3.1. Chemistry
General Methods: Commercially sourced solvents and starting materials, which included anhydrous solvents and chemicals, were utilized without undergoing any further purification. The reactions involving air- or moisture-sensitive reactants were taken into an argon atmosphere, and the solutions were transferred through oven-dried glassware and syringes. TLC analysis was carried out on silica gel plates (Huayao New Material Technology, Qingdao, China). Column chromatography was carried out on silica gel 200~300 mesh (Innochem Technology, Beijing, China). Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded in CDCl3 or methanol-d4 on a Bruker Avance 400 spectrometer (Bruker Company, Karlsruhe, Germany). The chemical shifts (δ values) and coupling constants (J values) are provided in ppm and Hz, respectively, and tetramethylsilane (TMS) serves as the internal standard. The splitting patterns are classified as s for singlet, d for doublet, t for triplet, q for quadruplet, m for multiplet, dd for double doublet, and brs for broad singlet. Room temperature was controlled at 23 ± 2 °C based on laboratory temperature.
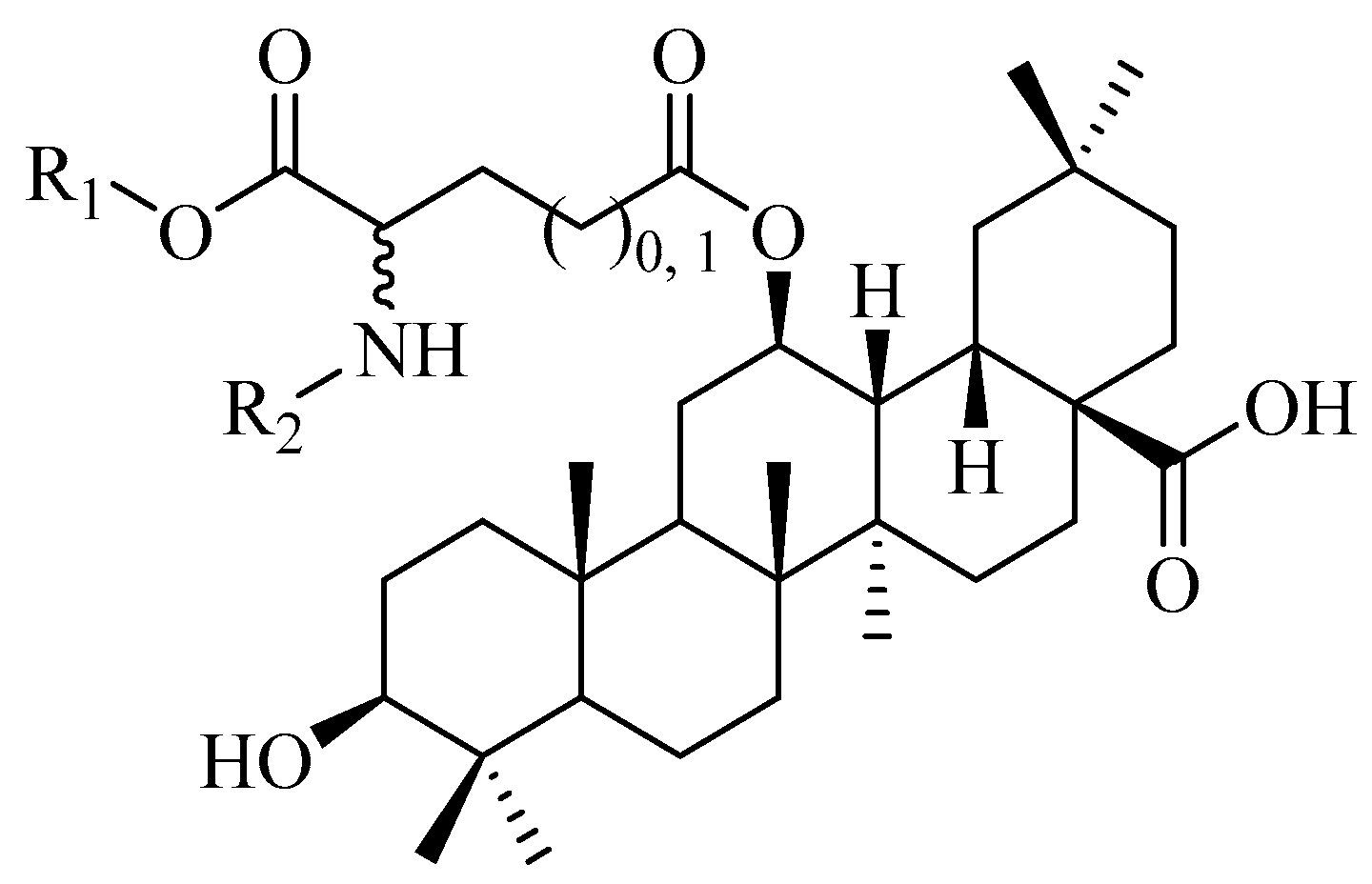
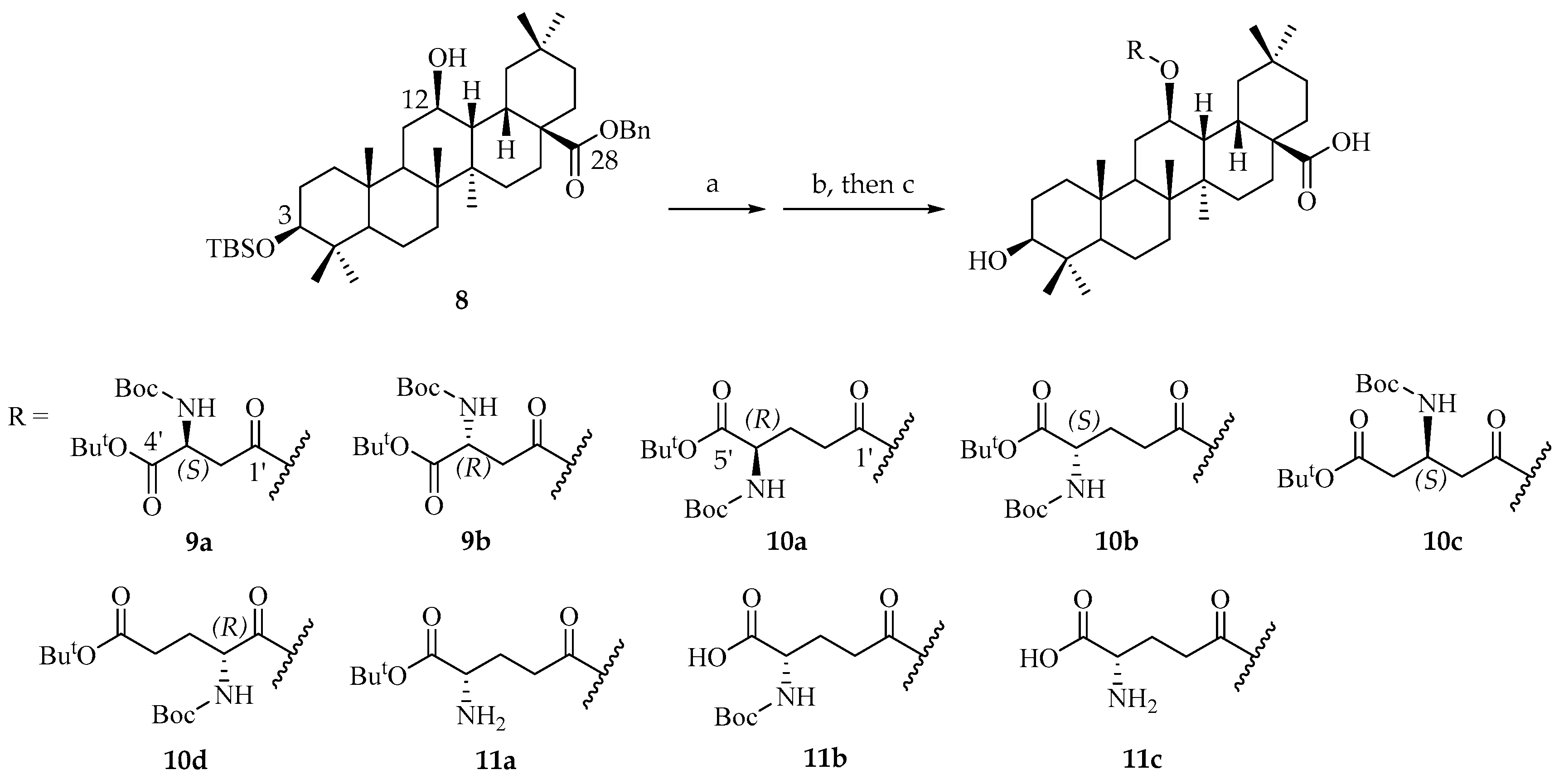
Prepare conditions of 12β-aspartyl/glutamyl OA derivatives. To a solution of key intermediate 8 (250 mg, 0.37 mmol, and 1.0 eq.) in 2 mL of toluene, 5 mL of EDC (1.47 mmol, 282 mg, and 4.0 eq.) and 4-DMAP (0.07 mmol, 9.0 mg, and 0.2 eq.) in toluene solution was added slowly. Then, the modified glutamic acid (4.0 eq.) was added in the reaction mixture and heated to 60 °C for stirring for 12 h. After TLC showed that the raw materials were completely consumed, the mixture was diluted with water and extracted with ethyl acetate. The organic layers were combined and dried over sodium sulfate and concentrated in vacuum to obtain crude product, and then directly subjected to flash chromatography on silica gel using PE/EtOAc (20:1~10:1) to obtain intermediates, which were directly used in the next step.
The intermediates obtained in the above steps were dissolved in 15 mL of a mixture of AcOH/THF/H2O 13:7:3 and stirred at 35 °C for 72 h. After TLC showed that most of the raw materials were consumed, NaHCO3 solution was added in the reaction mixture to remove acetic acid, and the solution was extracted with DCM. The organic layers were combined and dried over sodium sulfate and concentrated in vacuum to obtain crude product. Proceed to the next step without any further purification. The crude products were dissolved in 10 mL of methanol or THF, and 10% of the mass of palladium carbon was added, passing through hydrogen at 1 atmosphere. The reaction mixture was stirred at room temperature for 6 h. The reaction mixture was then filtered with diatomite and diluted with DCM. The organic layers were dried over sodium sulfate and concentrated in vacuum to obtain crude product and then directly subjected to flash chromatography on silica gel using PE/EtOAc (5:1~1:1). The organic layer was finally evaporated to dryness to obtain end products.
Benzyl 3β-(t-butyldimethylsiloxy)-12β-hydroxy-13H-olean-28-oate (8).
White solid 28 g, overall yield: 37.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.31–7.41 (m, 5H, Ph), 5.21 (d, 1H, J = 12.0 Hz, PhCHa), 5.06 (d, 1H, J = 12.0 Hz, PhCHb), 3.67 (ddd, 1H, J = 6.0, 10.0, 11.6 Hz, 12-H), 3.14 (dd, 1H, J = 4.8, 11.6 Hz, 3-H), 2.75 (dt, 1H, J = 5.2, 11.6 Hz, 18-H), 0.92 (s, 9H, 3Me), 0.88 (s, 9H, TBS-But), 0.87 (s, 3H, Me), 0.78 (s, 3H, Me), 0.71 (s, 3H, Me), 0.60 (s, 3H, Me), 0.59 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 177.8, 136.5, 128.5, 128.4, 128.1, 79.3, 68.3, 66.0, 55.3, 49.1, 47.4, 43.2, 41.5, 40.5, 39.4, 38.6, 36.8, 36.1, 34.5, 33.4, 33.1, 32.6, 31.8, 31.4, 30.5, 28.7, 28.4, 27.7, 25.9, 23.4, 23.2, 18.4, 18.1, 17.8, 16.2, 15.9, 15.8, −3.7, −4.9. HRMS (ESI) m/z Calcd for C43H70O4NaSi 701.4936 (M + Na)+, found 701.4936.
3-hydroxy-12β-(3′-((tert-butoxycarbonyl)amino)-4′-(tert-butoxy)-L-aspartate)oxy-13H-olean-28-oic acid (9a).
White solid 61 mg, overall yield: 70.0%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.73 (d, 1H, J = 8.4 Hz, NH), 4.99–5.05 (m, 1H, 12-H), 4.47 (dt, 1H, J = 4.0, 8.4 Hz, HαAsp), 3.20 (dd, 1H, J = 4.0, 11.6 Hz, 3-H), 2.97 (dd, 1H, J = 4.4, 17.2 Hz, HaβAsp), 2.77 (dd, 1H, J = 4.4, 17.2 Hz, HbβAsp), 2.50 (dt, 1H, J = 4.0, 14.0 Hz, 18-H), 2.11 (dd, 1H, J = 4.8, 11.6 Hz, 13-H), 1.48 (s, 9H, Boc), 1.43 (s, 9H, But), 1.00 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.87 (s, 3H, Me), 0.80 (s, 3H, Me), 0.74 (s, 3H, Me), 0.69 (d, 1H, J = 10.4 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.8, 170.5, 170.0, 155.8, 81.8, 79.6, 78.7, 72.6, 55.1, 50.3, 48.7, 47.2, 41.4, 40.5, 39.7, 38.8, 38.6, 37.3, 37.1, 36.0, 34.3, 33.2, 33.0, 32.3, 31.4, 30.5, 29.7, 28.8, 28.3, 28.0, 27.3, 27.1, 23.2, 22.7, 18.2, 17.7, 16.4, 15.7, 15.3. HRMS (ESI) m/z Calcd for C43H72NO9 746.5202 (M + H)+, found: 746.5196.
3-hydroxy-12β-(3′-((tert-butoxycarbonyl)amino)-4′-(tert-butoxy)-D-aspartate)oxy-13H-olean-28-oic acid (9b).
White solid 53 mg, overall yield: 68.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.53 (d, 1H, J = 8.8 Hz, NH), 4.98–5.04 (m, 1H, 12-H), 4.42 (dt, 1H, J = 4.0, 8.8 Hz, HαAsp), 3.20 (dd, 1H, J = 4.0, 11.2 Hz, 3-H), 2.99 (dd, 1H, J = 4.4, 17.6 Hz, HaβAsp), 2.74 (dd, 1H, J = 4.4, 17.6 Hz, HbβAsp), 2.48 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.10 (dd, 1H, J = 4.7, 11.4 Hz, 13-H), 1.47 (s, 9H, Boc), 1.43 (s, 9H, But), 1.00 (s, 3H, Me), 0.97 (s, 3H, Me), 0.91 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 6H, 2Me), 0.75 (s, 3H, Me), 0.66 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 184.0, 171.2, 170.1, 155.5, 82.1, 79.7, 78.7, 72.5, 55.2, 50.4, 48.8, 47.2, 41.4, 40.6, 39.7, 38.8, 38.5, 37.1, 36.9, 36.1, 34.3, 33.2, 33.1, 32.4, 31.3, 30.4, 29.7, 28.8, 28.3, 28.0, 27.9, 27.4, 27.2, 23.4, 22.9, 18.2, 17.6, 16.5, 15.8, 15.4. HRMS (ESI) m/z Calcd for C43H72NO9 746.5202 (M + H)+, found: 746.5196.
3-hydroxy-12β-(4′-((tert-butoxycarbonyl)amino)-5′-(tert-butoxy)-D-glutamate)oxy-13H-olean-28-oic acid (10a).
White solid 53 mg, overall yield: 52.0%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.15 (d, 1H, J = 8.0 Hz, NH), 4.97–5.03 (m, 1H, 12-H), 4.22–4.24 (m, 1H, HαGlu), 3.19 (dd, 1H, J = 4.5, 11.4 Hz, 3-H), 2.50 (dt, 1H, J = 4.4, 11.6 Hz, 18-H), 2.43 (ddd, 1H, J = 6.0, 10.0, 16.4 Hz, HaγGlu), 2.32 (ddd, 1H, J = 5.2, 10.4, 16.0 Hz, HbγGlu), 2.13–2.21 (m, 1H, HaβGlu), 2.07 (dd, 1H, J = 4.8, 12.0 Hz, 13-H), 1.47 (s, 9H, Boc), 1.44 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 9.6 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.2, 172.6, 171.5, 155.5, 82.1, 79.7, 78.8, 71.9, 55.2, 53.3, 48.7, 47.2, 41.4, 40.6, 40.0, 38.9, 38.6, 37.1, 36.0, 34.3, 33.3, 33.2, 32.4, 31.4, 30.6, 30.5, 28.8, 28.4, 27.4, 27.2, 23.4, 22.9, 18.2, 17.8, 16.5, 15.8, 15.4. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5349.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamte)oxy-13H-olean-28-oic acid (10b).
White solid 12 g, overall yield: 71.4%. [α = +8.3° (c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 5.09 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.16–4.22 (m, 1H, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.50 (dt, 1H, J = 4.0, 14.0 Hz, 18-H), 2.31–2.45 (m, 2H, 2HγGlu), 2.08 (dd, 1H, J = 4.0, 12.0 Hz, 13-H), 1.47 (s, 9H, Boc), 1.44 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 9.6 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.9, 172.6, 171.4, 155.4, 82.1, 79.8, 78.8, 72.0, 55.2, 53.6, 48.8, 47.2, 41.4, 40.6, 39.9, 38.8, 38.6, 37.1, 36.1, 34.3, 33.2, 32.4, 31.3, 30.7, 30.5, 28.8, 28.3, 28.0, 27.3, 27.1, 23.4, 22.8, 18.2, 17.7, 16.5, 15.9, 15.4. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5349.
3-hydroxy-12β-((S)-3′-((tert-butoxycarbonyl)amino)-5′-(tert-butoxy)-5-oxopentanoyl)oxy-13H-olean-28-oic acid (10c).
White solid 93 mg, overall yield: 46.0%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.37 (d, 1H, J = 8.8 Hz, NH), 4.98–5.04 (m, 1H, 12-H), 4.29 (br, 1H, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.56–2.72 (m, 4H, 2CH2), 2.46 (dt, 1H, J = 4.0, 14.0 Hz, 18-H), 2.08 (dd, 1H, J = 4.8, 11.2 Hz, 13-H), 1.45 (s, 9H, Boc), 1.42 (s, 9H, But), 1.00 (s, 3H, Me), 0.97 (s, 3H, Me), 0.91 (s, 3H, Me), 0.90 (s, 3H, Me), 0.83 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 9.6 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.6, 171.4, 170.4, 154.9, 81.0, 79.3, 78.8, 72.2, 55.2, 48.8, 47.2, 44.5, 41.4, 40.6, 39.8, 39.7, 38.8, 38.6, 37.9, 37.0, 36.1, 34.2, 33.3, 33.1, 32.3, 31.3, 30.4, 28.8, 28.4, 28.1, 28.0, 27.4, 27.1, 23.4, 22.8, 18.2, 17.7, 16.5, 15.8, 15.3. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5349.
3-hydroxy-12β-(2′-((tert-butoxycarbonyl)amino)-5′-(tert-butoxy)-D-glutamate)oxy-13H-olean-28-oic acid (10d).
White solid 6 mg, overall yield: 10.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.04–5.11 (m, 2H, NH, 12-H), 4.27–4.32 (m, 1H, HαGlu), 3.19 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.46 (dt, 1H, J = 4.0, 13.6 Hz, 18-H), 2.29–2.41 (m, 2H, 2HγGlu), 1.16–2.24 (m, 2H, 2HβGlu), 2.10 (dd, 1H, J = 4.4, 11.6 Hz, 13-H), 1.44 (s, 18H, 2But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.93 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.76 (s, 3H, Me), 0.67 (d, 1H, J = 9.6 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 172.1, 172.0, 155.4, 80.5, 79.7, 78.8, 73.2, 55.1, 53.4, 48.5, 47.2, 41.4, 40.6, 40.1, 38.8, 38.5, 37.1, 35.8, 33.4, 33.1, 32.3, 32.2, 31.5, 30.5, 29.7, 28.5, 28.3, 28.1, 28.0, 27.3, 27.2, 23.8, 22.6, 18.2, 17.8, 16.5, 15.7, 15.3. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5352.
3-hydroxy-12β-((S)-4′-amino-5′-(tert-butoxy)-5-oxopentanoyl)oxy-13H-olean-28-oic acid (11a).
White solid 35 mg, overall yield: 49.8%. 1H NMR (400 MHz, methanol-d4) δ (ppm): 5.00–5.07 (m, 1H, 12-H), 3.58 (t, 1H, J = 6.4 Hz, HαGlu), 3.21 (dd, 1H, J = 4.8, 10.8 Hz, 3-H), 2.45–2.54 (m, 3H, 18-H, 2HγGlu), 2.17 (dd, 1H, J = 4.8, 11.6 Hz, 13-H), 1.97–2.06 (m, 2H, 2HβGlu), 1.49 (s, 9H, But), 1.03 (s, 3H, Me), 0.96 (s, 3H, Me), 0.91 (s, 3H, Me), 0.89 (s, 3H, Me), 0.84 (s, 6H, 2Me), 0.74 (s, 3H, Me). HRMS (ESI) m/z Calcd for C39H66NO7 660.4834 (M + H)+, found: 660.4826.
3-hydroxy-12β-((S)-4′-((tert-butoxycarbonyl)amino)-4′-carboxybutanoyl)oxy-13H-olean-28-oic acid (11b).
White solid 77 mg, overall yield: 85.0%. 1H NMR (400 MHz, methanol-d4) δ (ppm): 4.95–5.00 (m, 1H, 12-H), 4.26 (br, 1H, HαGlu), 3.12 (dd, 1H, J = 5.6, 10.4 Hz, 3-H), 2.16–2.60 (m, 4H, 2HβGlu, 2HγGlu), 1.45 (s, 9H, Boc), 1.00 (s, 3H, Me), 0.98 (s, 3H, Me), 0.95 (s, 3H, Me), 0.85 (s, 3H, Me), 0.84 (s, 3H, Me), 0.80 (s, 3H, Me), 0.74 (s, 3H, Me). 13C NMR (100 MHz, methanol-d4) δ (ppm): 174.4, 80.5, 79.6, 79.4, 79.3, 78.9, 73.6, 56.6, 56.4, 42.7, 41.7, 39.9, 39.8, 38.2, 37.8, 35.9, 35.0, 34.1, 33.7, 33.3, 31.7, 30.7, 30.1, 28.9, 28.6, 28.4, 27.9, 24.3, 19.4, 18.3, 17.3, 16.5, 16.1. HRMS (ESI) m/z Calcd for C40H65NO9Na 726.4557 (M + H)+, found: 726.4546.
3-hydroxy-12β-((S)-4′-amino-4′-carboxybutanoyl)oxy-13H-olean-28-oic acid (11c).
To a solution of 50 mg of 10b in DCM, 50% TFA was added slowly. The mixture was stirred at 23 °C for 4 h. After the material was completely consumed, the mixture was concentrated with vacuum. Then, 1 mL of cold ethyl acetate was added, and cold petroleum ether was added dropwise until white solid precipitated out. The mixture was left to store in refrigerator at 4 °C for 2 h. The precipitate was filtered and rinsed with cold petroleum ether and dried to obtain 16.7 mg of pure white solid with yield of 42.0%. 1H NMR (400 MHz, methanol-d4) δ (ppm): 5.02–5.09 (m, 1H, 12-H), 4.70 (dd, 1H, J = 4.8, 11.6 Hz, 3-H), 3.97 (t, 1H, J = 6.0 Hz, HαGlu), 2.56–2.61 (m, 2H, 2HγGlu), 2.51 (dt, 1H, J = 4.4, 14.8 Hz, 18-H), 2.13–2.20 (m, 3H, 13-H, 2HβGlu), 1.06 (s, 3H, Me), 0.96 (s, 3H, Me), 0.91 (s, 9H, 3Me), 0.89 (s, 3H, Me), 0.84 (s, 3H, Me). 13C NMR (100 MHz, methanol-d4) δ (ppm): 181.9, 173.4, 171.7, 87.4, 73.6, 56.2, 53.5, 42.7, 41.8, 41.2, 39.1, 39.0, 38.1, 37.4, 35.3, 34.4, 33.7, 33.4, 32.8, 31.4, 31.2, 29.9, 28.4, 28.2, 27.0, 24.1, 24.0, 23.9, 19.1, 18.2, 16.9, 16.7, 16.3. HRMS (ESI) m/z Calcd for C35H58NO7 604.4208 (M + H)+, found: 604.4213.
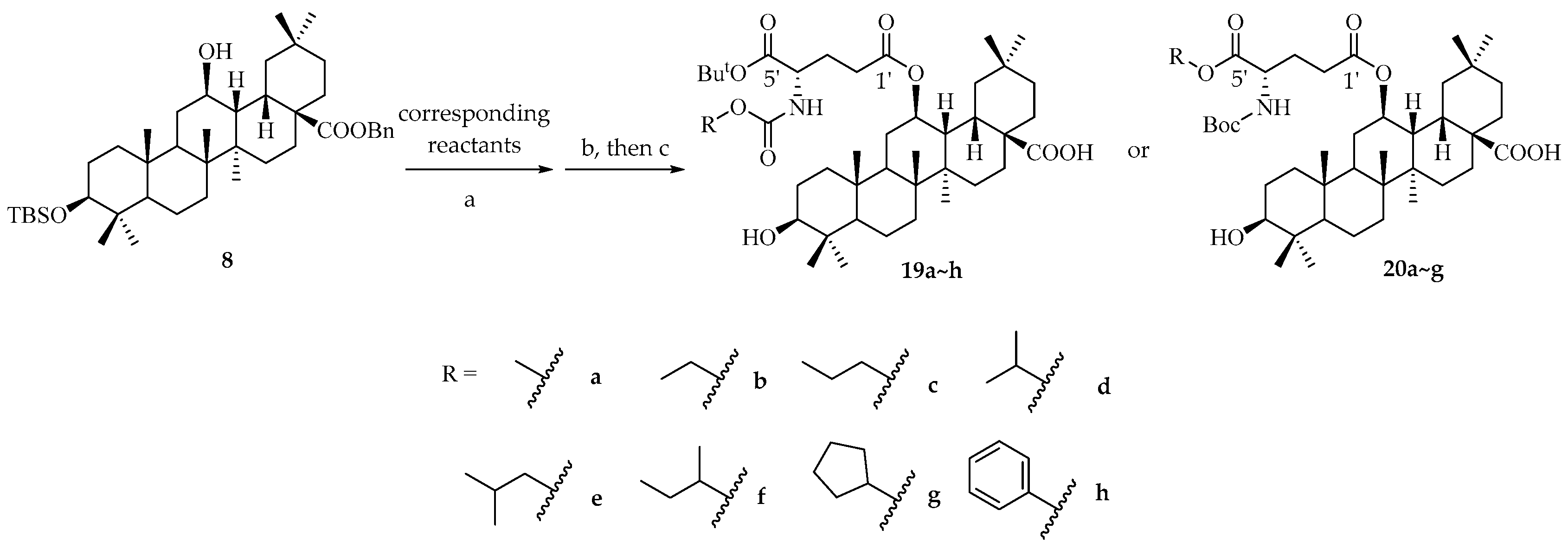
3β-hydroxy-12β-(4′-((methoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19a).
White solid 120 mg, overall yield: 47.2%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.34 (d, 1H, J = 8.4 Hz, NH), 4.95–5.03 (m, 1H, 12-H), 4.23–4.29 (m, 1H, HαGlu), 3.67 (s, 3H, OMe), 3.20 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 4.0, 12.8 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.14–2.21 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 11.6 Hz, 13-H), 1.94–2.02 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.6, 172.8 171.2, 156.8, 82.5, 78.9, 72.2, 55.3, 54.1, 52.5, 48.8, 47.3, 41.5, 40.7, 40.0, 39.0, 38.6, 37.2, 36.2, 34.4, 33.4, 33.3, 32.5, 31.4, 30.7, 30.6, 28.9, 28.1, 28.0, 27.4, 27.3, 23.5, 23.0, 18.3, 17.8, 16.6, 16.0, 15.5. HRMS (ESI) m/z Calcd for C41H68NO9 718.4889 (M + H)+, found: 718.4887.
3β-hydroxy-12β-(4′-((ethoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19b).
White solid 157 mg, overall yield: 54.1%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.28 (d, 1H, J = 8.0 Hz, NH), 4.95–5.03 (m, 1H, 12-H), 4.23–4.29 (m, 1H, HαGlu), 4.07–4.17 (m, 2H, OCH2CH3), 3.20 (dd, 1H, J = 4.0, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.14–2.21 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 11.6 Hz, 13-H), 1.94–2.00 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.24 (t, 3H, J = 7.2 Hz, OCH2CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.5, 172.7 171.3, 156.4, 82.4, 78.9, 72.2, 61.2, 55.3, 54.0, 52.5, 48.9, 47.3, 41.5, 40.7, 40.0, 39.0, 38.6, 37.2, 36.2, 34.4, 33.4, 33.3, 32.5, 31.4, 30.7, 30.6, 29.0, 28.1, 27.4, 27.3, 23.5, 23.0, 18.3, 17.8, 16.6, 16.0, 15.5, 14.7. HRMS (ESI) m/z Calcd for C42H70NO9 732.5045 (M + H)+, found: 732.5047.
3β-hydroxy-12β-(4′-((propoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19c).
White solid 120 mg, overall yield: 47.5%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.27 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.23–4.28 (m, 1H, HαGlu), 3.97–4.07 (m, 2H, OCH2CH2CH3), 3.20 (dd, 1H, J = 4.4, 12.0 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.14–2.21 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 12.0 Hz, 13-H), 1.94–2.01 (m, 1H, HbβGlu), 1.60–1.65 (m, 2H, OCH2CH2CH3), 1.47 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.93 (t, 3H, J = 7.6 Hz, OCH2CH2CH3), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.0, 155.6, 82.3, 78.8, 72.1, 66.8, 55.2, 53.9, 48.8, 47.2, 41.4, 40.6, 39.9, 38.9, 38.6, 37.1, 36.1, 34.3, 33.3, 33.2, 32.4, 31.4, 30.6, 30.5, 28.8, 28.0, 27.3, 27.2, 23.4, 22.9, 22.3, 18.2, 17.8, 16.5, 15.9, 15.4, 10.4. HRMS (ESI) m/z Calcd for C43H71NO9Na 768.5021 (M + Na)+, found: 768.5014.
3β-hydroxy-12β-(4′-((isopropoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19d).
White solid 153 mg, overall yield: 57.1%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.22 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.87–4.92 (m, 1H, OCH(CH3)2), 4.24 (dt, 1H, J = 6.0, 8.0 Hz, HαGlu), 3.20 (dd, 1H, J = 4.8, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 4.0, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.12–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 11.6 Hz, 13-H), 1.94–2.01 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.23 (d, 6H, J = 6.4 Hz, i-Pr-2CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.5, 172.6, 171.3, 155.9, 82.3, 78.8, 72.1, 68.5, 55.2, 53.8, 48.8, 47.2, 41.4, 40.6, 39.9, 38.9, 38.6, 37.1, 36.1, 34.3, 33.3, 33.2, 32.4, 31.3, 30.6, 30.5, 29.7, 28.8, 28.0, 27.3, 27.2, 23.4, 22.9, 22.2, 22.1, 18.2, 17.7, 16.5, 15.9, 15.4. HRMS (ESI) m/z Calcd for C43H72NO9 746.5201 (M + H)+, found: 746.5199.
3β-hydroxy-12β-(4′-((isobutoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19e).
White solid 139 mg, overall yield: 53.0%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.29 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.24 (dt, 1H, J = 5.6, 8.0 Hz, HαGlu), 3.78–3.92 (m, 2H, OCH2CH(CH3)2), 3.20 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.12–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 11.6 Hz, 13-H), 1.94–2.01 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.93 (d, 6H, J = 6.5 Hz, OCH2CH(CH3)2), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.5, 172.6, 171.2, 156.4, 82.3, 78.8, 72.1, 71.3, 55.2, 53.8, 48.8, 47.2, 41.4, 40.6, 39.9, 38.9, 38.6, 37.1, 36.1, 34.3, 33.3, 33.2, 32.4, 31.3, 30.6, 30.5, 29.7, 28.8, 28.0, 27.3, 27.2, 23.4, 22.9, 19.1, 18.2, 17.7, 16.5, 15.9, 15.4. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5357.
3β-hydroxy-12β-(4′-((sec-butoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19f).
White solid 103 mg, overall yield: 40.9%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.25 (d, 1H, J = 8.4 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.69–4.74 (m, 1H, OCH), 4.24 (dt, 1H, J = 6.0, 8.0 Hz, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.12–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 11.2 Hz, 13-H), 1.94–2.01 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.20 (d, 3H, J = 6.0 Hz, CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.89 (t, 3H, J = 4.0 Hz, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.1, 172.6, 171.2, 157.5, 82.3, 78.8, 73.1, 72.1, 55.2, 53.8, 48.8, 47.2, 41.4, 40.6, 39.9, 38.9, 38.6, 37.1, 36.1, 34.3, 33.3, 33.2, 32.4, 31.3, 30.6, 30.5, 29.7, 28.8, 28.0, 27.3, 27.2, 23.4, 22.9, 19.7, 18.2, 17.7, 16.5, 15.8, 15.4. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5361.
3β-hydroxy-12β-(4′-(((cyclopentyloxy)carbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19g).
White solid 140 mg, overall yield: 54.6%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.20 (d, 1H, J = 8.0 Hz, NH), 5.08 (br, 1H, Cyclopentyl-CH), 4.95–5.02 (m, 1H, 12-H), 4.24 (dt, 1H, J = 6.0, 8.0 Hz, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.31–2.44 (m, 2H, 2HγGlu), 2.12–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 11.4 Hz, 13-H), 1.94–2.01 (m, 1H, HbβGlu), 1.47 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.4, 172.6, 171.3, 156.1, 82.3, 78.8, 77.8, 72.1, 55.2, 53.8, 48.8, 47.2, 41.4, 40.6, 39.9, 38.9, 38.6, 37.1, 36.1, 34.3, 33.3, 33.2, 32.8, 32.7, 32.4, 31.3, 30.6, 30.5, 29.7, 28.8, 28.0, 27.9, 27.3, 27.2, 23.7, 23.6, 23.4, 22.9, 19.7, 18.2, 17.7, 16.5, 15.8, 15.4. HRMS (ESI) m/z Calcd for C45H74NO9 772.5358 (M + H)+, found: 772.5355.
3β-hydroxy-12β-(4′-((phenoxycarbonyl)amide)-5′-(tert-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (19h).
White solid 142 mg, overall yield: 47.5%. 1H NMR (400 MHz, CDCl3) δ (ppm): 7.34 (dd, 2H, J = 8.0, 8.4 Hz, Ph-2H), 7.19 (dd, 1H, J = 8.0, 8.4 Hz, Ph-H), 7.12 (d, 1H, J = 7.6 Hz, Ph-2H), 5.73 (d, 1H, J = 8.4 Hz, NH), 4.97–5.05 (m, 1H, 12-H), 4.32 (dt, 1H, J = 5.2, 7.6 Hz, HαGlu), 3.17 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.41–2.49 (m, 2H, 2HγGlu), 2.18–2.27 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 4.8, 12.0 Hz, 13-H), 2.01–2.08 (m, 1H, HbβGlu), 1.49 (s, 9H, But), 1.01 (s, 3H, Me), 0.96 (s, 3H, Me), 0.91 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.78 (s, 3H, Me), 0.74 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.4, 172.6, 170.8, 154.3, 129.3, 125.4, 121.6, 82.7, 78.8, 72.2, 55.2, 54.1, 48.8, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.1, 36.1, 34.3, 33.3, 33.2, 32.4, 31.4, 30.6, 30.5, 28.8, 28.0, 27.8, 27.3, 27.2, 23.4, 22.9, 18.2, 17.7, 16.5, 15.8, 15.4.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-methoxy-L-glutamate)oxy-13H-olean-28-oic acid (20a).
White solid 97 mg, overall yield: 33.2%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.20 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.30–4.33 (m, 1H, HαGlu), 3.75 (s, 3H, OMe), 3.20 (dd, 1H, J = 4.0, 11.6 Hz, 3-H), 2.49 (dt, 1H, J = 3.6, 13.6 Hz, 18-H), 2.40 (t, 2H, J = 8.0 Hz, 2HγGlu), 2.14–2.21 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 11.6 Hz, 13-H), 1.94–2.00 (m, 1H, HbβGlu), 1.44 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.8, 172.8 172.5, 155.4, 80.0, 78.8, 72.2, 55.2, 53.0, 52.4, 48.7, 47.2, 41.5, 41.4, 40.6, 39.9, 38.8, 38.5, 37.0, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.4, 28.8, 28.3, 28.0, 27.6, 27.3, 27.1, 23.3, 22.8, 18.2, 17.7, 16.6, 15.8, 15.3. HRMS (ESI) m/z Calcd for C41H67NO9Na 740.4708 (M + Na)+, found: 740.4706.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-ethoxy-L-glutamate)oxy-13H-olean-28-oic acid (20b).
White solid 159 mg, overall yield: 49.5%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.17 (d, 1H, J = 7.6 Hz, NH), 4.96–5.02 (m, 1H, 12-H), 4.30 (dt, 1H, J = 6.0, 6.0 Hz, HαGlu), 4.20 (q, 2H, J = 7.2 Hz, OCH2CH3), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.40 (t, 2H, J = 7.2 Hz, 2HγGlu), 2.14–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 11.2 Hz, 13-H), 1.95–2.00 (m, 1H, HbβGlu), 1.44 (s, 9H, But), 1.28 (t, 3H, J = 6.8 Hz, OCH2CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.7, 172.5 172.3, 155.4, 79.9, 78.8, 72.1, 61.5, 55.2, 53.0, 48.7, 47.2, 41.5, 41.4, 40.6, 39.9, 38.8, 38.5, 37.0, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.4, 28.8, 28.3, 28.0, 27.7, 27.3, 27.1, 23.3, 22.8, 18.2, 17.7, 16.5, 15.3, 14.2. HRMS (ESI) m/z Calcd for C42H69NO9Na 754.4865 (M + Na)+, found: 754.4858.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-propoxy-L-glutamate)oxy-13H-olean-28-oic acid (20c).
White solid 150 mg, overall yield: 89.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.16 (d, 1H, J = 8.0 Hz, NH), 4.96–5.02 (m, 1H, 12-H), 4.30 (dt, 1H, J = 6.0, 6.0 Hz, HαGlu), 4.10 (t, 2H, J = 6.4 Hz, OCH2CH2CH3), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.40 (t, 2H, J = 7.2 Hz, 2HγGlu), 2.14–2.20 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 11.2 Hz, 13-H), 1.95–2.00 (m, 1H, HbβGlu), 1.44 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.95 (t, 3H, J = 7.6 Hz, OCH2CH2CH3), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.6, 172.5 172.4, 155.4, 79.9, 78.8, 72.1, 67.0, 55.2, 53.0, 48.7, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.0, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.5, 30.4, 28.8, 28.3, 28.0, 27.7, 27.3, 27.1, 23.3, 22.8, 21.9, 18.2, 17.7, 16.5, 15.3, 10.4. HRMS (ESI) m/z Calcd for C43H71NO9Na 768.5021 (M + Na)+, found: 768.5012.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-isopropoxy-L-glutamate)oxy-13H-olean-28-oic acid (20d).
White solid 125 mg, overall yield: 74.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.15 (d, 1H, J = 8.0 Hz, NH), 5.04 (tt, 1H, J = 6.0, 6.0 Hz, OCHMe2), 4.96–5.00 (m, 1H, 12-H), 4.25 (dt, 1H, J = 6.4, 6.8 Hz, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.37–2.42 (m, 2H, 2HγGlu), 2.13–2.27 (m, 1H, HaβGlu), 2.08 (dd, 1H, J = 5.2, 11.2 Hz, 13-H), 1.95–2.00 (m, 1H, HbβGlu), 1.44 (s, 9H, But), 1.27 (d, 3H, J = 6.0 Hz, OCHMea), 1.25 (d, 3H, J = 6.0 Hz, OCHMeb), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.7, 172.5 171.8, 155.4, 79.9, 78.8, 72.1, 69.2, 55.2, 53.2, 48.7, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.0, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.5, 30.4, 28.8, 28.3, 28.0, 27.8, 27.3, 27.1, 23.3, 22.8, 21.8, 21.7, 18.2, 17.7, 16.5, 15.8, 15.3. HRMS (ESI) m/z Calcd for C43H71NO9Na 768.5021 (M + Na)+, found: 768.5012.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-isobutoxy-L-glutamate)oxy-13H-olean-28-oic acid (20e).
White solid 133 mg, overall yield: 43.4%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.13 (d, 1H, J = 8.0 Hz, NH), 4.96–5.02 (m, 1H, 12-H), 4.32 (dt, 1H, J = 6.0, 6.4 Hz, HαGlu), 3.92 (d, 2H, J = 6.8 Hz, OCH2CHMe2), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.40 (t, 2H, J = 7.6 Hz, 2HγGlu), 2.14–2.20 (m, 1H, HaβGlu), 2.09 (dd, 1H, J = 5.2, 11.2 Hz, 13-H), 1.94–2.02 (m, 2H, HbβGlu, OCH2CHMe2), 1.44 (s, 9H, But), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.95 (d, 6H, J = 6.8 Hz, OCH2CHMe2), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 182.6, 172.5 172.3, 155.4, 79.9, 78.8, 72.1, 71.5, 55.2, 53.1, 48.7, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.0, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.5, 30.4, 28.8, 28.3, 28.0, 27.8, 27.7, 27.3, 27.2, 23.3, 22.9, 19.0, 18.2, 17.7, 16.5, 15.8, 15.3. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5354.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-(sec-butoxy)-L-glutamate)oxy-13H-olean-28-oic acid (20f).
White solid 137 mg, overall yield: 44.8%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.13 (d, 1H, J = 8.4 Hz, NH), 4.96–5.02 (m, 1H, 12-H), 4.88 (ddq, 1H, J = 2.8, 6.0, 13.2 Hz, OCH(CH3)CH2CH3), 4.28 (dt, 1H, J = 6.0, 6.4 Hz, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.40 (t, 2H, J = 7.6 Hz, 2HγGlu), 2.14–2.20 (m, 1H, HaβGlu), 2.09 (dd, 1H, J = 5.2, 11.4 Hz, 13-H), 1.94–2.02 (m, 1H, HbβGlu), 1.44 (s, 9H, But), 1.23 (d, 3H, J = 6.0 Hz, OCH(CH3)CH2CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.90 (t, 3H, J = 6.4 Hz, OCH(CH3)CH2CH3), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.1, 172.5 171.9, 155.4, 79.9, 78.8, 73.8, 73.7, 72.1, 55.2, 53.3, 53.2, 48.7, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.1, 36.0, 34.3, 33.2, 33.1, 32.4, 31.3, 30.6, 30.4, 28.8, 28.7, 28.3, 28.0, 27.8, 27.3, 27.2, 23.4, 22.9, 19.4, 19.3, 18.2, 17.7, 16.5, 15.8, 15.3, 9.7. HRMS (ESI) m/z Calcd for C44H74NO9 760.5358 (M + H)+, found: 760.5359.
3β-hydroxy-12β-(4′-((tert-butoxycarbonyl)amide)-5′-cyclopentyloxy-L-glutamate)oxy-13H-olean-28-oic acid (20g).
White solid 179 mg, overall yield: 53.2%. 1H NMR (400 MHz, CDCl3) δ (ppm): 5.19–5.23 (m, 1H, cyclopentyl-CH), 5.12 (d, 1H, J = 8.0 Hz, NH), 4.95–5.02 (m, 1H, 12-H), 4.25 (dt, 1H, J = 6.0, 6.4 Hz, HαGlu), 3.20 (dd, 1H, J = 4.4, 11.2 Hz, 3-H), 2.49 (dt, 1H, J = 4.4, 14.0 Hz, 18-H), 2.36–2.41 (m, 2H, 2HγGlu), 2.08–2.20 (m, 1H, HaβGlu), 2.10 (dd, 1H, J = 5.2, 11.2 Hz, 13-H), 1.93–1.98 (m, 1H, HbβGlu), 1.55–1.68 (m, 8H, cyclopentyl-8H), 1.44 (s, 9H, But), 1.23 (d, 3H, J = 6.0 Hz, OCH(CH3)CH2CH3), 1.01 (s, 3H, Me), 0.97 (s, 3H, Me), 0.92 (s, 3H, Me), 0.89 (s, 3H, Me), 0.82 (s, 3H, Me), 0.81 (s, 3H, Me), 0.75 (s, 3H, Me), 0.67 (d, 1H, J = 10.0 Hz, 5-H). 13C NMR (100 MHz, CDCl3) δ (ppm): 182.8, 172.5 172.0, 155.4, 79.9, 78.8, 78.4, 72.1, 55.2, 53.1, 48.7, 47.2, 41.4, 40.6, 39.9, 38.8, 38.5, 37.1, 36.0, 34.3, 33.2, 33.1, 32.7, 32.6, 32.4, 31.3, 30.6, 30.4, 28.8, 28.7, 28.3, 28.0, 27.8, 27.3, 27.2, 23.7, 23.6, 23.4, 22.9, 18.2, 17.7, 16.5, 15.8, 15.3. HRMS (ESI) m/z Calcd for C45H74NO9 772.5358 (M + H)+, found: 772.5356.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}