville, B.; Woll, A.; Nagle, J.F.; Garcia, A.E.; Tristram-Nagle, S. HIV-1 Tat membrane interactions probed using X-ray and neutron scattering, CD spectroscopy and MD simulations. Biochim. Biophys. Acta 2014, 1838, 3078–3087. [Google Scholar] [CrossRef]
- Neale, C.; Huang, K.; Garcia, A.E.; Tristram-Nagle, S. Penetration of HIV-1 Tat47-57 into PC/PE Bilayers Assessed by MD Simulation and X-ray Scattering. Membranes 2015, 5, 473–494. [Google Scholar] [CrossRef]
- He, X.; Lin, M.; Sha, B.; Feng, S.; Shi, X.; Qu, Z.; Xu, F. Coarse-grained molecular dynamics studies of the translocation mechanism of polyarginines across asymmetric membrane under tension. Sci. Rep. 2015, 5, 12808. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; Hong, C.K.; Wang, Y.; Tieleman, D.P. Antimicrobial Peptide Simulations and the Influence of Force Field on the Free Energy for Pore Formation in Lipid Bilayers. J. Chem. Theory Comput. 2016, 12, 4524–4533. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; Sapay, N.; Tieleman, D.P. Atomistic simulations of pore formation and closure in lipid bilayers. Biophys. J. 2014, 106, 210–219. [Google Scholar] [CrossRef]
- Alaybeyoglu, B.; Sariyar Akbulut, B.; Ozkirimli, E. Insights into membrane translocation of the cell-penetrating peptide pVEC from molecular dynamics calculations. J. Biomol. Struct. Dyn. 2016, 34, 2387–2398. [Google Scholar] [CrossRef]
- Via, M.A.; Klug, J.; Wilke, N.; Mayorga, L.S.; Del Popolo, M.G. The interfacial electrostatic potential modulates the insertion of cell-penetrating peptides into lipid bilayers. Phys. Chem. Chem. Phys. 2018, 20, 5180–5189. [Google Scholar] [CrossRef]
- Ulmschneider, J.P. Charged Antimicrobial Peptides Can Translocate across Membranes without Forming Channel-like Pores. Biophys. J. 2017, 113, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, M.B.; Ulmschneider, J.P.; Freites, J.A.; von Heijne, G.; Tobias, D.J.; White, S.H. Transmembrane helices containing a charged arginine are thermodynamically stable. Eur. Biophys. J. 2017, 46, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Gumbart, J.C.; Ulmschneider, M.B.; Hazel, A.; White, S.H.; Ulmschneider, J.P. Computed Free Energies of Peptide Insertion into Bilayers are Independent of Computational Method. J. Membr. Biol. 2018, 251, 345–356, Erratum in J. Membr. Biol. 2018, 251, 357. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Kang, Z.; Yu, B.; Chen, Q.; Liu, Y.; Wang, Q. All-Factor Analysis and Correlations on the Transmembrane Process for Arginine-Rich Cell-Penetrating Peptides. Langmuir 2019, 35, 9286–9296. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.P.; Tada, S.; Yumoto, A.; Kitao, A.; Ito, Y.; Uzawa, T.; Tsuda, K. Using molecular dynamics simulations to prioritize and understand AI-generated cell penetrating peptides. Sci. Rep. 2021, 11, 10630. [Google Scholar] [CrossRef]
- Gimenez-Dejoz, J.; Numata, K. Molecular dynamics study of the internalization of cell-penetrating peptides containing unnatural amino acids across membranes. Nanoscale Adv. 2022, 4, 397–407. [Google Scholar] [CrossRef]
- Grasso, G.; Muscat, S.; Rebella, M.; Morbiducci, U.; Audenino, A.; Danani, A.; Deriu, M.A. Cell penetrating peptide modulation of membrane biomechanics by Molecular dynamics. J. Biomech. 2018, 73, 137–144. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Bennett, W.F.; Tieleman, D.P. The importance of membrane defects-lessons from simulations. Acc. Chem. Res. 2014, 47, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Czajlik, A.; Mesko, E.; Penke, B.; Perczel, A. Investigation of penetratin peptides. Part 1. The environment dependent conformational properties of penetratin and two of its derivatives. J. Pept. Sci. 2002, 8, 151–171. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Daly, N.L.; Plan, M.R.; Waine, C.; Craik, D.J. Twists, knots, and rings in proteins. Structural definition of the cyclotide framework. J. Biol. Chem. 2003, 278, 8606–8616. [Google Scholar] [CrossRef] [PubMed]
- Korsinczky, M.L.; Schirra, H.J.; Rosengren, K.J.; West, J.; Condie, B.A.; Otvos, L.; Anderson, M.A.; Craik, D.J. Solution structures by 1H NMR of the novel cyclic trypsin inhibitor SFTI-1 from sunflower seeds and an acyclic permutant. J. Mol. Biol. 2001, 311, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Heitz, A.; Hernandez, J.F.; Gagnon, J.; Hong, T.T.; Pham, T.T.; Nguyen, T.M.; Le-Nguyen, D.; Chiche, L. Solution structure of the squash trypsin inhibitor MCoTI-II. A new family for cyclic knottins. Biochemistry 2001, 40, 7973–7983. [Google Scholar] [CrossRef] [PubMed]
- Letoha, T.; Gaal, S.; Somlai, C.; Venkei, Z.; Glavinas, H.; Kusz, E.; Duda, E.; Czajlik, A.; Petak, F.; Penke, B. Investigation of penetratin peptides. Part 2. In vitro uptake of penetratin and two of its derivatives. J. Pept. Sci. 2005, 11, 805–811. [Google Scholar] [CrossRef]
- Dom, G.; Shaw-Jackson, C.; Matis, C.; Bouffioux, O.; Picard, J.J.; Prochiantz, A.; Mingeot-Leclercq, M.-P.; Brasseur, R.; Rezsohazy, R. Cellular uptake of Antennapedia penetratin peptides is a two-step process in which phase transfer precedes a tryptophan-dependent translocation. Nucleic Acids Res. 2003, 31, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Kar, R.K.; Mondal, S.; Pahan, K.; Bhunia, A. Structural Elucidation of the Cell-Penetrating Penetratin Peptide in Model Membranes at the Atomic Level: Probing Hydrophobic Interactions in the Blood-Brain Barrier. Biochemistry 2016, 55, 4982–4996. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. Cytotoxicity of organic compounds against ovarian cancer cells: A quantitative structure-activity relationship study. Mol. Pharm. 2006, 3, 441–450. [Google Scholar] [CrossRef]
- Machatha, S.G.; Bustamante, P.; Yalkowsky, S.H. Deviation from linearity of drug solubility in ethanol/water mixtures. Int. J. Pharm. 2004, 283, 83–88. [Google Scholar] [CrossRef]
- Kong, Z.; Sun, D.; Jiang, Y.; Hu, Y. Design, synthesis, and evaluation of 1,4-benzodioxan-substituted chalcones as selective and reversible inhibitors of human monoamine oxidase B. J. Enzyme Inhib. Med. Chem. 2020, 35, 1513–1523. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.; Poso, A.; Bartos, P.; Miettinen, M.S. Structure of POPC Lipid Bilayers in OPLS3e Force Field. J. Chem. Inf. Model. 2022, 62, 6462–6474. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.; Ivanova, A. Testing the limits of model membrane simulations-bilayer composition and pressure scaling. J. Comput. Chem. 2018, 39, 387–396. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Ramachandran, B.; Muthupandian, S.; Jeyaraman, J.; Lopes, B.S. Computational exploration of molecular flexibility and interaction of meropenem analogs with the active site of oxacillinase-23 in Acinetobacter baumannii. Front. Chem. 2023, 11, 1090630. [Google Scholar] [CrossRef]










| Peptide | PDB ID | Sequence | Reference |
|---|---|---|---|
| Penetratin | 1KZ0 | RQIKIWFQNRRMKWKK | [52] |
| 6,14-Phe-penetratin | 1KZ2 | RQIKIFFQNRRMKFKK | [52] |
| Dodeca-penetratin | 1KZ5 | RQIKIWFRKWKK | [52] |
| Kalata B1 | 1NB1 | [CGETCVGGTCNTPGCTCSWPVCTRNGLPV] | [53] |
| SFTI-1 | 1JBL | [GRCTKSIPPICFPD] | [54] |
| MCoTI-II | 1HA9 | [SGSDGGVCPKILKKCRRDSDCPGACICRGNGYCG] | [55] |
| Entry | Peptide | Conjugate | H-Bond | π-Cation | Salt Bridge |
|---|---|---|---|---|---|
| 1 | penetratin (1KZ0) | unconjugated | 11 | 0 | 4 |
| 2 | penetratin (1KZ0) | doxorubicin | 0 | 0 | 0 |
| 3 | penetratin (1KZ0) | rasagiline | 12 | 1 | 7 |
| 4 | penetratin (1KZ0) | zidovudine | 1 | 0 | 2 |
| 5 | 6,14-Phe-penetratin (1KZ2) | unconjugated | 7 | 1 | 5 |
| 6 | 6,14-Phe-penetratin (1KZ2) | doxorubicin | 8 | 0 | 3 |
| 7 | 6,14-Phe-penetratin (1KZ2) | rasagiline | 10 | 0 | 4 |
| 8 | 6,14-Phe-penetratin (1KZ2) | zidovudine | 0 | 0 | 0 |
| 9 | dodeca-penetratin (1KZ5) | unconjugated | 6 | 0 | 1 |
| 10 | dodeca-penetratin (1KZ5) | doxorubicin | 2 | 1 | 3 |
| 11 | dodeca-penetratin (1KZ5) | rasagiline | 5 | 0 | 4 |
| 12 | dodeca-penetratin (1KZ5) | zidovudine | 4 | 0 | 2 |
| 13 | SFTI-1 (1NB1) | unconjugated | 0 | 0 | 0 |
| 14 | SFTI-1 (1NB1) | doxorubicin | 10 | 0 | 2 |
| 15 | SFTI-1 (1NB1) | rasagiline | 6 | 0 | 2 |
| 16 | SFTI-1 (1NB1) | zidovudine | 2 | 0 | 2 |
| 17 | Kalata B1 (1JBL) | unconjugated | 2 | 0 | 0 |
| 18 | Kalata B1 (1JBL) | doxorubicin | 6 | 0 | 3 |
| 19 | Kalata B1 (1JBL) | rasagiline | 4 | 0 | 3 |
| 20 | Kalata B1 (1JBL) | zidovudine | 3 | 0 | 1 |
| 21 | MCoTI-II (1HA9) | unconjugated | 0 | 0 | 0 |
| 22 | MCoTI-II (1HA9) | doxorubicin | 8 | 0 | 11 |
| 23 | MCoTI-II (1HA9) | rasagiline | 6 | 0 | 8 |
| 24 | MCoTI-II (1HA9) | zidovudine | 16 | 0 | 6 |
| Doxorubicin | Rasagiline | Zidovudine | |
|---|---|---|---|
| penetratin and analogues |  |  |  |
| Kalata B1 |  |  |  |
| SFTI-1 |  |  |  |
| MCoTI-II |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivánczi, M.; Balogh, B.; Kis, L.; Mándity, I. Molecular Dynamics Simulations of Drug-Conjugated Cell-Penetrating Peptides. Pharmaceuticals 2023, 16, 1251. https://doi.org/10.3390/ph16091251
Ivánczi M, Balogh B, Kis L, Mándity I. Molecular Dynamics Simulations of Drug-Conjugated Cell-Penetrating Peptides. Pharmaceuticals. 2023; 16(9):1251. https://doi.org/10.3390/ph16091251
Chicago/Turabian StyleIvánczi, Márton, Balázs Balogh, Loretta Kis, and István Mándity. 2023. "Molecular Dynamics Simulations of Drug-Conjugated Cell-Penetrating Peptides" Pharmaceuticals 16, no. 9: 1251. https://doi.org/10.3390/ph16091251