



Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
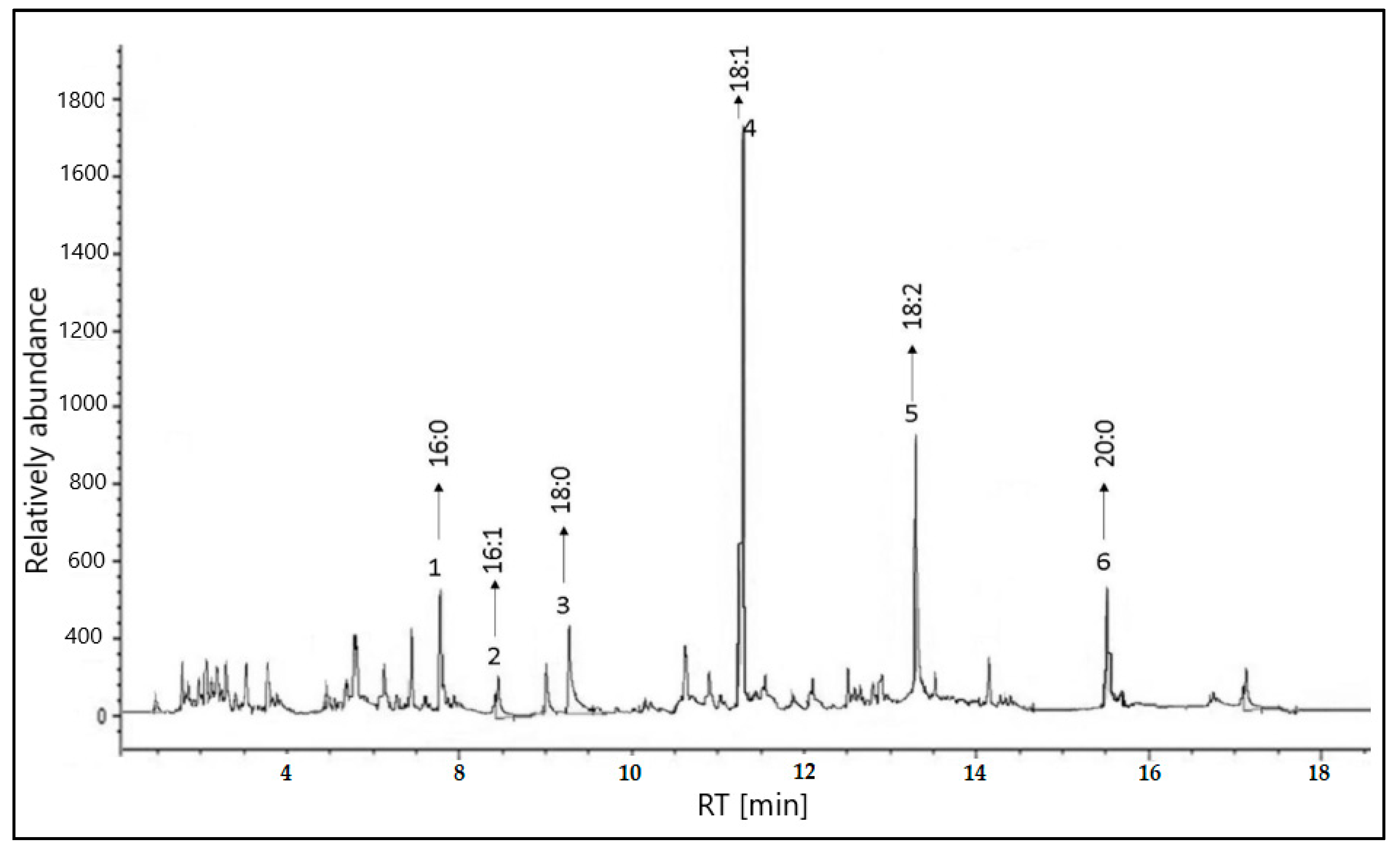
2.1. GC–MS Analysis of the Extract
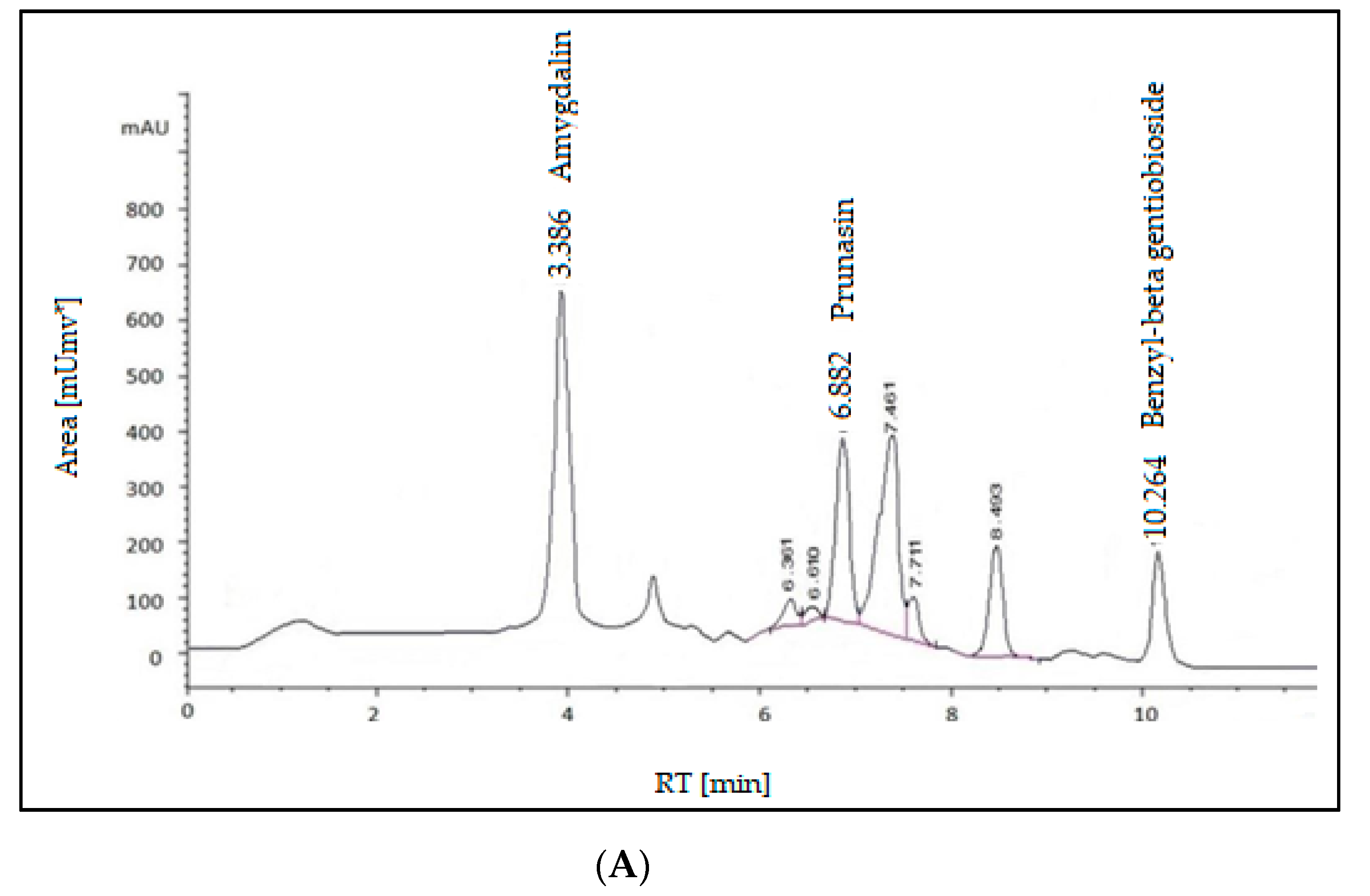
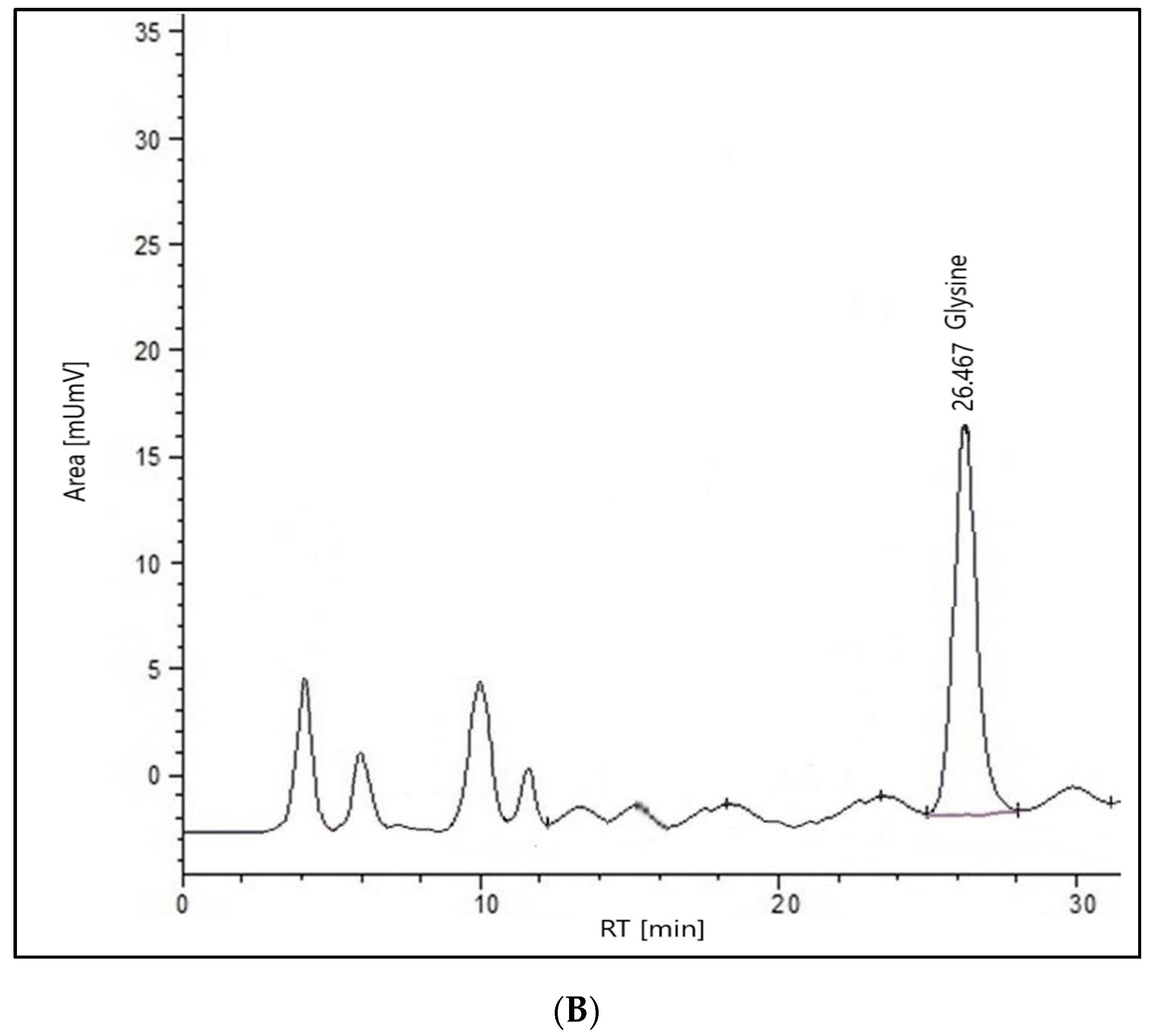
2.2. HPLC Analysis of the Extract
2.2.1. Glycine
2.2.2. Glycosides
2.2.3. Flavonoids
2.2.4. Steroids
2.3. LC-MS/MS Descriptions
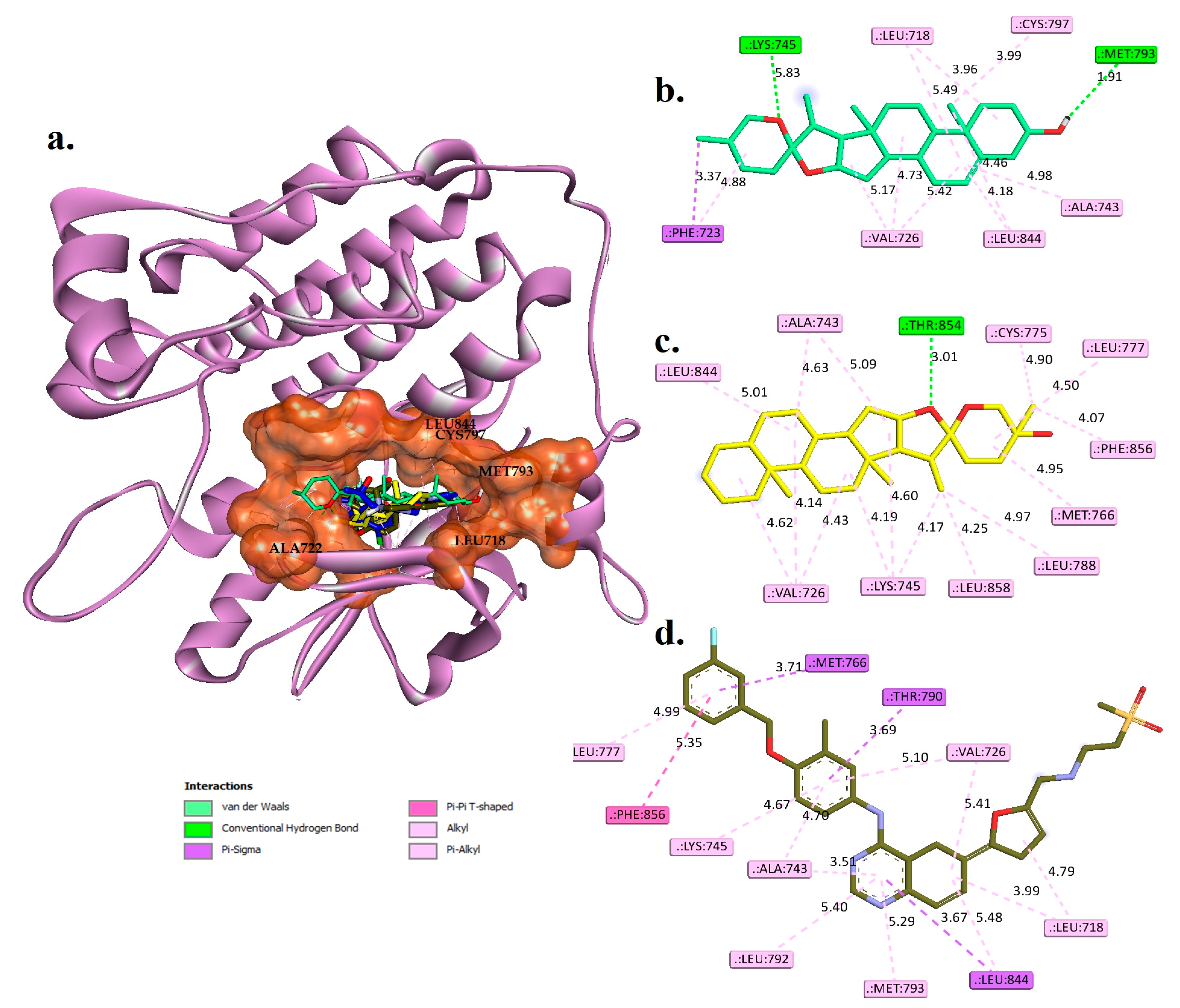
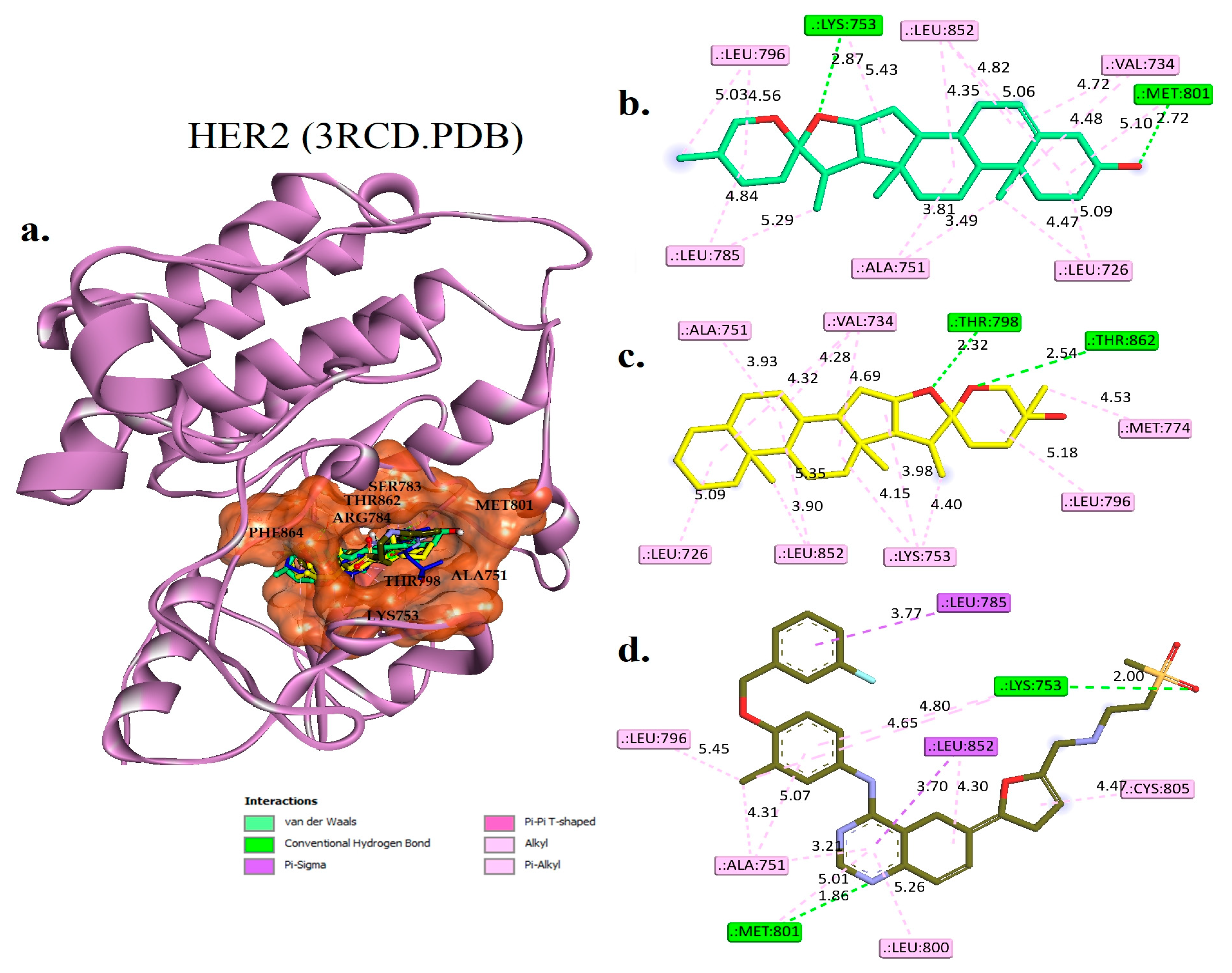
2.4. Molecular Docking Studies
Interaction Examines the Hit Phytocompounds
2.5. ADMET Prediction
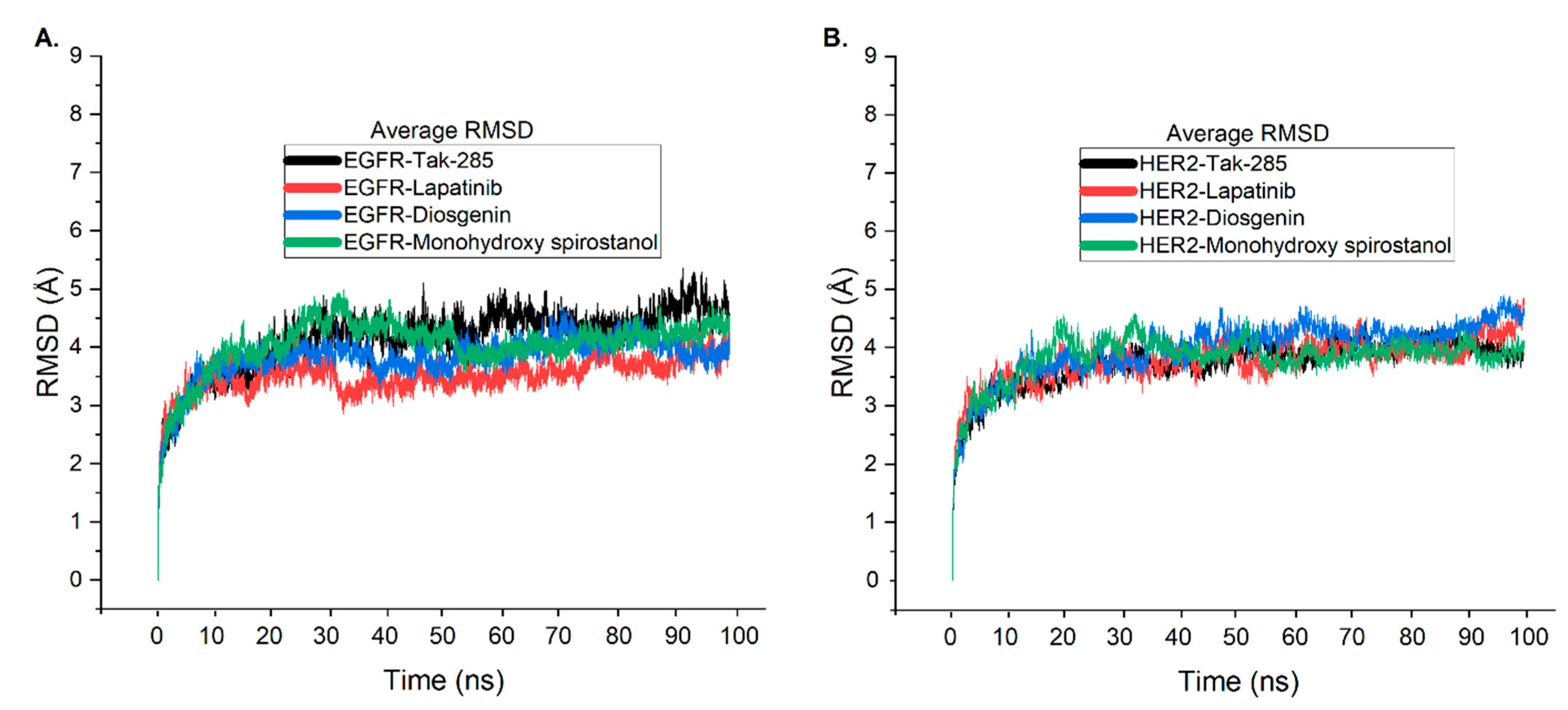
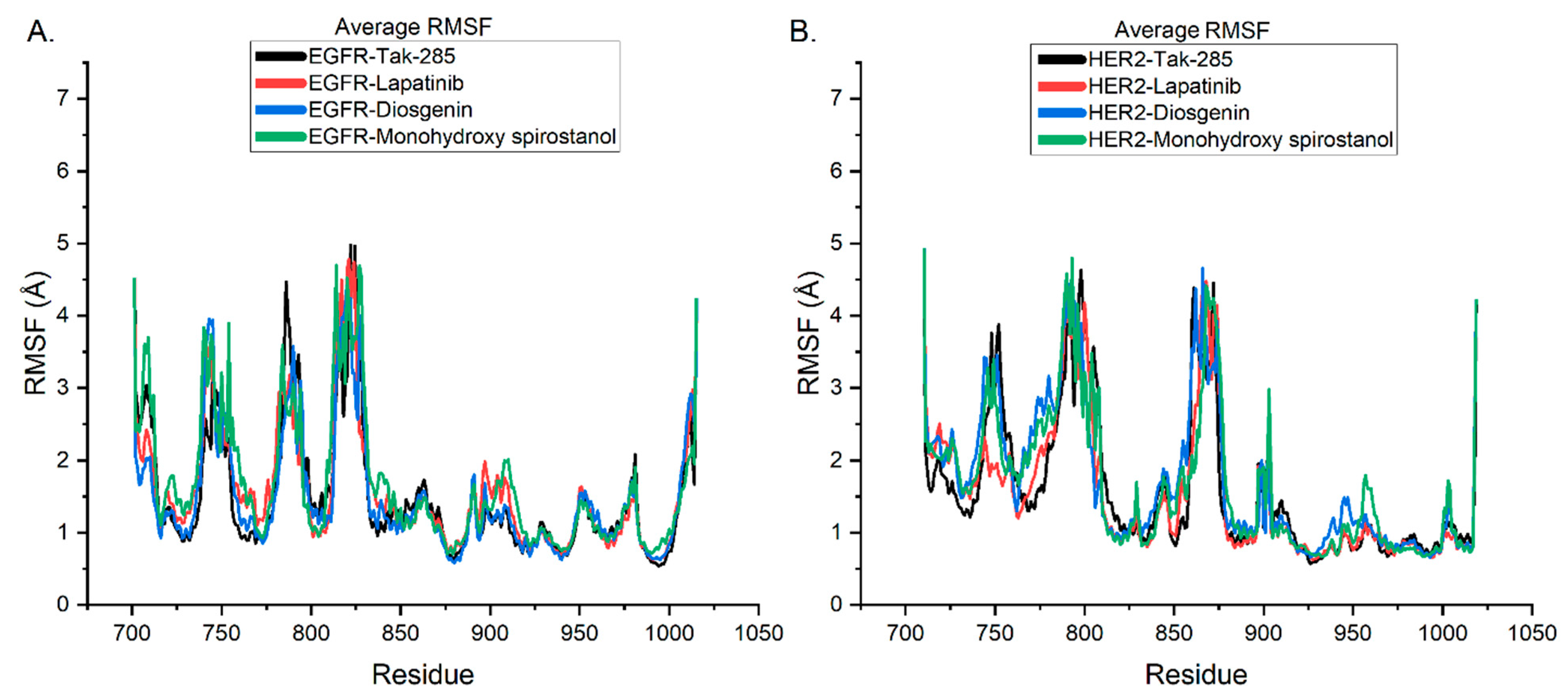
2.6. Molecular Dynamic Simulations
3. Materials and Methods
3.1. Collection and Preparation of Prunus amygdalus var amara Seeds Extract
3.2. Instruments and Preparation of Prunus amygdalus var amara Seed Extraction
3.2.1. GC-MS Analysis
3.2.2. HPLC System-Glycine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flow Rate 0.4 mL/min | A | B |
| Time | Methanol: Formic acid (10%: 0.1%) | Methanol: Formic acid (50%: 0.1%) |
| 0–6.5 | 10–30 | 90–70 |
| 6.5–7 | 30–100 | 70–0 |
| 8–8.5 | 100–10 | 0–90 |
| 8.5–12.5 | 10 | 90 |
3.2.3. Glycosides
| Flow Rate 0.8 mL/min | A % | B % |
| Time | 0.1% phosphoric acid: water | Methanol 100% HPLC—Grade |
| 0 | 95 | 5 |
| 0–0.5 | 5–25 | 95–75 |
| 0.5–2 | 80–90 | 20–10 |
| 2–4.5 | 10–60 | 90–40 |
| 4.5–8 | 50–60 | 50–40 |
| 8–14 | 0 | 100 |
3.2.4. Flavonoids
3.2.5. Steroids
3.3. Molecular Docking
3.3.1. Protein Structure Preparation
3.3.2. Ligand Preparation
3.3.3. Molecular Docking Preparation
3.4. ADMET Prediction
3.5. Molecular Dynamics
3.6. Binding Free Energy Calculation Using MM/PBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K. Various types and management of breast cancer: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109. [Google Scholar] [PubMed]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.K.; Wang, X.; Gundamaraju, R.; Joon, S.; Tsagkaris, C.; Behzad, S.; Khan, J.; Gautam, R.; Goyal, R.; Rakmai, J. Natural products derived from medicinal plants and microbes might act as a game-changer in breast cancer: A comprehensive review of preclinical and clinical studies. Crit. Rev. Food Sci. Nutr. 2022, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi-Majd, M.; Hassanshahi, G.; Vatanparast, M.; Karimabad, M.N. Investigation of the Effect of Prunus amygdalus amara on the Expression of some Genes of Apoptosis and Immortality in Breast Cancer Cells (MCF-7). Curr. Drug Res. Rev. Former. Curr. Drug Abus. Rev. 2022, 14, 73–79. [Google Scholar]
- Saati, S.; Dehghan, P.; Azizi-Soleiman, F.; Mobasseri, M. The effect of bitter almond (Amygdalus communis L. var. Amara) gum as a functional food on metabolic profile, inflammatory markers, and mental health in type 2 diabetes women: A blinded randomized controlled trial protocol. Trials 2023, 24, 35. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Gohil, K.J.; Rajput, R.T.; Sharma, V. Almond (Prunus amygdalus Batsch.): A Latest Review on Pharmacology and Medicinal uses. Res. J. Pharm. Technol. 2022, 15, 3301–3308. [Google Scholar] [CrossRef]
- Moradi, B.; Heidari-Soureshjani, S.; Asadi-Samani, M.; Yang, Q. A systematic review of phytochemical and phytotherapeutic characteristics of bitter almond. Int. J. Pharm. Phytopharm. Res. 2017, 7, 1–9. [Google Scholar]
- Keser, S.; Demir, E.; Yilmaz, O. Some Bioactive Compounds and Antioxidant Activities of the Bitter Almond Kernel (Prunus dulcis var. amara). J. Chem. Soc. Pak. 2014, 36, 922–930. [Google Scholar]
- Guici El Kouacheur, K.; Cherif, H.S.; Saidi, F.; Bensouici, C.; Fauconnier, M.L. Prunus amygdalus var. amara (bitter almond) seed oil: Fatty acid composition, physicochemical parameters, enzyme inhibitory activity, antioxidant and anti-inflammatory potential. J. Food Meas. Charact. 2023, 17, 371–384. [Google Scholar] [CrossRef]
- Mericli, F.; Becer, E.; Kabadayı, H.; Hanoglu, A.; Yigit Hanoglu, D.; Ozkum Yavuz, D.; Ozek, T.; Vatansever, S. Fatty acid composition and anticancer activity in colon carcinoma cell lines of Prunus dulcis seed oil. Pharm. Biol. 2017, 55, 1239–1248. [Google Scholar] [CrossRef]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in cancer: Signaling mechanisms, drugs, and acquired resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef] [PubMed]
- Murphrey, M.B.; Quaim, L.; Varacallo, M. Biochemistry, Epidermal Growth Factor Receptor; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Rubin, I.; Yarden, Y. The basic biology of HER2. Ann. Oncol. 2001, 12, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Schneider, M.R.; Yarden, Y. The EGFR-HER2 module: A stem cell approach to understanding a prime target and driver of solid tumors. Oncogene 2016, 35, 2949–2960. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Peckys, D.B.; de Jonge, N. EGFR expression in HER2-driven breast cancer cells. Int. J. Mol. Sci. 2020, 21, 9008. [Google Scholar] [CrossRef]
- Riecke, K.; Witzel, I. Targeting the human epidermal growth factor receptor family in breast cancer beyond HER2. Breast Care 2020, 15, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ghamande, S.; Liu, H.; Xue, L.; Zhao, S.; Tan, W.; Zhao, L.; Tang, S.-C.; Wu, D.; Korkaya, H. Targeting EGFR/HER2/HER3 with a three-in-one aptamer-siRNA chimera confers superior activity against HER2+ breast cancer. Mol. Ther. Nucleic Acids 2018, 10, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Borrero-García, L.D.; del Mar Maldonado, M.; Medina-Velázquez, J.; Troche-Torres, A.L.; Velazquez, L.; Grafals-Ruiz, N.; Dharmawardhane, S. Rac inhibition as a novel therapeutic strategy for EGFR/HER2 targeted therapy resistant breast cancer. BMC Cancer 2021, 21, 652. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Montemurro, F.; Valabrega, G.; Aglietta, M. Lapatinib: A dual inhibitor of EGFR and HER2 tyrosine kinase activity. Expert Opin. Biol. Ther. 2007, 7, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yu, H.; Ma, Q.; Shen, S.; Das, U.N. Linoleic acid suppresses colorectal cancer cell growth by inducing oxidant stress and mitochondrial dysfunction. Lipids Health Dis. 2010, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Ito, H.; Mukainaka, T.; Tokuda, H.; Nishino, H.; Yoshida, T. Anti-tumor promoting effect of glycosides from Prunus persica seeds. Biol. Pharm. Bull. 2003, 26, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Newmark, J.; Brady, R.O.; Grimley, P.M.; Gal, A.E.; Waller, S.G.; Thistlethwaite, J.R. Amygdalin (Laetrile) and prunasin beta-glucosidases: Distribution in germ-free rat and in human tumor tissue. Proc. Natl. Acad. Sci. USA 1981, 78, 6513–6516. [Google Scholar] [CrossRef]
- Kashyap, D.; Sharma, A.; Tuli, H.S.; Sak, K.; Punia, S.; Mukherjee, T.K. Kaempferol–A dietary anticancer molecule with multiple mechanisms of action: Recent trends and advancements. J. Funct. Foods 2017, 30, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Arya, P.; Kumar, P. Diosgenin a steroidal compound: An emerging way to cancer management. J. Food Biochem. 2021, 45, e14005. [Google Scholar] [CrossRef]
- El Sayed, A.M.; Basam, S.M.; Marzouk, H.S.; El-Hawary, S. LC–MS/MS and GC–MS profiling as well as the antimicrobial effect of leaves of selected Yucca species introduced to Egypt. Sci. Rep. 2020, 10, 17778. [Google Scholar] [CrossRef]
- Shukla, R.; Tripathi, T. Molecular dynamics simulation of protein and protein–ligand complexes. In Computer-Aided Drug Design; Springer: Singapore, 2020; pp. 133–161. [Google Scholar]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.; Suárez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of oligopeptides in high yield under simple programmable conditions. Nat. Commun. 2015, 6, 8385. [Google Scholar] [CrossRef]
- Bolarinwa, I.F.; Orfila, C.; Morgan, M.R. Amygdalin content of seeds, kernels and food products commercially-available in the UK. Food Chem. 2014, 152, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, C.; Ravaglia, D.; Ragaini, A.; Costa, G. Phenolic compounds in peach (Prunus persica) cultivars at harvest and during fruit maturation. Ann. Appl. Biol. 2008, 153, 11–23. [Google Scholar] [CrossRef]
- Król-Kogus, B.; Głód, D.; Krauze-Baranowska, M. Qualitative and quantitative HPLC-ELSD-ESI-MS analysis of steroidal saponins in fenugreek seed. Acta Pharm. 2020, 70, 89–99. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.-C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo [3,2-d] pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The protein data bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. In Small Molecules in Oncology; Springer International Publishing: Cham, Switzerlands, 2018; pp. 19–44. [Google Scholar]
- Dassault Systèmes. BIOVIA Discovery Studio 2020; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering: Methods and Protocols; Humana: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Lin, Z.; Van Gunsteren, W.F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theory Comput. 2011, 7, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Rühle, V. Pressure coupling/barostats. J. Club 2008, 1–5. [Google Scholar]
- Berendsen, H.J.; Postma, J.v.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic interactions are a key to MDM2 inhibition by polyphenols as revealed by molecular dynamics simulations and MM/PBSA free energy calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. The Potential Anti-Cancerous Activity of Prunus amygdalus var. amara Extract. Processes 2023, 11, 1277. [Google Scholar] [CrossRef]







| Peak no. | Retention Time (TR) | Type | Area % | Component Name | Percentage Contains % |
|---|---|---|---|---|---|
| 1 | 7.832 | (C16:0) | 6.550 | Palmitic | 6.550 |
| 2 | 8.488 | (C16:1) | 0.812 | Palmitoleic | 0.812 |
| 3 | 9.267 | (C18:0) | 4.843 | Stearic | 4.843 |
| 4 | 11.316 | (C18:1) | 52.042 | Oleic | 52.042 |
| 5 | 13.233 | (C18:2) | 27.512 | Linoleic | 27.512 |
| 6 | 15.512 | (C20:0) | 8.241 | Arachidic | 8.241 |
| # | Target | EGFR (3POZ.PDB) | HER2 (3RCD.PDB) |
|---|---|---|---|
| Docking Score (kcal/mol) | Docking Score (kcal/mol) | ||
| 1 | Amygdalin | −7.57 | −6.15 |
| 2 | Arachidic Acid | −6.46 | −5.87 |
| 3 | Benzyl-beta gentiobioside | −6.8 | −6.03 |
| 4 | Centaureidin | −8.26 | −7.07 |
| 5 | Diosgenin | −9.77 | −10.1 |
| 6 | Glycine | −5.03 | −4.27 |
| 7 | Kaempferol | −8.46 | −7.1 |
| 8 | Linoleic Acid | −6.99 | −5.73 |
| 9 | Monohydroxy spirostanol | −9.74 | −9.19 |
| 10 | Oleic Acid | −6.45 | −5.84 |
| 11 | Palmitic Acid | −5.99 | −5.47 |
| 12 | Palmitoleic Acid | −5.92 | −5.5 |
| 13 | Prunasin | −8.14 | −6.14 |
| 14 | Stearic Acid | −6.2 | −5.53 |
| Tak-285 (control) | −9.71 | −9.21 | |
| Lapatinib (control) | −9.64 | −9.76 |
| Phytocompounds | Hydrogen Bond Interactions | Pi-Sigma | Hydrophobic Interaction | |
|---|---|---|---|---|
| Residues | Distances (Å) | |||
| Diosgenin | LYS745 and MET793 | 1.81 and 1.91 | PHE723 | LEU718, VAL726, PHE723, CYS799, LEU844, and ALA743 |
| Monohydroxy spirostanol | THR854 | 2.01 | ----- | VAL726, LYS745, MET766, ALA743, LEU777, LEU844, CYS775, LEU788, PHE856, and LEU858 |
| Tak-285 (control) | LEU777, THR790, and MET793 | 3.45, 3.60, and 2.78 | LEU718 and LEU844 | VAL726, ALA743, LYS745, PHE766, CYS775, LEU788, LEU792, PHE856, and LEU858 |
| Lapatinib | ---- | ---- | MET766, THR790, and LEU844 | LEU718, VAL726, ALA743, LYS745, LEU777, LEU792, MET793, LEU844, and PHE856 |
| Phytocompounds | Hydrogen Bond Interactions | Pi-Sigma | Hydrophobic Interaction | |
|---|---|---|---|---|
| Residues | Distances (Å) | |||
| Diosgenin | LYS745 and MET793 | 1.81 and 1.91 | PHE723 | LEU718, PHE723, VAL726, ALA743, CYS799, and LEU844 |
| Monohydroxy spirostanol | THR854 | 2.01 | ----- | VAL726, ALA743, MET766, LYS745, CYS775, LEU844, LEU777, LEU788, PHE856, and LEU858 |
| Tak-285 (control) | LEU777, THR790, and MET793 | 3.45, 3.60, and 2.78 | LEU718 and LEU844 | VAL726, ALA743, LYS745, PHE766, CYS775, LEU788, LEU792, PHE856, and LEU858 |
| Lapatinib | ---- | ---- | MET766, THR790, and LEU844 | LEU718, VAL726, ALA743, LYS745, LEU777, LEU792, MET793, LEU844, and PHE856 |
| ADMET Prediction | Compound | |||
|---|---|---|---|---|
| Diosgenin | Monohydroxy Spirostanol | Tak-285 | Lapatinib | |
| Fsp3 | 0.926 | 1 | 0.269 | 0.172 |
| Lipinski Rule | Accepted | Accepted | Accepted | Accepted |
| PAINS | 0 | 0 | 0 | 0 |
| BMS Rule | 0 | 0 | 0 | 0 |
| Caco-2 Permeability | −4.805 | −4.805 | −5.505 | −5.707 |
| Pgp-inhibitor | 0.281 | 0.993 | 0.992 | 0.999 |
| Pgp-substrate | 0.001 | 0.001 | 0.973 | 0.995 |
| HIA | 0.005 | 0.002 | 0.009 | 0.006 |
| BBB Penetration | 0.701 | 0.187 | 0.912 | 0.043 |
| CL | 23.332 | 22.718 | 8.791 | 3.997 |
| T1/2 | 0.023 | 0.08 | 0.59 | 0.066 |
| AMES Toxicity | 0.053 | 0.019 | 0.632 | 0.536 |
| Rat Oral Acute Toxicity | 0.748 | 0.67 | 0.637 | 0.388 |
| Carcinogenicity | 0.188 | 0.274 | 0.108 | 0.022 |
| System | ΔGBinding (kJ/mol) | Electrostatic (kJ/mol) | Van der Waal (kJ/mol) | Polar Salvation (kJ/mol) | Non-Polar Salvation (kJ/mol) |
|---|---|---|---|---|---|
| EGFR-tak-285 | −68.33 ± 0.16 | −28.05 ± 0.16 | −50.93 ± 0.34 | 29.98 ± 0.27 | −11.33 ± 0.19 |
| EGFR-lapatinib | −71.73 ± 0.24 | −13.74 ± 0.18 | −64.99 ± 0.31 | 25.59 ± 0.28 | −18.59 ± 0.14 |
| EGFR-diosgenin | −67.65 ± 0.13 | −23.25 ± 0.33 | −54.92 ± 0.23 | 26.98 ± 0.31 | −16.46 ± 0.16 |
| EGFR-monohydroxy spirostanol | −62.73 ± 0.31 | −18.74 ± 0.34 | −53.99 ± 0.19 | 24.23 ± 0.30 | −14.23 ± 0.14 |
| HER2-tak-285 | −72.08 ± 0.23 | −11.15 ± 0.26 | −69.93 ± 0.32 | 34.98 ± 0.23 | −25.98 ± 0.16 |
| HER2-lapatinib | −80.24 ± 0.21 | −7.72 ± 0.22 | −78.99 ± 0.15 | 30.87 ± 0.19 | −24.40 ± 0.15 |
| HER2-diosgenin | −78.85 ± 0.33 | −10.25 ± 0.16 | −72.68 ± 0.24 | 27.76 ± 0.24 | −23.68 ± 0.17 |
| HER2-monohydroxy spirostanol | −69.31 ± 0.19 | −10.87 ± 0.18 | −65.36 ± 0.34 | 29.11 ± 0.28 | −22.19 ± 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalayel, M.H.F.; Al-Mazaideh, G.M.; Alanezi, A.A.; Almuqati, A.F.; Alotaibi, M. Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach. Pharmaceuticals 2023, 16, 704. https://doi.org/10.3390/ph16050704
Shalayel MHF, Al-Mazaideh GM, Alanezi AA, Almuqati AF, Alotaibi M. Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach. Pharmaceuticals. 2023; 16(5):704. https://doi.org/10.3390/ph16050704
Chicago/Turabian StyleShalayel, Mohammed Helmy Faris, Ghassab M. Al-Mazaideh, Abdulkareem A. Alanezi, Afaf F. Almuqati, and Meshal Alotaibi. 2023. "Diosgenin and Monohydroxy Spirostanol from Prunus amygdalus var amara Seeds as Potential Suppressors of EGFR and HER2 Tyrosine Kinases: A Computational Approach" Pharmaceuticals 16, no. 5: 704. https://doi.org/10.3390/ph16050704