
Development of Novel Small-Molecule Activators of Pyruvate Kinase Muscle Isozyme 2, PKM2, to Reduce Photoreceptor Apoptosis
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Synthesis of PKM2 Activators for Intraocular Delivery
2.2. Small Molecules Activate Recombinant PKM2 and Increase PK Activity In Vitro and In Vivo
2.3. X-ray Structure of PKM2 in Complex with Compound 2
2.4. Compound 2 Treatment Reduces Apoptosis and Prevents Cell Death In Vitro
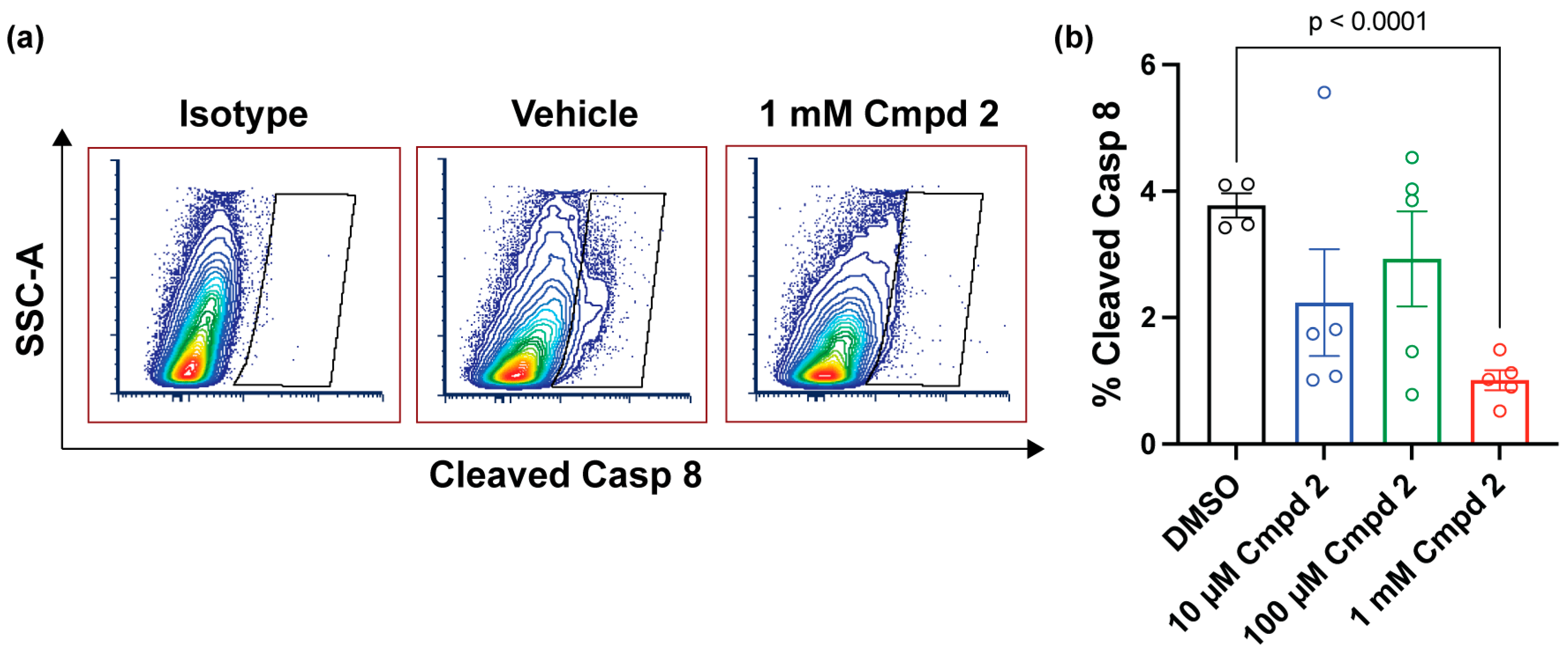
2.5. Compound 2 Reduces Apoptosis after Experimental Retinal Detachment
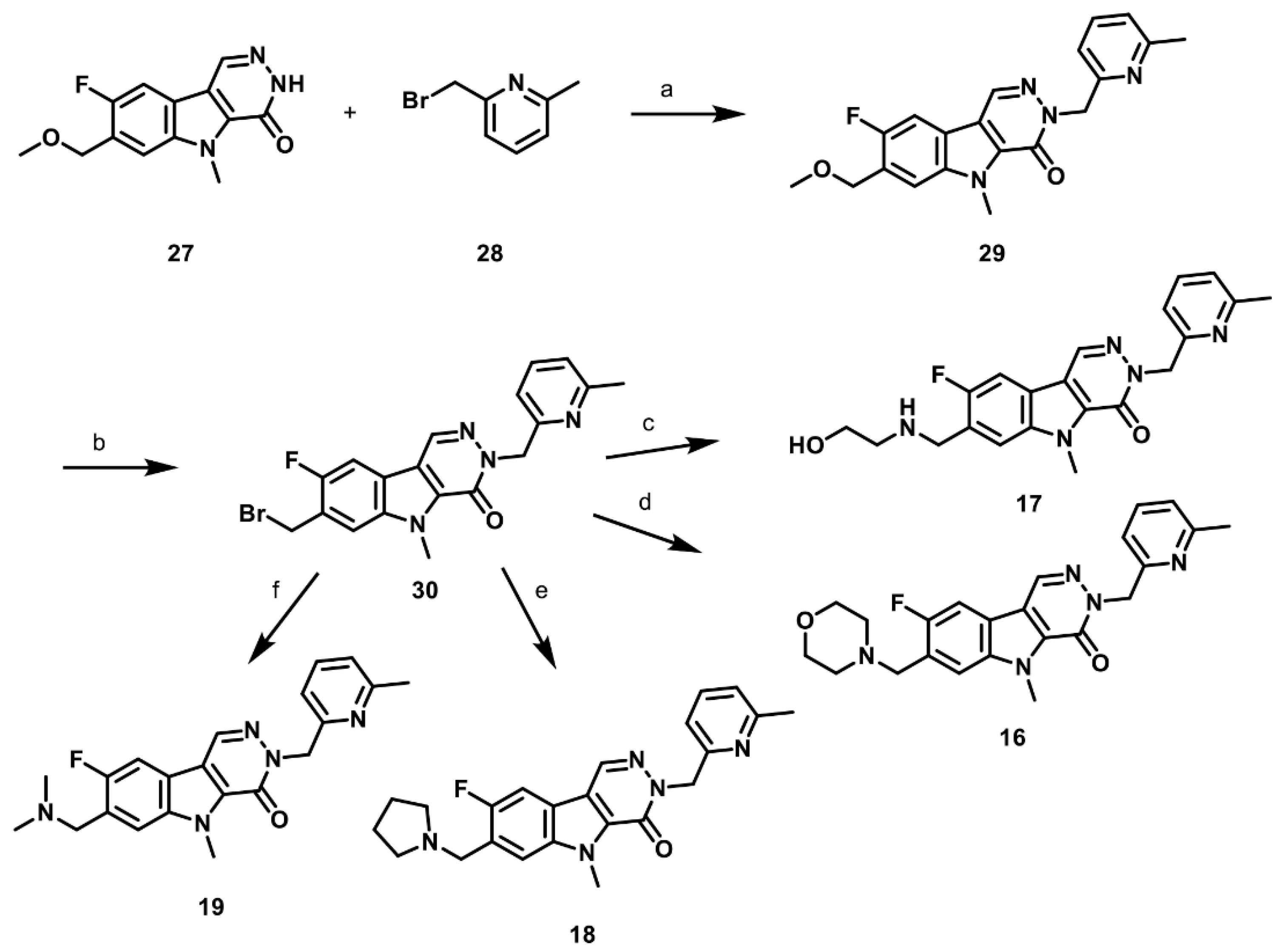
2.6. Synthesis of Structurally Diverse PKM2 Activators with Improved Solubility and Retained Potency
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Pyruvate Kinase Activity Enzyme Assay
4.4. Aqueous Solubility of Small Molecule PKM2 Activators
4.5. PKM2 Protein Expression and Purification
4.6. PKM2 Crystallization and Structure Determination
4.7. Western Blot
4.8. Caspase Activity Assay
4.9. Cell Viability
4.10. Experimental Model of Retinal Detachment
4.11. Flow Cytometric Assessment of Cleaved Caspase 8 after Experimental Retinal Detachment
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the Insulin/MTOR Pathway Delays Cone Death in a Mouse Model of Retinitis Pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Rüegg, M.A.; Punzo, C. Activated MTORC1 Promotes Long-Term Cone Survival in Retinitis Pigmentosa Mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical Adaptations of the Retina and Retinal Pigment Epithelium Support a Metabolic Ecosystem in the Vertebrate Eye. eLife 2017, 6, e28899. [Google Scholar] [CrossRef]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.-Y.; Zieger, M.; Hay, N.; Punzo, C. Aerobic Glycolysis Is Essential for Normal Rod Function and Controls Secondary Cone Death in Retinitis Pigmentosa. Cell Rep. 2018, 23, 2629–2642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Justus, S.; Hsu, C.-W.; Bonet-Ponce, L.; Wu, W.-H.; Tsai, Y.-T.; Wu, W.-P.; Jia, Y.; Duong, J.K.; et al. Reprogramming Metabolism by Targeting Sirtuin 6 Attenuates Retinal Degeneration. J. Clin. Investig. 2016, 126, 4659–4673. [Google Scholar] [CrossRef]
- Chinchore, Y.; Begaj, T.; Wu, D.; Drokhlyansky, E.; Cepko, C.L. Glycolytic Reliance Promotes Anabolism in Photoreceptors. eLife 2017, 6, e25946. [Google Scholar] [CrossRef]
- Wubben, T.J.; Pawar, M.; Smith, A.; Toolan, K.; Hager, H.; Besirli, C.G. Photoreceptor Metabolic Reprogramming Provides Survival Advantage in Acute Stress While Causing Chronic Degeneration. Sci. Rep. 2017, 7, 17863. [Google Scholar] [CrossRef] [PubMed]
- Wubben, T.J.; Pawar, M.; Weh, E.; Smith, A.; Sajjakulnukit, P.; Zhang, L.; Dai, L.; Hager, H.; Pai, M.P.; Lyssiotis, C.A.; et al. Small Molecule Activation of Metabolic Enzyme Pyruvate Kinase Muscle Isozyme 2, PKM2, Circumvents Photoreceptor Apoptosis. Sci. Rep. 2020, 10, 2990. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Han, G.; Wood, J.P.M. An Explanation for the Warburg Effect in the Adult Mammalian Retina. Clin. Experiment. Ophthalmol. 2013, 41, 517. [Google Scholar] [CrossRef]
- Chertov, A.O.; Holzhausen, L.; Kuok, I.T.; Couron, D.; Parker, E.; Linton, J.D.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Roles of Glucose in Photoreceptor Survival. J. Biol. Chem. 2011, 286, 34700–34711. [Google Scholar] [CrossRef]
- Ng, S.K.; Wood, J.P.M.; Chidlow, G.; Han, G.; Kittipassorn, T.; Peet, D.J.; Casson, R.J. Cancer-like Metabolism of the Mammalian Retina. Clin. Exp. Ophthalmol. 2015, 43, 367–376. [Google Scholar] [CrossRef]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 Contributes to Cancer Metabolism. Cancer Lett. 2015, 356, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Wood, J.P.M.; Han, G.; Kittipassorn, T.; Peet, D.J.; Chidlow, G. M-Type Pyruvate Kinase Isoforms and Lactate Dehydrogenase A in the Mammalian Retina: Metabolic Implications. Investig. Ophthalmol. Vis. Sci. 2016, 57, 66–80. [Google Scholar] [CrossRef]
- Gui, D.Y.; Lewis, C.A.; Vander Heiden, M.G. Allosteric Regulation of PKM2 Allows Cellular Adaptation to Different Physiological States. Sci. Signal. 2013, 6, pe7. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrústegui, J.; Hurley, J.B. Pyruvate Kinase and Aspartate-Glutamate Carrier Distributions Reveal Key Metabolic Links between Neurons and Glia in Retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed]
- Rajala, R.V.S.; Rajala, A.; Kooker, C.; Wang, Y.; Anderson, R.E. The Warburg Effect Mediator Pyruvate Kinase M2 Expression and Regulation in the Retina. Sci. Rep. 2016, 6, 37727. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Z. Pyruvate Kinase M2 at a Glance. J. Cell Sci. 2015, 128, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Walsh, M.J.; Brimacombe, K.R.; Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Hong, B.-S.; Tempel, W.; Dimov, S.; Veith, H.; et al. ML265: A Potent PKM2 Activator Induces Tetramerization and Reduces Tumor Formation and Size in a Mouse Xenograft Model. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate Kinase M2 Activators Promote Tetramer Formation and Suppress Tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Boxer, M.B.; Vander Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of Thieno[3,2-b]Pyrrole[3,2-d]Pyridazinones as Activators of the Tumor Cell Specific M2 Isoform of Pyruvate Kinase. Bioorg. Med. Chem. Lett. 2010, 20, 3387–3393. [Google Scholar] [CrossRef]
- Sodano, T.M.; Combee, L.A.; Stephenson, C.R.J. Recent Advances and Outlook for the Isosteric Replacement of Anilines. ACS Med. Chem. Lett. 2020, 11, 1785–1788. [Google Scholar] [CrossRef]
- Xue, J.; Zhang, Y.-S.; Huan, Z.; Yang, J.-D.; Cheng, J.-P. Catalytic Vilsmeier-Haack Reactions for C1-Deuterated Formylation of Indoles. J. Org. Chem. 2022, 87, 15539–15546. [Google Scholar] [CrossRef]
- Tan, E.; Ding, X.-Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of Cone-Photoreceptor-Specific Antigens in a Cell Line Derived from Retinal Tumors in Transgenic Mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Al-Ubaidi, M.R.; Matsumoto, H.; Kurono, S.; Singh, A. Proteomics Profiling of the Cone Photoreceptor Cell Line, 661W. Adv. Exp. Med. Biol. 2008, 613, 301–311. [Google Scholar] [CrossRef] [PubMed]
- al-Ubaidi, M.R.; Font, R.L.; Quiambao, A.B.; Keener, M.J.; Liou, G.I.; Overbeek, P.A.; Baehr, W. Bilateral Retinal and Brain Tumors in Transgenic Mice Expressing Simian Virus 40 Large T Antigen under Control of the Human Interphotoreceptor Retinoid-Binding Protein Promoter. J. Cell Biol. 1992, 119, 1681–1687. [Google Scholar] [CrossRef]
- Parikh, R.; Ross, J.S.; Sangaralingham, L.R.; Adelman, R.A.; Shah, N.D.; Barkmeier, A.J. Trends of Anti-Vascular Endothelial Growth Factor Use in Ophthalmology Among Privately Insured and Medicare Advantage Patients. Ophthalmology 2017, 124, 352–358. [Google Scholar] [CrossRef]
- Patel, S. Medicare Spending on Anti-Vascular Endothelial Growth Factor Medications. Ophthalmol. Retin. 2018, 2, 785–791. [Google Scholar] [CrossRef]
- Vezina, M.; Bussieres, M.; Glazier, G.; Gagnon, M.-P.; Martel, D. Determination of Injectable Intravitreous Volumes in Rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3219. [Google Scholar]
- Dureau, P.; Bonnel, S.; Menasche, M.; Dufier, J.L.; Abitbol, M. Quantitative Analysis of Intravitreal Injections in the Rat. Curr. Eye Res. 2001, 22, 74–77. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Smart, O.S.; Horský, V.; Gore, S.; Svobodová Vařeková, R.; Bendová, V.; Kleywegt, G.J.; Velankar, S. Validation of Ligands in Macromolecular Structures Determined by X-Ray Crystallography. Acta Crystallogr. Sect. Struct. Biol. 2018, 74, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Zacks, D.N.; Zheng, Q.-D.; Han, Y.; Bakhru, R.; Miller, J.W. FAS-Mediated Apoptosis and Its Relation to Intrinsic Pathway Activation in an Experimental Model of Retinal Detachment. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4563–4569. [Google Scholar] [CrossRef]
- Zacks, D.N.; Boehlke, C.; Richards, A.-L.; Zheng, Q.-D. Role of the Fas-Signaling Pathway in Photoreceptor Neuroprotection. Arch. Ophthalmol. Chic. Ill 1960 2007, 125, 1389–1395. [Google Scholar] [CrossRef]
- Kanan, Y.; Moiseyev, G.; Agarwal, N.; Ma, J.-X.; Al-Ubaidi, M.R. Light Induces Programmed Cell Death by Activating Multiple Independent Proteases in a Cone Photoreceptor Cell Line. Investig. Ophthalmol. Vis. Sci. 2007, 48, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Besirli, C.G.; Chinskey, N.D.; Zheng, Q.-D.; Zacks, D.N. Inhibition of Retinal Detachment-Induced Apoptosis in Photoreceptors by a Small Peptide Inhibitor of the Fas Receptor. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2177–2184. [Google Scholar] [CrossRef]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Besirli, C.G.; Zheng, Q.-D.; Reed, D.M.; Zacks, D.N. ERK-Mediated Activation of Fas Apoptotic Inhibitory Molecule 2 (Faim2) Prevents Apoptosis of 661W Cells in a Model of Detachment-Induced Photoreceptor Cell Death. PLoS ONE 2012, 7, e46664. [Google Scholar] [CrossRef] [PubMed]
- Zacks, D.N.; Hänninen, V.; Pantcheva, M.; Ezra, E.; Grosskreutz, C.; Miller, J.W. Caspase Activation in an Experimental Model of Retinal Detachment. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1262–1267. [Google Scholar] [CrossRef]
- Hisatomi, T.; Sakamoto, T.; Goto, Y.; Yamanaka, I.; Oshima, Y.; Hata, Y.; Ishibashi, T.; Inomata, H.; Susin, S.A.; Kroemer, G. Critical Role of Photoreceptor Apoptosis in Functional Damage after Retinal Detachment. Curr. Eye Res. 2002, 24, 161–172. [Google Scholar] [CrossRef]
- Le Goff, M.M.; Bishop, P.N. Adult Vitreous Structure and Postnatal Changes. Eye 2008, 22, 1214–1222. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; Womack, T.O. BUSTER Version 2.10.3; Global Phasing Ltd.: Cambridge, UK, 2011. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Weh, E.; Lutrzykowska, Z.; Smith, A.; Hager, H.; Pawar, M.; Wubben, T.J.; Besirli, C.G. Hexokinase 2 Is Dispensable for Photoreceptor Development but Is Required for Survival during Aging and Outer Retinal Stress. Cell Death Dis. 2020, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Besirli, C.G.; Chinskey, N.D.; Zheng, Q.-D.; Zacks, D.N. Autophagy Activation in the Injured Photoreceptor Inhibits Fas-Mediated Apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4193–4199. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
| Recombinant PKM2 † | 661W Cells † | In Vivo | |||||||
| Cmpd ID | X | R1 | R2 | AC50 (nM) | Max Activation (%) | AC50 (nM) | Max Activation (%) | PK Activation, Fold Change & | Aqueous Solubility (M) * |
| ML-265 |  |  |  | 70 ± 17 | 292 ± 21 | 108 ± 32 | 370 ± 21 | 1.00 ± 0.06 | 153.4 |
| 1 |  |  |  | 23 ± 4 | 224 ± 9 | 175 ± 64 | 219 ± 16 | 0.04 ± 0.09 | <0.01 |
| 2 |  |  |  | 50 ± 12 | 292 ± 20 | 120 ± 45 | 355 ± 26 | 0.95 ± 0.13 | 99.8 |
| 3 |  |  |  | 47 ± 10 | 279 ± 16 | 104 ± 32 | 280 ± 17 | 0.50 ± 0.16 | 12.1 |
| 4 |  |  |  | 48 ± 10 | 216 ± 12 | 66 ± 19 | 256 ± 13 | 0.20 ± 0.09 | <0.01 |
| 5 |  |  |  | 61 ± 12 | 249 ± 14 | 132 ± 47 | 236 ± 16 | 0.13 ± 0.06 | 12.7 |
| PKM2-Compound 2 | |
|---|---|
| Data Collection | |
| Space group | I222 |
| Unit cell dimensions | |
| a, b, c (Å) | 84.81, 115.97, 131.84 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) & | 1.84 (1.87–1.84) |
| Rsym # | 0.038 (0.137) |
| <I/I> * | 29.5 (10.9) |
| % Completeness $ | 94.4 (95.3) |
| Redundancy | 3.9 (3.8) |
| Refinement | |
| Resolution (Å) | 1.84 |
| No. of unique reflections | 53,262 |
| R-factor + | 0.163 |
| Rfree ## | 0.190 |
| RMSD ! | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.89 |
| MolProbity Score %$ | 0.91 |
| Clash Score %$ | 1.61 |
| RSCC ** | 0.96 |
| RSR ** | 0.14 |
| Recombinant PKM2 † | 661W Cells † | ||||||
|---|---|---|---|---|---|---|---|
| Cmpd ID | R | AC50 (nM) | Max Activation (%) | AC50 (nM) | Max Activation (%) | Aqueous Solubility (M) * | Chemical Scaffold |
| 16 |  | 65 ± 8 | 224 ± 8 | 162 ± 46 | 268 ± 15 | 396.6 |  |
| 17 |  | 58 ± 5 | 213 ± 6 | 1616 ± 428 | 430 ± 33 | 1116.5 | |
| 18 |  | 64 ± 4 | 245 ± 5 | 106 ± 27 | 257 ± 12 | 858.1 | |
| 19 |  | 83 ± 9 | 266 ± 9 | 222 ± 81 | 297 ± 22 | 1013.8 | |
| 20 |  | 62 ± 11 | 239 ± 12 | 93 ± 29 | 288 ± 17 | 462.2 |  |
| 21 |  | 66 ± 10 | 339 ± 16 | 72 ± 24 | 299 ± 18 | 333.7 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wubben, T.J.; Chaudhury, S.; Watch, B.T.; Stuckey, J.A.; Weh, E.; Fernando, R.; Goswami, M.; Pawar, M.; Rech, J.C.; Besirli, C.G. Development of Novel Small-Molecule Activators of Pyruvate Kinase Muscle Isozyme 2, PKM2, to Reduce Photoreceptor Apoptosis. Pharmaceuticals 2023, 16, 705. https://doi.org/10.3390/ph16050705
Wubben TJ, Chaudhury S, Watch BT, Stuckey JA, Weh E, Fernando R, Goswami M, Pawar M, Rech JC, Besirli CG. Development of Novel Small-Molecule Activators of Pyruvate Kinase Muscle Isozyme 2, PKM2, to Reduce Photoreceptor Apoptosis. Pharmaceuticals. 2023; 16(5):705. https://doi.org/10.3390/ph16050705
Chicago/Turabian StyleWubben, Thomas J., Sraboni Chaudhury, Brennan T. Watch, Jeanne A. Stuckey, Eric Weh, Roshini Fernando, Moloy Goswami, Mercy Pawar, Jason C. Rech, and Cagri G. Besirli. 2023. "Development of Novel Small-Molecule Activators of Pyruvate Kinase Muscle Isozyme 2, PKM2, to Reduce Photoreceptor Apoptosis" Pharmaceuticals 16, no. 5: 705. https://doi.org/10.3390/ph16050705