

Current Management and Future Perspectives in the Treatment of Lp(a) with a Focus on the Prevention of Cardiovascular Diseases
 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
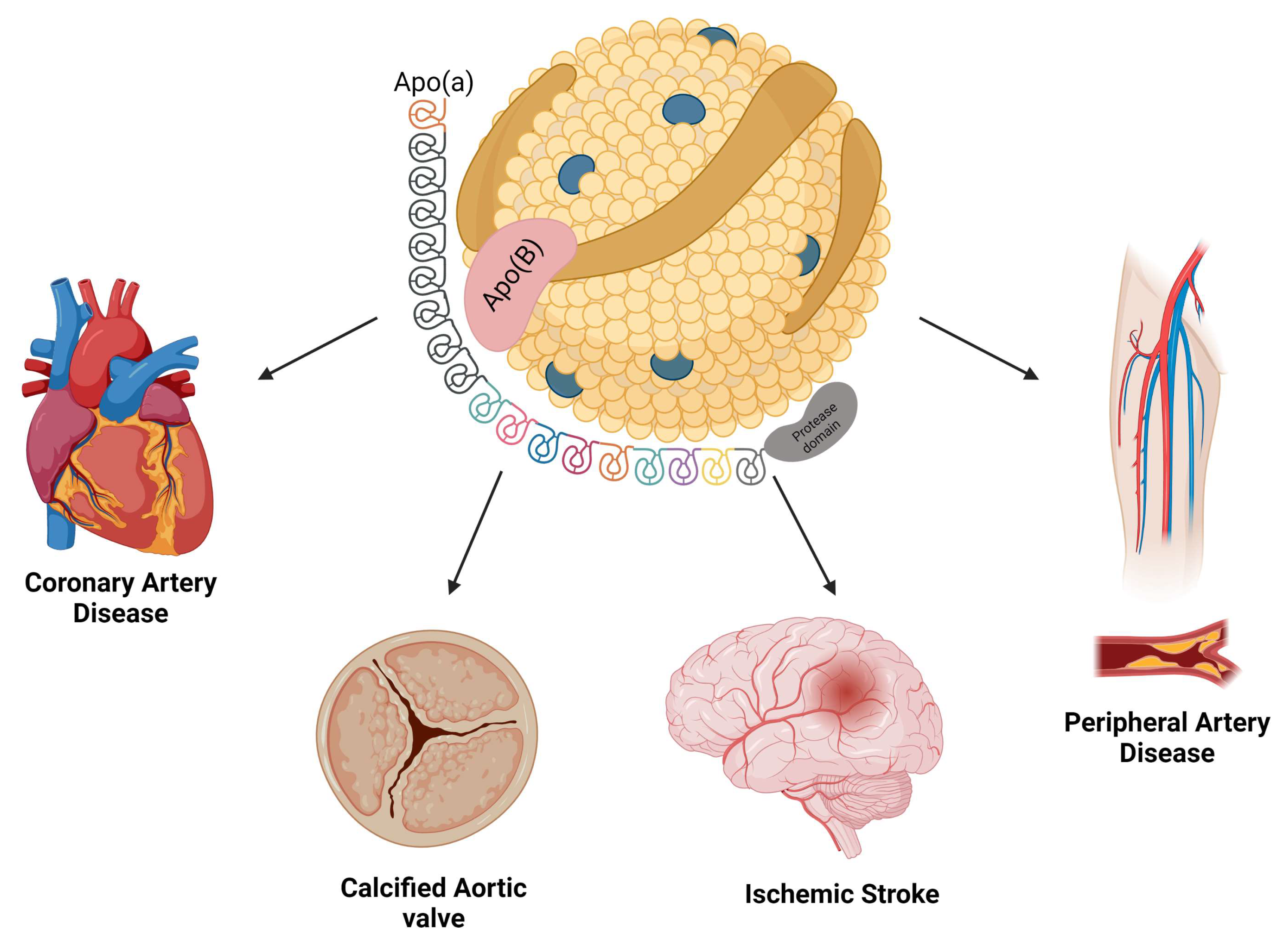
2. Biochemistry of Lp(a)
3. Association between Lp(a) and Cardiovascular Diseases
3.1. Atherosclerosis and Coronary Artery Disease
3.2. Cerebrovascular Disease
3.3. Calcific Aortic Valve Stenosis
3.4. Aortic Dissection
3.5. Peripheral Arterial Disease
3.6. Heart Failure
3.7. Diabetes Mellitus
3.8. Thrombosis
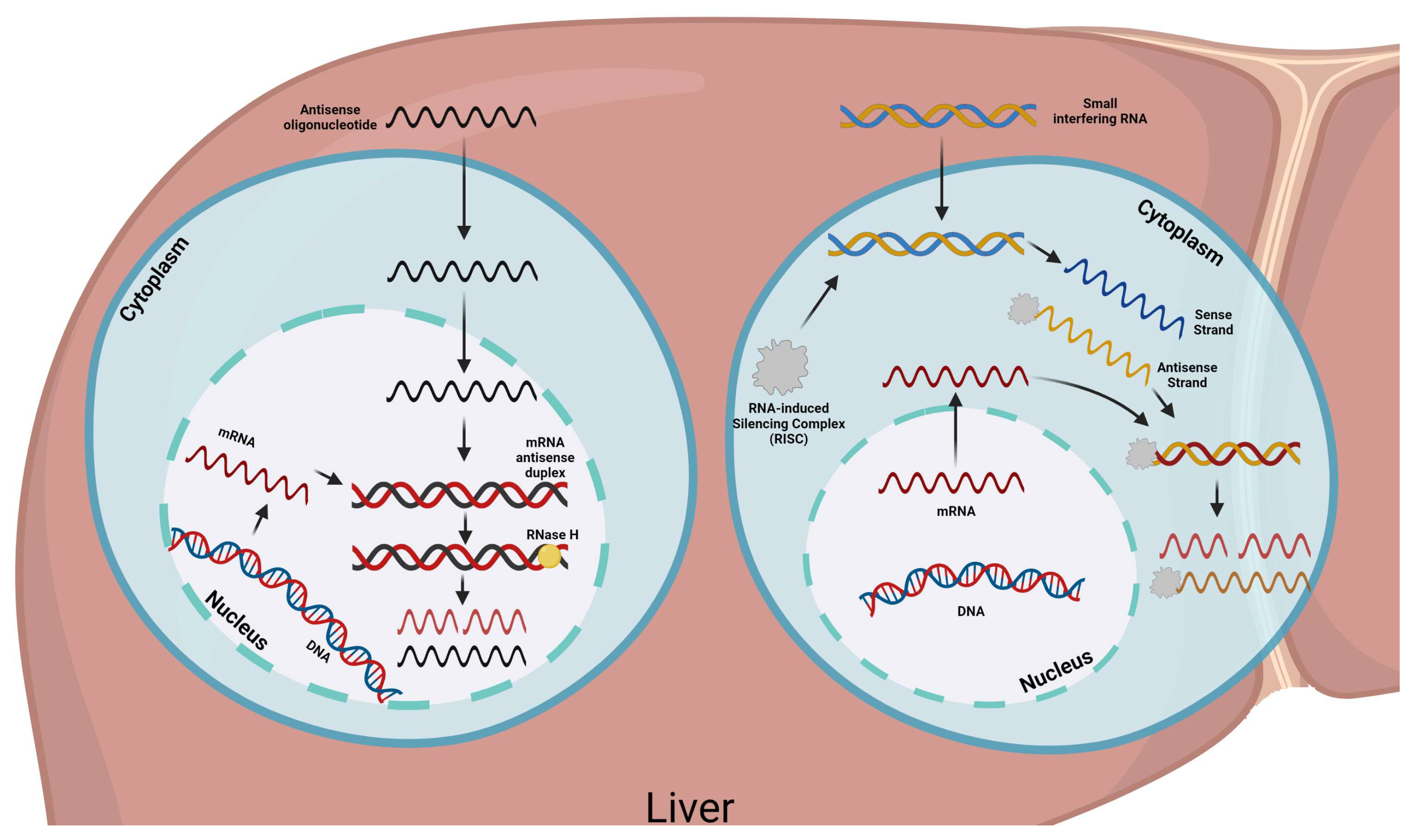
4. Lp(a) Treatment
4.1. The Effect of Currently Available Therapies on Lp(a)
4.2. Inclisiran
4.3. Novel Therapies
4.3.1. Olpasiran
4.3.2. Pelacarsen
4.3.3. SLN360
4.3.4. LY3819469
5. Future Perspectives
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cholesterol Treatment Trialists Collabtion. Efficacy and safety of statin therapy in older people: A meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 2019, 393, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, S.; Tang, M.; Liu, F.; Xia, P.; Shu, M.; Wu, X. Ezetimibe for the prevention of cardiovascular disease and all-cause mortality events. Cochrane Database Syst. Rev. 2018, 11, CD012502. [Google Scholar] [CrossRef]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-Gonzalez, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef]
- Dhindsa, D.S.; Sandesara, P.B.; Shapiro, M.D.; Wong, N.D. The Evolving Understanding and Approach to Residual Cardiovascular Risk Management. Front. Cardiovasc. Med. 2020, 7, 88. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein (a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Shaya, G.E.; Leucker, T.M.; Jones, S.R.; Martin, S.S.; Toth, P.P. Coronary heart disease risk: Low-density lipoprotein and beyond. Trends Cardiovasc. Med. 2022, 32, 181–194. [Google Scholar] [CrossRef]
- Fujino, M.; Nicholls, S.J. Lipoprotein (a): Cardiovascular risk and emerging therapies. Expert. Rev. Cardiovasc. Ther. 2023, 21, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Malick, W.A.; Goonewardena, S.N.; Koenig, W.; Rosenson, R.S. Clinical Trial Design for Lipoprotein (a)-Lowering Therapies: JACC Focus Seminar 2/3. J. Am. Coll. Cardiol. 2023, 81, 1633–1645. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein (a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein (a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arter. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. Lipoprotein (a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arter. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimikas, S.; Moriarty, P.M.; Stroes, E.S. Emerging RNA Therapeutics to Lower Blood Levels of Lp(a): JACC Focus Seminar 2/4. J. Am. Coll. Cardiol. 2021, 77, 1576–1589. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Matsubara, M.; Shibuya, H.; Fuda, H.; Chiba, H. Effect of ageing on plasma lipoprotein (a) levels. Ann. Clin. Biochem. 2002, 39 Pt 3, 237–240. [Google Scholar] [CrossRef] [Green Version]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein (a) levels from childhood to adulthood: Data in nearly 3,000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid changes during the menopause transition in relation to age and weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352–1361. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Soffer, G. The impact of race and ethnicity on lipoprotein (a) levels and cardiovascular risk. Curr. Opin. Lipidol. 2021, 32, 163–166. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arter. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein (a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein (a) molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaubatz, J.W.; Chari, M.V.; Nava, M.L.; Guyton, J.R.; Morrisett, J.D. Isolation and characterization of the two major apoproteins in human lipoprotein [a]. J. Lipid Res. 1987, 28, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Vavuranakis, M.A.; Jones, S.R.; Cardoso, R.; Gerstenblith, G.; Leucker, T.M. The role of Lipoprotein (a) in cardiovascular disease: Current concepts and future perspectives. Hellenic. J. Cardiol. 2020, 61, 398–403. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Gabel, B.; Koschinsky, M.L.; Gaur, V.P. Effect of the number of apolipoprotein (a) kringle 4 domains on immunochemical measurements of lipoprotein (a). Clin. Chem. 1995, 41, 246–255. [Google Scholar] [CrossRef]
- Gries, A.; Nimpf, J.; Nimpf, M.; Wurm, H.; Kostner, G.M. Free and Apo B-associated Lpa-specific protein in human serum. Clin. Chim. Acta 1987, 164, 93–100. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Lipoprotein (a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef]
- Maloberti, A.; Fabbri, S.; Colombo, V.; Gualini, E.; Monticelli, M.; Daus, F.; Busti, A.; Galasso, M.; De Censi, L.; Algeri, M.; et al. Lipoprotein (a): Cardiovascular Disease, Aortic Stenosis and New Therapeutic Option. Int. J. Mol. Sci. 2022, 24, 170. [Google Scholar] [CrossRef]
- Berman, A.N.; Blankstein, R. Current and future role of lipoprotein (a) in preventive cardiology. Curr. Opin. Cardiol. 2019, 34, 514–518. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Boekholdt, S.M.; Dube, M.P.; Rheaume, E.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein (a) levels, genotype, and incident aortic valve stenosis: A prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Lamina, C.; Ward, N.C. Lipoprotein (a) and diabetes mellitus. Atherosclerosis 2022, 349, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hong, Y.; Yang, W.; Zheng, Z. Association of lipoprotein (a) with aortic dissection. Clin. Cardiol. 2022, 45, 908–912. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically elevated lipoprotein (a) and increased risk of myocardial infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Morrow, D.A.; Tsimikas, S.; Sloan, S.; Ren, A.F.; Hoffman, E.B.; Desai, N.R.; Solomon, S.D.; Domanski, M.; Arai, K.; et al. Lipoprotein (a) for risk assessment in patients with established coronary artery disease. J. Am. Coll. Cardiol. 2014, 63, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugovsek, S.; Sebestjen, M. Lipoprotein (a)-The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein (a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huded, C.P.; Shah, N.P.; Puri, R.; Nicholls, S.J.; Wolski, K.; Nissen, S.E.; Cho, L. Association of Serum Lipoprotein (a) Levels and Coronary Atheroma Volume by Intravascular Ultrasound. J. Am. Heart Assoc. 2020, 9, e018023. [Google Scholar] [CrossRef]
- Kaiser, Y.; Daghem, M.; Tzolos, E.; Meah, M.N.; Doris, M.K.; Moss, A.J.; Kwiecinski, J.; Kroon, J.; Nurmohamed, N.S.; van der Harst, P.; et al. Association of Lipoprotein (a) with Atherosclerotic Plaque Progression. J. Am. Coll. Cardiol. 2022, 79, 223–233. [Google Scholar] [CrossRef]
- Mehta, A.; Vasquez, N.; Ayers, C.R.; Patel, J.; Hooda, A.; Khera, A.; Blumenthal, R.S.; Shapiro, M.D.; Rodriguez, C.J.; Tsai, M.Y.; et al. Independent Association of Lipoprotein (a) and Coronary Artery Calcification with Atherosclerotic Cardiovascular Risk. J. Am. Coll. Cardiol. 2022, 79, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Schreiner, P.J.; Morrisett, J.D.; Chambless, L.E.; Rosamond, W.D.; Folsom, A.R. Lipoprotein (a) and incident ischemic stroke: The Atherosclerosis Risk in Communities (ARIC) study. Stroke 2006, 37, 1407–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of lipoprotein (a) and the risk of future stroke. JAMA 1995, 273, 1269–1273. [Google Scholar] [CrossRef]
- Kumar, P.; Swarnkar, P.; Misra, S.; Nath, M. Lipoprotein (a) level as a risk factor for stroke and its subtype: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 15660. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.K.L.M.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein (a) and Oxidized Phospholipids Promote Valve Calcification in Patients with Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Hafiane, A.; Thanassoulis, G.; Ott, L.; Filwood, N.; Cerruti, M.; Gourgas, O.; Shum-Tim, D.; Al Kindi, H.; de Varennes, B.; et al. Lipoprotein (a) Induces Human Aortic Valve Interstitial Cell Calcification. JACC Basic Transl. Sci. 2017, 2, 358–371. [Google Scholar] [CrossRef]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.W.; et al. Lipoprotein (a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef]
- Kaiser, Y.; van der Toorn, J.E.; Singh, S.S.; Zheng, K.H.; Kavousi, M.; Sijbrands, E.J.G.; Stroes, E.S.G.; Vernooij, M.W.; de Rijke, Y.B.; Boekholdt, S.M.; et al. Lipoprotein (a) is associated with the onset but not the progression of aortic valve calcification. Eur. Heart J. 2022, 43, 3960–3967. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein (a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [Green Version]
- Aboyans, V.; Criqui, M.H.; Denenberg, J.O.; Knoke, J.D.; Ridker, P.M.; Fronek, A. Risk factors for progression of peripheral arterial disease in large and small vessels. Circulation 2006, 113, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Volpato, S.; Vigna, G.B.; McDermott, M.M.; Cavalieri, M.; Maraldi, C.; Lauretani, F.; Bandinelli, S.; Zuliani, G.; Guralnik, J.M.; Fellin, R.; et al. Lipoprotein (a), inflammation, and peripheral arterial disease in a community-based sample of older men and women (the InCHIANTI study). Am. J. Cardiol. 2010, 105, 1825–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurdasani, D.; Sjouke, B.; Tsimikas, S.; Hovingh, G.K.; Luben, R.N.; Wainwright, N.W.; Pomilla, C.; Wareham, N.J.; Khaw, K.T.; Boekholdt, S.M.; et al. Lipoprotein (a) and risk of coronary, cerebrovascular, and peripheral artery disease: The EPIC-Norfolk prospective population study. Arter. Thromb. Vasc. Biol. 2012, 32, 3058–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laschkolnig, A.; Kollerits, B.; Lamina, C.; Meisinger, C.; Rantner, B.; Stadler, M.; Peters, A.; Koenig, W.; Stöckl, A.; Dähnhardt, D.; et al. Lipoprotein (a) concentrations, apolipoprotein (a) phenotypes, and peripheral arterial disease in three independent cohorts. Cardiovasc. Res. 2014, 103, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Forbang, N.I.; Criqui, M.H.; Allison, M.A.; Ix, J.H.; Steffen, B.T.; Cushman, M.; Tsai, M.Y. Sex and ethnic differences in the associations between lipoprotein (a) and peripheral arterial disease in the Multi-Ethnic Study of Atherosclerosis. J. Vasc. Surg. 2016, 63, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Rowbotham, S.; Velu, R.; Quigley, F.; Jenkins, J.; Bourke, M.; Bourke, B.; Thanigaimani, S.; Chan, D.C.; Watts, G.F. Association of Serum Lipoprotein (a) with the Requirement for a Peripheral Artery Operation and the Incidence of Major Adverse Cardiovascular Events in People with Peripheral Artery Disease. J. Am. Heart Assoc. 2020, 9, e015355. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein (a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Agarwala, A.; Pokharel, Y.; Saeed, A.; Sun, W.; Virani, S.S.; Nambi, V.; Ndumele, C.; Shahar, E.; Heiss, G.; Boerwinkle, E.; et al. The association of lipoprotein (a) with incident heart failure hospitalization: Atherosclerosis Risk in Communities study. Atherosclerosis 2017, 262, 131–137. [Google Scholar] [CrossRef]
- Eraikhuemen, N.; Lazaridis, D.; Dutton, M.T. Emerging Pharmacotherapy to Reduce Elevated Lipoprotein (a) Plasma Levels. Am. J. Cardiovasc. Drugs 2021, 21, 255–265. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein (a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Farmakis, I.; Doundoulakis, I.; Pagiantza, A.; Zafeiropoulos, S.; Antza, C.; Karvounis, H.; Giannakoulas, G. Lipoprotein (a) Reduction with Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Systematic Review and Meta-analysis. J. Cardiovasc. Pharmacol. 2021, 77, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein (a) concentrations, isoform size, and risk of type 2 diabetes: A Mendelian randomisation study. Lancet Diabetes Endocrinol. 2013, 1, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Kostner, K.M.; Kostner, G.M. Lp(a) and the Risk for Cardiovascular Disease: Focus on the Lp(a) Paradox in Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3584. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein (a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein (a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Galvano, F.; Malaguarnera, M.; Vacante, M.; Motta, M.; Russo, C.; Malaguarnera, G.; D’Orazio, N.; Malaguarnera, L. The physiopathology of lipoprotein (a). Front. Biosci. 2010, 2, 866–875. [Google Scholar]
- Helgadottir, A.; Gretarsdottir, S.; Thorleifsson, G.; Holm, H.; Patel, R.S.; Gudnason, T.; Jones, G.T.; van Rij, A.M.; Eapen, D.J.; Baas, A.F.; et al. Apolipoprotein (a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J. Am. Coll. Cardiol. 2012, 60, 722–729. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein (a) associates with atherosclerotic stenosis rather than venous thrombosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, D.I.; Rider, D.A.; Yavari, A.; Wikstrom Lindholm, M.; Campion, G.V.; Nissen, S.E. Treatment and prevention of lipoprotein (a)-mediated cardiovascular disease: The emerging potential of RNA interference therapeutics. Cardiovasc. Res. 2022, 118, 1218–1231. [Google Scholar] [CrossRef]
- Ray, K.K.; Ference, B.A.; Severin, T.; Blom, D.; Nicholls, S.J.; Shiba, M.H.; Almahmeed, W.; Alonso, R.; Daccord, M.; Ezhov, M.; et al. World Heart Federation Cholesterol Roadmap. Glob. Heart 2022, 17, 75. [Google Scholar] [CrossRef]
- Liu, C.; Chen, J.; Chen, H.; Zhang, T.; He, D.; Luo, Q.; Chi, J.; Hong, Z.; Liao, Y.; Zhang, S.; et al. PCSK9 Inhibition: From Current Advances to Evolving Future. Cells 2022, 11, 2972. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein (a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Szarek, M.; Bittner, V.A.; Aylward, P.; Baccara-Dinet, M.; Bhatt, D.L.; Diaz, R.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Harrington, R.A.; et al. Lipoprotein (a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur. Heart J. 2020, 41, 4245–4255. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Landmesser, U.; López-Jaramillo, P.; Manvelian, G.; Pordy, R.; et al. Lipoprotein (a) and Benefit of PCSK9 Inhibition in Patients with Nominally Controlled LDL Cholesterol. J. Am. Coll. Cardiol. 2021, 78, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Blom, D.; Seidah, N.G.; Honarpour, N.; Lira, A.; Xue, A.; Chiruvolu, P.; et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: An analysis of 10 clinical trials and the LDL receptor’s role. J. Lipid Res. 2016, 57, 1086–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants with Risk of Coronary Disease and the Implications for Lipoprotein (a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, K.K.; Vallejo-Vaz, A.J.; Ginsberg, H.N.; Davidson, M.H.; Louie, M.J.; Bujas-Bobanovic, M.; Minini, P.; Eckel, R.H.; Cannon, C.P. Lipoprotein (a) reductions from PCSK9 inhibition and major adverse cardiovascular events: Pooled analysis of alirocumab phase 3 trials. Atherosclerosis 2019, 288, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein (a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Deshmukh, H.A.; Colhoun, H.M.; Johnson, T.; McKeigue, P.M.; Betteridge, D.J.; Durrington, P.N.; Fuller, J.H.; Livingstone, S.; Charlton-Menys, V.; Neil, A.; et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: Importance of Lp(a). J. Lipid Res. 2012, 53, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Berg, K.; Dahlen, G.; Christophersen, B.; Cook, T.; Kjekshus, J.; Pedersen, T. Lp(a) lipoprotein level predicts survival and major coronary events in the Scandinavian Simvastatin Survival Study. Clin. Genet. 1997, 52, 254–261. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein (a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.; Phillips, C.O.; Foody, J.M.; Wang, Y.; Mangalmurti, S.; Ko, D.T.; Krumholz, H.M. Risks associated with statin therapy: A systematic overview of randomized clinical trials. Circulation 2006, 114, 2788–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholesterol Treatment Trialists Collaboration. Effect of statin therapy on muscle symptoms: An individual participant data meta-analysis of large-scale, randomised, double-blind trials. Lancet 2022, 400, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Ganga, H.V.; Slim, H.B.; Thompson, P.D. A systematic review of statin-induced muscle problems in clinical trials. Am. Heart J. 2014, 168, 6–15. [Google Scholar] [CrossRef]
- Thomas, T.; Zhou, H.; Karmally, W.; Ramakrishnan, R.; Holleran, S.; Liu, Y.; Jumes, P.; Wagner, J.A.; Hubbard, B.; Previs, S.F.; et al. CETP (Cholesteryl Ester Transfer Protein) Inhibition with Anacetrapib Decreases Production of Lipoprotein (a) in Mildly Hypercholesterolemic Subjects. Arter. Thromb. Vasc. Biol. 2017, 37, 1770–1775. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein (a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527 e4. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar]
- Albers, J.J.; Slee, A.; O’Brien, K.D.; Robinson, J.G.; Kashyap, M.L.; Kwiterovich, P.O., Jr.; Xu, P.; Marcovina, S.M. Relationship of apolipoproteins A-1 and B, and lipoprotein (a) to cardiovascular outcomes: The AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J. Am. Coll. Cardiol. 2013, 62, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, P.M.; Gray, J.V.; Gorby, L.K. Lipoprotein apheresis for lipoprotein (a) and cardiovascular disease. J. Clin. Lipidol. 2019, 13, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Leebmann, J.; Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Maerz, W.; Lehmacher, W.; Heibges, A. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein (a)-hyperlipoproteinemia, and progressive cardiovascular disease: Prospective observational multicenter study. Circulation 2013, 128, 2567–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, B.; Julius, U.; Lansberg, P.; Jaeger, B.; Mellwig, K.P.; Weiss, N.; Graehlert, X.; Roeder, I.; Ramlow, W. Rationale and design of MultiSELECt: A European Multicenter Study on the Effect of Lipoprotein (a) Elimination by lipoprotein apheresis on Cardiovascular outcomes. Atheroscler. Suppl. 2017, 30, 180–186. [Google Scholar] [CrossRef]
- Mickiewicz, A.; Borowiec-Wolna, J.; Bachorski, W.; Gilis-Malinowska, N.; Gałąska, R.; Raczak, G.; Chmara, M.; Wasąg, B.; Jaguszewski, M.J.; Fijałkowski, M. Long-term lipoprotein apheresis in the treatment of severe familial hypercholesterolemia refractory to high intensity statin therapy: Three year experience at a lipoprotein apheresis centre. Cardiol. J. 2019, 26, 669–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korneva, V.A.; Kuznetsova, T.Y.; Julius, U. Modern Approaches to Lower Lipoprotein (a) Concentrations and Consequences for Cardiovascular Diseases. Biomedicines 2021, 9, 1271. [Google Scholar] [CrossRef]
- Katsiki, N.; Vrablik, M.; Banach, M.; Gouni-Berthold, I. Inclisiran, Low-Density Lipoprotein Cholesterol and Lipoprotein (a). Pharmaceuticals 2023, 16, 577. [Google Scholar] [CrossRef]
- Ray, K.K.; Stoekenbroek, R.M.; Kallend, D.; Leiter, L.A.; Landmesser, U.; Wright, R.S.; Wijngaard, P.; Kastelein, J.J.P.; Ray, K.K.; Stoekenbroek, R.M. Effect of an siRNA Therapeutic Targeting PCSK9 on Atherogenic Lipoproteins: Prespecified Secondary End Points in ORION. Circulation 2018, 138, 1304–1316. [Google Scholar] [CrossRef]
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Scott, W.R.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Ray, K.K.; Kallend, D.; Leiter, L.A.; Raal, F.J.; Koenig, W.; Jaros, M.J.; Schwartz, G.G.; Landmesser, U.; Garcia, C.L.; Wright, R.S. Effect of inclisiran on lipids in primary prevention: The ORION-11 trial. Eur. Heart J. 2022, 43, 5047–5057. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic. Acid. Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanoli, T.; Yue, P.; Yablonskiy, D.; Schonfeld, G. Fatty liver in familial hypobetalipoproteinemia: Roles of the APOB defects, intra-abdominal adipose tissue, and insulin sensitivity. J. Lipid Res. 2004, 45, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, G.; Barba, A.A. Drug Delivery of siRNA Therapeutics. Pharmaceutics 2020, 12, 178. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Sosnowska, B.; Surma, S.; Banach, M. Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy. Pharmaceuticals 2022, 15, 1573. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein (a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. A Study to Evaluate the Pharmacokinetics, Safety, and Pharmacodynamics of Olpasiran in Participants with Various Degrees of Hepatic Impairment clinicaltrials.gov: Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05481411. (accessed on 15 May 2023).
- Clinicaltrials.gov. A Study to Evaluate the Pharmacokinetics, Safety, and Pharmacodynamics of Olpasiran in Participants with Normal Renal Function and Participants with Various Degrees of Renal Impairment Clinicaltrials.gov: Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05489614 (accessed on 20 May 2023).
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F. Small Interfering RNA to Reduce Lipoprotein (a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Olpasiran Trials of Cardiovascular Events and Lipoprotein (a) Reduction (OCEAN(a))—Outcomes Trial ClinicalTrials.gov: ClinicalTrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05581303 (accessed on 18 May 2023).
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M. Antisense oligonucleotides targeting apolipoprotein (a) in people with raised lipoprotein (a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein (a) Production in Individuals with Elevated Plasma Lipoprotein (a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Study to Investigate Safety, Tolerability, PK and PD Response of SLN360 in Subjects with Elevated Lipoprotein (a) Clinicaltrials.gov: Clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04606602 (accessed on 1 June 2023).
- Clinicaltrials.gov. Evaluate SLN360 in Participants with Elevated Lipoprotein (a) at High Risk of Atherosclerotic Cardiovascular Disease Events Clinicaltrials.gov: Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05537571 (accessed on 1 June 2023).
- Clinicaltrials.gov. A Study of LY3819469 in Healthy Participants Clinicaltrials.gov: Clinicaltrials.gov. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04914546 (accessed on 1 June 2023).
- Clinicaltrials.gov. A Study of LY3819469 in Participants with Elevated Lipoprotein (a) [Lp(a)] Clinicaltrials.gov: Clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05565742 (accessed on 1 June 2023).
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Prasad, A.; Choi, Y.S.; Xing, C.; Clopton, P.; Witztum, J.L.; Tsimikas, S. LPA Gene, Ethnicity, and Cardiovascular Events. Circulation 2017, 135, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, S.E.; Nicholls, S.J.; Sipahi, I.; Libby, P.; Raichlen, J.S.; Ballantyne, C.M.; Davignon, J.; Erbel, R.; Fruchart, J.C.; Tardif, J.C. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: The ASTEROID trial. JAMA 2006, 295, 1556–1565. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Agent | Phase of Development | Study Name | Efficacy Data | Safety Data |
|---|---|---|---|---|
| Olpasiran (GalNAc–siRNA) | Phase 3 (Ongoing) | OCEAN(a)—Outcomes NCT05581303 | Not yet available. Inclusion Criteria Elevated Lp(a) levels (≥200 nmol/L) with a history of either a myocardial infarction and/or coronary revascularization. Primary Outcome Time to coronary heart disease death, myocardial infarction, or urgent coronary revascularization. | Not yet available. |
| Phase 2 (Completed) | OCEAN(a)-DOSE NCT04270760 | At 36 weeks, Olpasiran significantly reduced the Lp(a) concentrations in a dose-dependent manner (from 70% to 100%). | The incidence of adverse events was similar across all the groups. The most common adverse events were injection-site reactions. | |
| Phase 1 (Completed) | “Safety, Tolerability, Pharmacokinetics and Pharmacodynamics Study of AMG 890 in Subjects with Elevated Plasma Lipoprotein(a)” NCT03626662 | Olpasiran reduced Lp(a) levels by 71% to 96% (in patients with Lp(a) levels between 70 and 199 nmol/L) and by 75% to 89% (in patients with Lp(a) concentrations higher than 200 nmol/L). | No serious safety concerns were identified. | |
| Phase 1 (Completed) | “A Study to Evaluate the Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of Olpasiran in Chinese Participants with Elevated Serum Lipoprotein(a)” NCT04987320 | The maximal Lp(a) reduction occurred at day 57, with mean Lp(a) reductions ranging from 56% to 99% | Adverse events were mild in severity, with no serious or fatal adverse events. No relevant changes in tolerability-related laboratory analytes or vital signs were observed. | |
| Phase 1 (Ongoing) | “A Study to Evaluate the Pharmacokinetics, Safety, and Pharmacodynamics of Olpasiran in Participants with Various Degrees of Hepatic Impairment” NCT05481411 | Not yet available. | Not yet available. | |
| Phase 1 (Ongoing) | “A Study to Evaluate the Pharmacokinetics, Safety, and Pharmacodynamics of Olpasiran in Participants with Normal Renal Function and Participants with Various Degrees of Renal Impairment” NCT05489614 | Not yet available. | Not yet available. | |
| Pelacarsen (ASO) | Phase 3 (Ongoing) | HORIZON Trial NCT04023552 | Not yet available. Inclusion Criteria Lp(a) ≥ 70 mg/dL and a history of myocardial infarction, ischemic stroke, or clinically significant symptomatic peripheral artery disease. Primary outcome Time to first occurrence of major adverse cardiovascular events. | Not yet available. |
| Phase 3 (Ongoing) | “A Multicenter Trial Assessing the Impact of Lipoprotein(a) Lowering with Pelacarsen (TQJ230) on the Rate of Weekly Lipoprotein Apheresis Sessions in Patients with Hyperlipoproteinemia(a) and Established Cardiovascular Disease in Germany” NCT05305664 | Not yet available. Inclusion Criteria Patients undergoing lipoprotein apheresis for isolated Lp(a) with Lp(a) >60 mg/dL and prior myocardial infarction, ischemic stroke, and/or clinically significant symptomatic peripheral artery disease. Primary outcome Rate of lipoprotein apheresis sessions performed over 52 weeks normalized to the weekly lipoprotein apheresis schedule. | ||
| Phase 2 (Completed) | “Phase 2 Study of ISIS 681257 (AKCEA-APO(a)-LRx) in Participants with Hyperlipoproteinemia(a) and Cardiovascular Disease” NCT03070782 | Pelacarsen resulted in dose-dependent decreases in lp(a) levels from 35% to 80%. | Injection-site reactions were the most frequently reported adverse event and occurred in 27% of the patients who received Pelacarsen. | |
| Phase 1/2a (Completed) | “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of IONIS APO(a)-LRx in Healthy Volunteers with Elevated Lipoprotein(a)” NCT02414594 | Significant dose-dependent reductions in mean Lp(a) concentrations were noted in patients treated with Pelacarsen (single dose 26% to 85%; multiple doses 66% to 92%). | No significant differences between any Pelacarsen dose and placebo were seen with respect to side effects. | |
| Phase 1 (Completed) | “A Study to Assess Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Pelacarsen (ISIS 681257) in Healthy Japanese Participants” NCT05337878 | Not available. | Not available. | |
| Phase 1 (Completed) | “Study to Assess the Pharmacokinetics of Pelacarsen (TQJ230) in Participants with Mild Hepatic Impairment Compared to Matched Healthy Participants” NCT05026996 | Not available. | Not available. | |
| SLN360 (GalNAc–siRNA) | Phase 2 (Active, not recruiting) | “Evaluate SLN360 in Participants with Elevated Lipoprotein(a) at High Risk of Atherosclerotic Cardiovascular Disease Events” NCT05537571 | Not yet available. | Not yet available. |
| Phase 1 (Completed) | “Study to Investigate Safety, Tolerability, PK and PD Response of SLN360 in Subjects with Elevated Lipoprotein(a)” NCT04606602 | Significant dose-dependent lowering of plasma Lp(a) concentrations was observed with SLN360 treatment (from 46% to 98%). | No significant side effects. One participant experienced two serious adverse event episodes judged to be unrelated to study drug. | |
| LY3819469 (GalNAc–siRNA) | Phase 1 (Completed) | “A Study of LY3819469 in Healthy Participants” NCT04914546 | Not available. | Not available. |
| Phase 2 (Active, not recruiting) | “A Study of LY3819469 in Participants with Elevated Lipoprotein(a) [Lp(a)]” NCT05565742 | Not yet available. | Not yet available. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farina, J.M.; Pereyra, M.; Mahmoud, A.K.; Chao, C.-J.; Barry, T.; Halli Demeter, S.M.; Ayoub, C.; Arsanjani, R. Current Management and Future Perspectives in the Treatment of Lp(a) with a Focus on the Prevention of Cardiovascular Diseases. Pharmaceuticals 2023, 16, 919. https://doi.org/10.3390/ph16070919
Farina JM, Pereyra M, Mahmoud AK, Chao C-J, Barry T, Halli Demeter SM, Ayoub C, Arsanjani R. Current Management and Future Perspectives in the Treatment of Lp(a) with a Focus on the Prevention of Cardiovascular Diseases. Pharmaceuticals. 2023; 16(7):919. https://doi.org/10.3390/ph16070919
Chicago/Turabian StyleFarina, Juan M., Milagros Pereyra, Ahmed K. Mahmoud, Chieh-Ju Chao, Timothy Barry, Susan M. Halli Demeter, Chadi Ayoub, and Reza Arsanjani. 2023. "Current Management and Future Perspectives in the Treatment of Lp(a) with a Focus on the Prevention of Cardiovascular Diseases" Pharmaceuticals 16, no. 7: 919. https://doi.org/10.3390/ph16070919