
Advances in Drug Discovery Targeting Lysosomal Membrane Proteins


Abstract
:1. Introduction
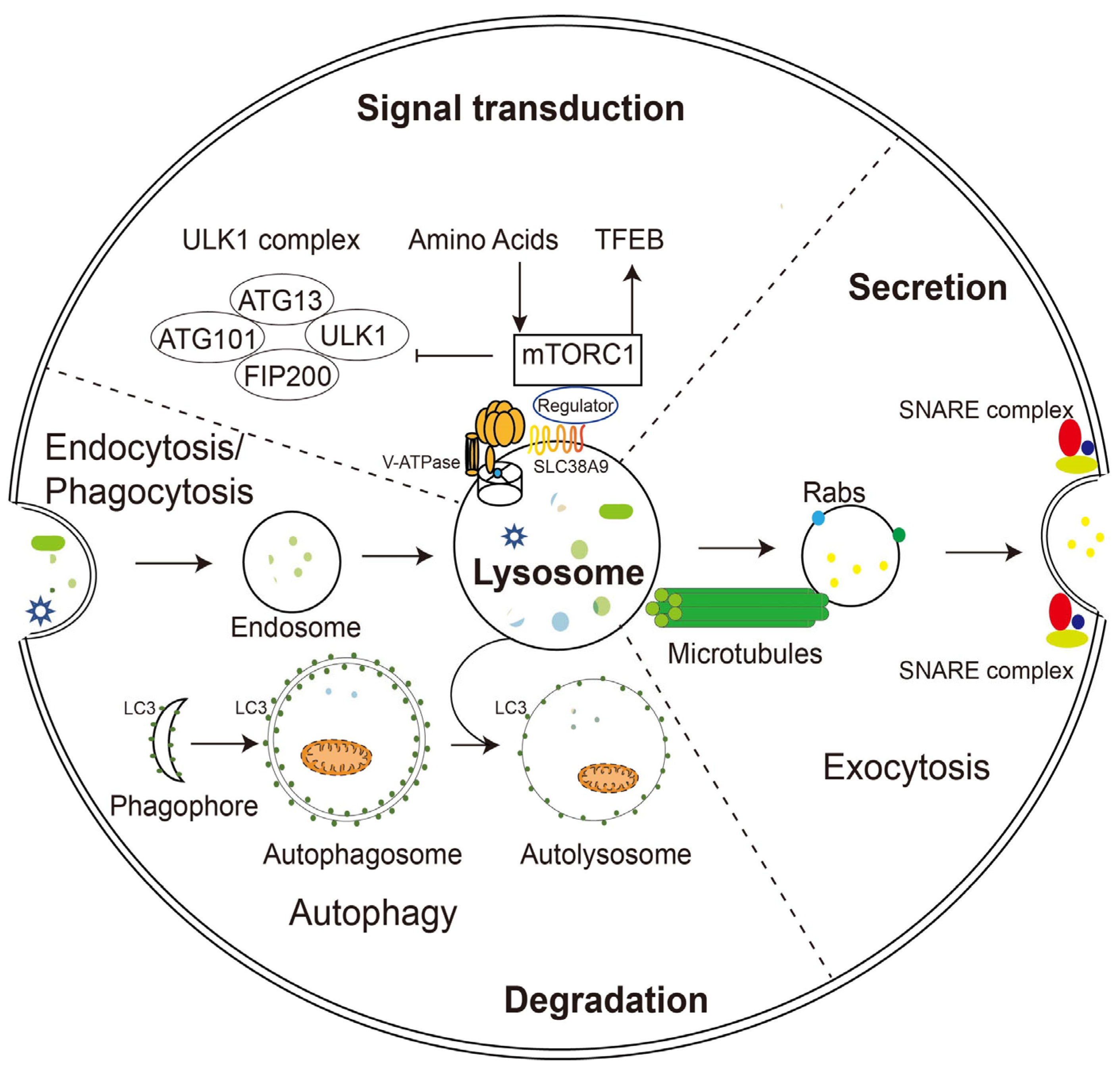
2. Functions of Lysosomes
2.1. Lysosomal Degradation
2.2. Lysosomal Exocytosis
2.3. Signaling Function of Lysosomes
2.4. Restoration of Lysosomal Function
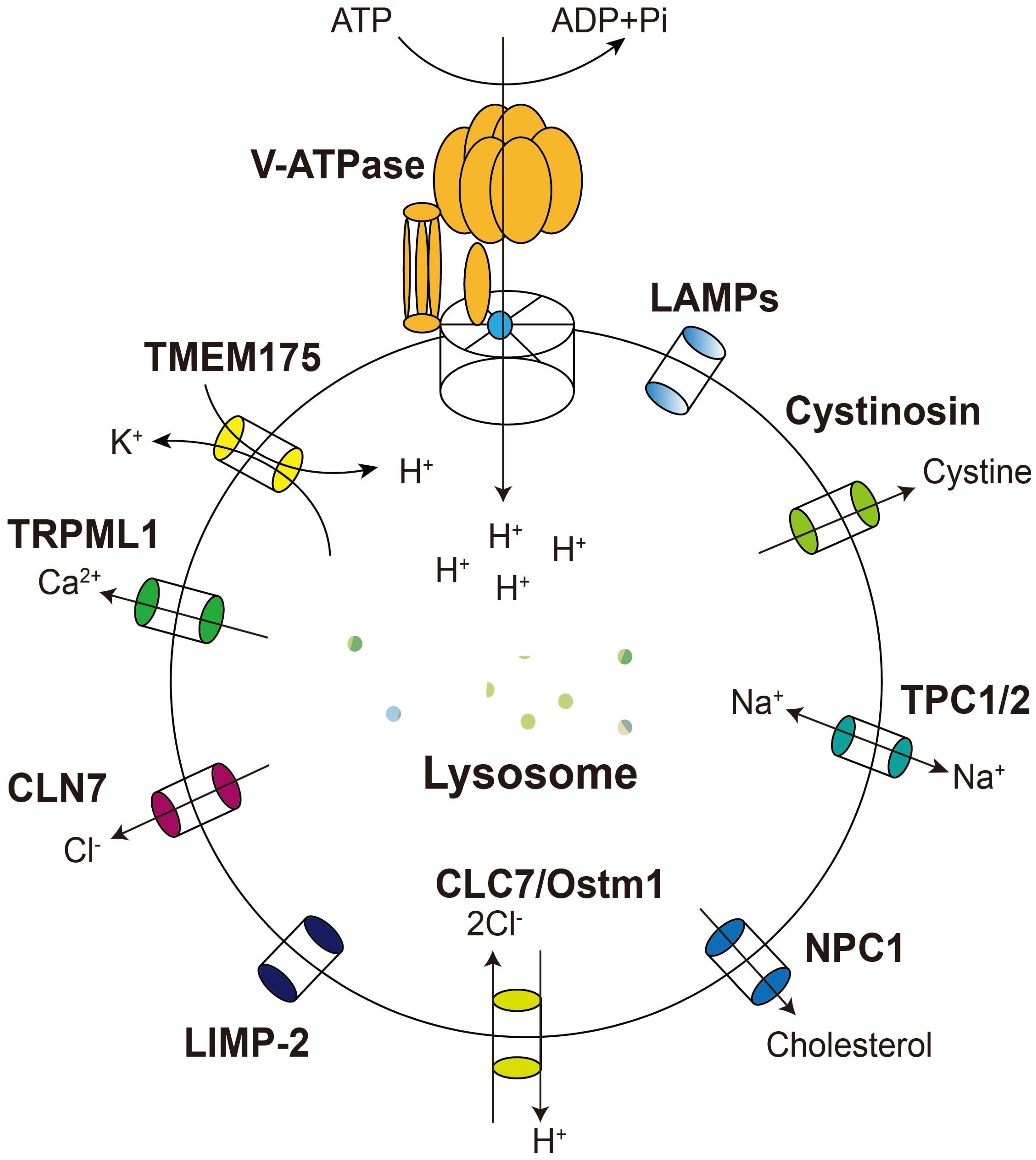
3. Lysosomal Membrane Proteins and Their Relationship with Diseases
3.1. The Vacuolar-Type ATPase
3.2. Lysosomal Calcium Channel TRPML1
3.3. Lysosomal Potassium Channel TMEM175
3.4. CLC7
3.5. NPC
3.6. TPCs
3.7. CLN7
3.8. LAMP1 and LAMP2
3.9. Cystinosin
3.10. LIMP-2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Bar-Peled, L. Lysosome: The Metabolic Signaling Hub. Traffic 2019, 20, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the Stores: Lysosomal Diseases and Therapeutic Strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Zoncu, R.; Perera, R.M. Built to Last: Lysosome Remodeling and Repair in Health and Disease. Trends Cell Biol. 2022, 32, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of Lysosomal Acidification—The Emerging Role of v-ATPase in Aging and Neurodegenerative Disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef]
- de Duve, C. The Lysosome Turns Fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Saftig, P.; Haas, A. Turn up the Lysosome. Nat. Cell Biol. 2016, 18, 1025–1027. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Tanaka, Y.; Saftig, P. At the Acidic Edge: Emerging Functions for Lysosomal Membrane Proteins. Trends Cell Biol. 2003, 13, 137–145. [Google Scholar] [CrossRef]
- Birgisdottir, Å.B.; Johansen, T. Autophagy and Endocytosis—Interconnections and Interdependencies. J. Cell Sci. 2020, 133, jcs228114. [Google Scholar] [CrossRef]
- Saffi, G.T.; Botelho, R.J. Lysosome Fission: Planning for an Exit. Trends Cell Biol. 2019, 29, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs—Engines for Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Verhage, M.; Toonen, R.F. Regulated Exocytosis: Merging Ideas on Fusing Membranes. Curr. Opin. Cell Biol. 2007, 19, 402–408. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and Functions of Lysosome Positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Griffiths, G.M. Secretory Mechanisms in Cell-Mediated Cytotoxicity. Annu. Rev. Cell Dev. Biol. 2007, 23, 495–517. [Google Scholar] [CrossRef]
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The Cell Biology of Osteoclast Function. J. Cell Sci. 2000, 113, 377–381. [Google Scholar] [CrossRef]
- Wesolowski, J.; Paumet, F. The Impact of Bacterial Infection on Mast Cell Degranulation. Immunol. Res. 2011, 51, 215–226. [Google Scholar] [CrossRef]
- Logan, M.R.; Odemuyiwa, S.O.; Moqbel, R. Understanding Exocytosis in Immune and Inflammatory Cells: The Molecular Basis of Mediator Secretion. J. Allergy Clin. Immunol. 2003, 111, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Ye, S.; Whiteheart, S.W. The Platelet Release Reaction: Just When You Thought Platelet Secretion Was Simple. Curr. Opin. Hematol. 2008, 15, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Tulsiani, D.R.P.; Abou-Haila, A.; Loeser, C.R.; Pereira, B.M.J. The Biological and Functional Significance of the Sperm Acrosome and Acrosomal Enzymes in Mammalian Fertilization. Exp. Cell Res. 1998, 240, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Di Malta, C.; Ballabio, A. Non-Canonical MTORC1 Signaling at the Lysosome. Trends Cell Biol. 2022, 32, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Sabatini, D.M. Nutrient Regulation of MTORC1 at a Glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the MTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a Glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome Biogenesis in Health and Disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Yao, R.Q.; Li, Y.X.; Zhao, P.Y.; Ren, C.; Du, X.H.; Yao, Y.M. Lysosomal Quality Control of Cell Fate: A Novel Therapeutic Target for Human Diseases. Cell Death Dis. 2020, 11, 817. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The Many Functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Yang, H.; Tan, J.X. Lysosomal Quality Control: Molecular Mechanisms and Therapeutic Implications. Trends Cell Biol. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a Glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef]
- Jia, J.; Claude-Taupin, A.; Gu, Y.; Choi, S.W.; Peters, R.; Bissa, B.; Mudd, M.H.; Allers, L.; Pallikkuth, S.; Lidke, K.A.; et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 2020, 52, 69–87.e8. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B.; Wrocklage, C.; Pan, C.; Jäger, R.; Kösters, B.; Schäfer, H.; Elsässer, H.P.; Mann, M.; Hasilik, A. Integral and Associated Lysosomal Membrane Proteins. Traffic 2007, 8, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Ruivo, R.; Anne, C.; Sagné, C.; Gasnier, B. Molecular and Cellular Basis of Lysosomal Transmembrane Protein Dysfunction. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 636–649. [Google Scholar] [CrossRef]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 Targets Both Autophagy and Apoptosis Pathways in Pediatric B-Cell Acute Lymphoblastic Leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Yang, S.; Zhang, L.; Yang, T. V-ATPases and Osteoclasts: Ambiguous Future of V-ATPases Inhibitors in Osteoporosis. Theranostics 2018, 8, 5379–5399. [Google Scholar] [CrossRef]
- Lebreton, S.; Jaunbergs, J.; Roth, M.G.; Ferguson, D.A.; De Brabander, J.K. Evaluating the Potential of Vacuolar ATPase Inhibitors as Anticancer Agents and Multigram Synthesis of the Potent Salicylihalamide Analog Saliphenylhalamide. Bioorganic Med. Chem. Lett. 2008, 18, 5879–5883. [Google Scholar] [CrossRef]
- Scheeff, S.; Rivière, S.; Ruiz, J.; Abdelrahman, A.; Schulz-Fincke, A.C.; Köse, M.; Tiburcy, F.; Wieczorek, H.; Gütschow, M.; Müller, C.E.; et al. Synthesis of Novel Potent Archazolids: Pharmacology of an Emerging Class of Anticancer Drugs. J. Med. Chem. 2020, 63, 1684–1698. [Google Scholar] [CrossRef]
- Kim, D.; Hwang, H.Y.; Kim, J.Y.; Lee, J.Y.; Yoo, J.S.; Marko-Varga, G.; Kwon, H.J. FK506, an Immunosuppressive Drug, Induces Autophagy by Binding to the V-ATPase Catalytic Subunit A in Neuronal Cells. J. Proteome Res. 2017, 16, 55–64. [Google Scholar] [CrossRef]
- Chen, F.; Kang, R.; Liu, J.; Tang, D. The V-ATPases in Cancer and Cell Death. Cancer Gene Ther. 2022, 29, 1529–1541. [Google Scholar] [CrossRef]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The Activation of Mucolipin TRP Channel 1 (TRPML1) Protects Motor Neurons from L-BMAA Neurotoxicity by Promoting Autophagic Clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef]
- Pollmanns, M.R.; Beer, J.; Rosignol, I.; Rodriguez-Muela, N.; Falkenburger, B.H.; Dinter, E. Activated Endolysosomal Cation Channel TRPML1 Facilitates Maturation of α-Synuclein-Containing Autophagosomes. Front. Cell. Neurosci. 2022, 16, 861202. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels as Decoders of Cellular Signals. Trends Biochem. Sci. 2019, 44, 110–124. [Google Scholar] [CrossRef]
- Oh, S.C.; Stix, R.; Zhou, W.; Faraldo-Gómez, J.D.; Hite, R.K. Mechanism of 4-Aminopyridine Inhibition of the Lysosomal Channel TMEM175. Proc. Natl. Acad. Sci. USA 2022, 119, e2208882119. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Wang, C.; Feng, X.; Geng, Q.; Chen, W.; Marthi, M.; Zhang, W.; Gao, C.; Reid, W.; et al. Parkinson’s Disease-Risk Protein TMEM175 Is a Proton-Activated Proton Channel in Lysosomes. Cell 2022, 185, 2292–2308.e20. [Google Scholar] [CrossRef]
- Schaller, S.; Henriksen, K.; Sveigaard, C.; Heegaard, A.M.; Hélix, N.; Stahlhut, M.; Ovejero, M.C.; Johansen, J.V.; Solberg, H.; Andersen, T.L.; et al. The Chloride Channel Inhibitor N53736 Prevents Bone Resorption in Ovariectomized Rats without Changing Bone Formation. J. Bone Miner. Res. 2004, 19, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Furtado, D.; Cortez-Jugo, C.; Hung, Y.H.; Bush, A.I.; Caruso, F. MRNA Treatment Rescues Niemann–Pick Disease Type C1 in Patient Fibroblasts. Mol. Pharm. 2022, 19, 3987–3999. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-schott, C.; Biel, M.; Davey, R.A. Two-Pore Channels Control Ebola Virus Host Cell Entry and Are Drug Targets for Disease Treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. CLN7 Gene Therapy: Hope for an Ultra-Rare Condition. J. Clin. Investig. 2022, 132, 5–8. [Google Scholar] [CrossRef]
- Manso, A.M.; Hashem, S.I.; Nelson, B.C.; Gault, E.; Soto-Hermida, A.; Villarruel, E.; Brambatti, M.; Bogomolovas, J.; Bushway, P.J.; Chen, C.; et al. Systemic AAV9.LAMP2B Injection Reverses Metabolic and Physiologic Multiorgan Dysfunction in a Murine Model of Danon Disease. Sci. Transl. Med. 2020, 12, eaax1744. [Google Scholar] [CrossRef]
- Kerem, E. ELX-02: An Investigational Read-through Agent for the Treatment of Nonsense Mutation-Related Genetic Disease. Expert Opin. Investig. Drugs 2020, 29, 1347–1354. [Google Scholar] [CrossRef]
- Jeźégou, A.; Llinares, E.; Anne, C.; Kieffer-Jaquinod, S.; O’Regan, S.; Aupetit, J.; Chabli, A.; Sagné, C.; Debacker, C.; Chadefaux-Vekemans, B.; et al. Heptahelical Protein PQLC2 Is a Lysosomal Cationic Amino Acid Exporter Underlying the Action of Cysteamine in Cystinosis Therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E3434–E3443. [Google Scholar] [CrossRef] [PubMed]
- Besouw, M.; Masereeuw, R.; Van Den Heuvel, L.; Levtchenko, E. Cysteamine: An Old Drug with New Potential. Drug Discov. Today 2013, 18, 785–792. [Google Scholar] [CrossRef]
- Rothaug, M.; Zunke, F.; Mazzulli, J.R.; Schweizer, M.; Altmeppen, H.; Lüllmann-Rauche, R.; Kallemeijn, W.W.; Gaspar, P.; Aerts, J.M.; Glatzel, M.; et al. LIMP-2 Expression Is Critical for β-Glucocerebrosidase Activity and α-Synuclein Clearance. Proc. Natl. Acad. Sci. USA 2014, 111, 15573–15578. [Google Scholar] [CrossRef] [PubMed]
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-Type ATPase: A Proton Pump to Lysosomal Trafficking. Proc. Jpn. Acad. Ser. B 2019, 95, 261–277. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H+-ATPase (V-ATPase): From Proton Pump to Signaling Complex in Health and Disease. Am. J. Physiol.-Cell Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Dubos, A.; Castells-Nobau, A.; Meziane, H.; Oortveld, M.A.W.; Houbaert, X.; Iacono, G.; Martin, C.; Mittelhaeuser, C.; Lalanne, V.; Kramer, J.M.; et al. Conditional Depletion of Intellectual Disability and Parkinsonism Candidate Gene ATP6AP2 in Fly and Mouse Induces Cognitive Impairment and Neurodegeneration. Hum. Mol. Genet. 2015, 24, 6736–6755. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Cabrera-Socorro, A.; Chitayat, D.; Lemonnier, T.; Féraud, O.; Cifuentes-Diaz, C.; Gervasi, N.; Mombereau, C.; Ghosh, T.; Stoica, L.; et al. ATP6AP2 Variant Impairs CNS Development and Neuronal Survival to Cause Fulminant Neurodegeneration. J. Clin. Investig. 2019, 129, 2145–2162. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin Signalling in Neurodegeneration: Mechanisms and Therapeutic Opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- Wallings, R.; Connor-Robson, N.; Wade-Martins, R. LRRK2 Interacts with the Vacuolar-Type H+-ATPase Pump A1 Subunit to Regulate Lysosomal Function. Hum. Mol. Genet. 2019, 28, 2696–2710. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Jiang, L.S.; Zhang, Y.; Tian, Y.; Li, L.S.; Lu, Y.L.; Yang, W.J.; Shi, J.S. Dendrobium Nobile Lindl. Alkaloids Decreases the Level of Intracellular β-Amyloid by Improving Impaired Autolysosomal Proteolysis in APP/PS1 Mice. Front. Pharmacol. 2018, 9, 1479. [Google Scholar] [CrossRef] [PubMed]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-Atpases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Pérez-Sayáns, M.; Somoza-Martín, J.M.; Barros-Angueira, F.; Rey, J.M.G.; García-García, A. V-ATPase Inhibitors and Implication in Cancer Treatment. Cancer Treat. Rev. 2009, 35, 707–713. [Google Scholar] [CrossRef]
- Nishihara, T.; Akifusa, S.; Koseki, T.; Kato, S.; Muro, M.; Hanada, N. Specific Inhibitors of Vacuolar Type H+-ATPases Induce Apoptotic Cell Death. Biochem. Biophys. Res. Commun. 1995, 212, 255–262. [Google Scholar] [CrossRef]
- Visentin, L.; Dodds, R.A.; Valente, M.; Misiano, P.; Bradbeer, J.N.; Oneta, S.; Liang, X.; Gowen, M.; Farina, C. A Selective Inhibitor of the Osteoclastic V-H+-ATPase Prevents Bone Loss in Both Thyroparathyroidectomized and Ovariectomized Rats. J. Clin. Investig. 2000, 106, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Supino, R.; Scovassi, A.I.; Croce, A.C.; Bo, L.D.; Favini, E.; Corbelli, A.; Farina, C.; Misiano, P.; Zunino, F. BIological Effects of a New Vacuolar-H,+-ATPase Inhibitor in Colon Carcinoma Cell Lines. Ann. N. Y. Acad. Sci. 2009, 1171, 606–616. [Google Scholar] [CrossRef]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca(2+)Retaker of the Lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, M.; Yang, Y.; Xu, H. Organellar TRP Channels. Nat. Struct. Mol. Biol. 2018, 25, 1009–1018. [Google Scholar] [CrossRef]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The Hidden Potential of Lysosomal Ion Channels: A New Era of Oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-Mediated Autophagy Is Defective in Mucolipidosis Type IV. J. Cell. Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Venugopal, B.; Browning, M.F.; Curcio-Morelli, C.; Varro, A.; Michaud, N.; Nanthakumar, N.; Walkley, S.U.; Pickel, J.; Slaugenhaupt, S.A. Neurologic, Gastric, and Opthalmologic Pathologies in a Murine Model of Mucolipidosis Type IV. Am. J. Hum. Genet. 2007, 81, 1070–1083. [Google Scholar] [CrossRef]
- Micsenyi, M.C.; Dobrenis, K.; Stephney, G.; Pickel, J.; Vanier, M.T.; Slaugenhaupt, S.A.; Walkley, S.U. Neuropathology of the Mcoln1-/- Knockout Mouse Model of Mucolipidosis Type IV. J. Neuropathol. Exp. Neurol. 2009, 68, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L. TRPML1 and TFEB, an Intimate Affair. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar] [CrossRef]
- Puertollano, R.; Kiselyov, K. TRPMLs: In Sickness and in Health. Am. J. Physiol. Ren. Physiol. 2009, 296, F1245–F1254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide Isoforms Determine Compartment-Specific Ion Channel Activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef]
- Pi, T.; Gan, N.; Han, Y. Structural Mechanism of Allosteric Activation of TRPML1 by PI(3,5)P2 and Rapamycin. Proc. Natl. Acad. Sci. USA 2022, 119, e2120404119. [Google Scholar] [CrossRef]
- Martin, S.; Harper, C.B.; May, L.M.; Coulson, E.J.; Meunier, F.A.; Osborne, S.L. Inhibition of PIKfyve by YM-201636 Dysregulates Autophagy and Leads to Apoptosis-Independent Neuronal Cell Death. PLoS ONE 2013, 8, e60152. [Google Scholar] [CrossRef]
- Fine, M.; Schmiege, P.; Li, X. Structural Basis for PtdInsP2-Mediated Human TRPML1 Regulation. Nat. Commun. 2018, 9, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a Therapeutic Target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Schmiege, P.; Fine, M.; Blobel, G.; Li, X. Human TRPML1 Channel Structures in Open and Closed Conformations. Nature 2017, 550, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K+ Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; Bingol, B.; Sheng, M.; et al. A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, M.; Wang, P.; Syeda, A.K.R.; Huang, P.; Dong, X.P. Lysosomal Potassium Channels. Cell Calcium 2022, 102, 102536. [Google Scholar] [CrossRef] [PubMed]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.K.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 Deficiency Impairs Lysosomal and Mitochondrial Function and Increases α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef]
- Jinn, S.; Blauwendraat, C.; Toolan, D.; Gretzula, C.A.; Drolet, R.E.; Smith, S.; Nalls, M.A.; Marcus, J.; Singleton, A.B.; Stone, D.J. Functionalization of the TMEM175 p.M393T Variant as a Risk Factor for Parkinson Disease. Hum. Mol. Genet. 2019, 28, 3244–3254. [Google Scholar] [CrossRef]
- Hu, M.; Chen, J.; Liu, S.; Xu, H. The Acid Gate in the Lysosome. Autophagy 2022, 19, 1368–1370. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Schrecker, M.; Korobenko, J.; Hite, R.K. Cryo-Em Structure of the Lysosomal Chloride-Proton Exchanger Clc-7 in Complex with Ostm1. eLife 2020, 9, e59555. [Google Scholar] [CrossRef]
- Majumdar, A.; Capetillo-Zarate, E.; Cruz, D.; Gouras, G.K.; Maxfield, F.R. Degradation of Alzheimer’s Amyloid Fibrils by Microglia Requires Delivery of CIC-7 to Lysosomes. Mol. Biol. Cell 2011, 22, 1664–1676. [Google Scholar] [CrossRef]
- Lange, P.F.; Wartosch, L.; Jentsch, T.J.; Fuhrmann, J.C. ClC-7 Requires Ostm1 as a β-Subunit to Support Bone Resorption and Lysosomal Function. Nature 2006, 440, 220–223. [Google Scholar] [CrossRef]
- Zifarelli, G. The Role of the Lysosomal Cl−/H+ Antiporter ClC-7 in Osteopetrosis and Neurodegeneration. Cells 2022, 11, 366. [Google Scholar] [CrossRef]
- Zifarelli, G. A Tale of Two CLCs: Biophysical Insights toward Understanding ClC-5 and ClC-7 Function in Endosomes and Lysosomes. J. Physiol. 2015, 593, 4139–4150. [Google Scholar] [CrossRef]
- Bose, S.; He, H.; Stauber, T. Neurodegeneration Upon Dysfunction of Endosomal/Lysosomal CLC Chloride Transporters. Front. Cell Dev. Biol. 2021, 9, 639231. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Jabs, S.; Hohensee, S.; Chan, W.L.; Kornak, U.; Jentsch, T.J. Transport Activity and Presence of ClC-7/Ostm1 Complex Account for Different Cellular Functions. EMBO Rep. 2014, 15, 784–791. [Google Scholar] [CrossRef]
- Pfeffer, S.R. NPC Intracellular Cholesterol Transporter 1 (NPC1)-Mediated Cholesterol Export from Lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Louwette, S.; Régal, L.; Wittevrongel, C.; Thys, C.; Vandeweeghde, G.; Decuyper, E.; Leemans, P.; De vos, R.; Van geet, C.; Jaeken, J.; et al. NPC1 Defect Results in Abnormal Platelet Formation and Function: Studies in Niemann-Pick Disease Type C1 Patients and Zebrafish. Hum. Mol. Genet. 2013, 22, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Nübling, G.; Castrop, F.; Jochim, A.; Schulte, E.C.; Mollenhauer, B.; Lichtner, P.; Peters, A.; Gieger, C.; Marquardt, T.; et al. Niemann-Pick C Disease Gene Mutations and Age-Related Neurodegenerative Disorders. PLoS ONE 2013, 8, e82879. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Miglustat: A Review of Its Use in Niemann-Pick Disease Type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, Y.; Osaka, H.; Kouga, T.; Jimbo, E.; Muramatsu, K.; Nakamura, S.; Takayanagi, Y.; Onaka, T.; Muramatsu, S.I.; Yamagata, T. Gene Therapy in a Mouse Model of Niemann-Pick Disease Type C1. Hum. Gene Ther. 2021, 32, 589–598. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational Changes in the Ebola Virus Membrane Fusion Machine Induced by PH, Ca2+, and Receptor Binding. PLoS Biol. 2020, 18, e3000626. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Wu, X.; Du, X.; Yao, X.; Zhao, X.; Lee, J.; Yang, H.; Yan, N. Structural Basis of Low-PH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 2020, 182, 98–111.e18. [Google Scholar] [CrossRef]
- Gong, X.; Qian, H.; Zhou, X.; Wu, J.; Wan, T.; Cao, P.; Huang, W.; Zhao, X.; Wang, X.; Wang, P.; et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016, 165, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.E.; Kelu, J.J.; Miller, A.L. Role of Two-Pore Channels in Embryonic Development and Cellular Differentiation. Cold Spring Harb. Perspect. Biol. 2020, 12, a035170. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC Proteins Are Phosphoinositide-Activated Sodium-Selective Ion Channels in Endosomes and Lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP Mobilizes Calcium from Acidic Organelles through Two-Pore Channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. MTOR Regulates Lysosomal ATP-Sensitive Two-Pore Na+ Channels to Adapt to Metabolic State. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.C.; Jiang, Y. Structural Mechanisms of Phospholipid Activation of the Human TPC2 Channel. eLife 2019, 8, e45222. [Google Scholar] [CrossRef]
- Marchant, J.S.; Gunaratne, G.S.; Cai, X.; Slama, J.T.; Patel, S. NAADP-Binding Proteins Find Their Identity. Trends Biochem. Sci. 2022, 47, 235–249. [Google Scholar] [CrossRef]
- Hermann, J.; Bender, M.; Schumacher, D.; Woo, M.S.; Shaposhnykov, A.; Rosenkranz, S.C.; Kuryshev, V.; Meier, C.; Guse, A.H.; Friese, M.A.; et al. Contribution of NAADP to Glutamate-Evoked Changes in Ca2+ Homeostasis in Mouse Hippocampal Neurons. Front. Cell Dev. Biol. 2020, 8, 496. [Google Scholar] [CrossRef] [PubMed]
- Prat Castro, S.; Kudrina, V.; Jaślan, D.; Böck, J.; Scotto Rosato, A.; Grimm, C. Neurodegenerative Lysosomal Storage Disorders: TPC2 Comes to the Rescue! Cells 2022, 11, 2807. [Google Scholar] [CrossRef]
- Nguyen, O.N.P.; Grimm, C.; Schneider, L.S.; Chao, Y.K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, A.F.; Parrington, J. Endolysosomal Ca2+ Signaling in Cancer: The Role of TPC2, From Tumorigenesis to Metastasis. Front. Cell Dev. Biol. 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Johnson, T.B.; Cain, J.T.; White, K.A.; Ramirez-Montealegre, D.; Pearce, D.A.; Weimer, J.M. Therapeutic Landscape for Batten Disease: Current Treatments and Future Prospects. Nat. Rev. Neurol. 2019, 15, 161–178. [Google Scholar] [CrossRef]
- Brudvig, J.J.; Weimer, J.M. On the Cusp of Cures: Breakthroughs in Batten Disease Research. Curr. Opin. Neurobiol. 2022, 72, 48–54. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, W.; Lin, B.; Yao, Y.; Li, C.; Hu, W.; Wu, H.; Huang, J.; Zhang, M.; Xue, T.; et al. CLN7 Is an Organellar Chloride Channel Regulating Lysosomal Function. Sci. Adv. 2021, 7, eabj9608. [Google Scholar] [CrossRef]
- Chen, X.; Dong, T.; Hu, Y.; Shaffo, F.C.; Belur, N.R.; Mazzulli, J.R.; Gray, S.J. AAV9/MFSD8 Gene Therapy Is Effective in Preclinical Models of Neuronal Ceroid Lipofuscinosis Type 7 Disease. J. Clin. Investig. 2022, 132, e146286. [Google Scholar] [CrossRef] [PubMed]
- Lunding, L.P.; Krause, D.; Stichtenoth, G.; Stamme, C.; Lauterbach, N.; Hegermann, J.; Ochs, M.; Schuster, B.; Sedlacek, R.; Saftig, P.; et al. LAMP3 Deficiency Affects Surfactant Homeostasis in Mice. PLoS Genet. 2021, 17, e1009619. [Google Scholar] [CrossRef] [PubMed]
- Schwake, M.; Schröder, B.; Saftig, P. Lysosomal Membrane Proteins and Their Central Role in Physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Andrejewski, N.; Punnonen, E.L.; Guhde, G.; Tanaka, Y.; Lüllmann-Rauch, R.; Hartmann, D.; Von Figura, K.; Saftig, P. Normal Lysosomal Morphology and Function in LAMP-1-Deficient Mice. J. Biol. Chem. 1999, 274, 12692–12701. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Surabhi, S.; Rojo-Cortés, F.; Dulac, A.; Jenny, A.; Birman, S. Lamp1 Deficiency Enhances Sensitivity to α-Synuclein and Oxidative Stress in Drosophila Models of Parkinson Disease. Int. J. Mol. Sci. 2022, 23, 3078. [Google Scholar] [CrossRef] [PubMed]
- Cawley, N.X.; Sojka, C.; Cougnoux, A.; Lyons, A.T.; Nicoli, E.R.; Wassif, C.A.; Porter, F.D. Abnormal LAMP1 Glycosylation May Play a Role in Niemann-Pick Disease, Type C Pathology. PLoS ONE 2020, 15, e0227829. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W.; Bagshaw, R.D.; Mahuran, D.J. The Integral Membrane of Lysosomes: Its Proteins and Their Roles in Disease. J. Proteom. 2009, 72, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the Lysosome in Cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Gude, R.P.; Kalraiya, R.D. Regulation of Melanoma Metastasis to Lungs by Cell Surface Lysosome Associated Membrane Protein-1 (LAMP1) via Galectin-3. Biochem. Biophys. Res. Commun. 2014, 449, 332–337. [Google Scholar] [CrossRef]
- Guo, X.; Schmiege, P.; Assafa, T.E.; Wang, R.; Xu, Y.; Donnelly, L.; Fine, M.; Ni, X.; Jiang, J.; Millhauser, G.; et al. Structure and Mechanism of Human Cystine Exporter Cystinosin. Cell 2022, 185, 3739–3752.e18. [Google Scholar] [CrossRef]
- Kalatzis, V.; Cherqui, S.; Antignac, C.; Gasnier, B. Cystinosin, the Protein Defective in Cystinosis, Is a H+-Driven Lysosomal Cystine Transporter. EMBO J. 2001, 20, 5940–5949. [Google Scholar] [CrossRef] [PubMed]
- Brasell, E.J.; Chu, L.L.; Akpa, M.M.; Eshkar-Oren, I.; Alroy, I.; Corsini, R.; Gilfix, B.M.; Yamanaka, Y.; Huertas, P.; Goodyer, P. The Novel Aminoglycoside, ELX-02, Permits CTNSW138X Translational Read-through and Restores Lysosomal Cystine Efflux in Cystinosis. PLoS ONE 2019, 14, e0223954. [Google Scholar] [CrossRef] [PubMed]
- Löbel, M.; Salphati, S.P.; El Omari, K.; Wagner, A.; Tucker, S.J.; Parker, J.L.; Newstead, S. Structural Basis for Proton Coupled Cystine Transport by Cystinosin. Nat. Commun. 2022, 13, 4845. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Malini, E.; Zampieri, S.; Deganuto, M.; Romanello, M.; Sechi, A.; Bembi, B.; Dardis, A. Role of LIMP-2 in the Intracellular Trafficking of β-Glucosidase in Different Human Cellular Models. FASEB J. 2015, 29, 3839–3852. [Google Scholar] [CrossRef]
- Heybrock, S.; Kanerva, K.; Meng, Y.; Ing, C.; Liang, A.; Xiong, Z.J.; Weng, X.; Ah Kim, Y.; Collins, R.; Trimble, W.; et al. Lysosomal Integral Membrane Protein-2 (LIMP-2/SCARB2) Is Involved in Lysosomal Cholesterol Export. Nat. Commun. 2019, 10, 3521. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Functions | Chemicals/Therapies | Mechanism | Stage of Development | Disease/Potential Application |
|---|---|---|---|---|---|
| V-ATPase | Acidification of lysosome | Bafilomycin A1, Concanamycin A, INDO L0 | Inhibit c subunit of V0 domain in V-ATPase | Tool compound/Preclinical | Cancer [37] |
| SB242784 | With high potency and selectivity for the c, a, or V0 domain of osteoclast V-ATPase | Preclinical | Osteoporosis [38] | ||
| Salicylihalamide A, SaliPhe, Archazolid | Inhibit V0 domain of V-ATPase | Preclinical | Cancer [39,40] | ||
| FK506 | Protect nervous system through binding with ATP6V1A and inducing autophagy | Preclinical | Neuronal cells [41] | ||
| NiK12192 | Indole derivative that causes a reduction in the volume and acidity of lysosomes, leads to antimetastatic effect | Preclinical | Cancer [42] | ||
| TRPML1 | Regulate lysosomal calcium concentration | ML-SA1 | TRPML1 activator | Preclinical | Neurodegenerative diseases, MLIV [43,44,45] |
| PI (4,5) P2 | TRPML1 activator | ||||
| PI (3,5) P2 | TRPML1 agonists | ||||
| SF-22 | TRPML1 activator | ||||
| MK6-83 | TRPML1 activator | ||||
| TMEM175 | Modulate lysosomal potassium and proton | 4-AP | Inhibit the K+ channel activity of TMEM175 [46] | Phase III | Spinal muscular atrophy (NCT01645787), Guillain-Barre Syndrome (NCT00056810) |
| ArA | Activate TMEM175 and enhance the permeability of K+ and H+ | Preclinical | Neurodegenerative disease [47] | ||
| DCPIB | Ion channel activator | ||||
| ML67-33 | Ion channel activator | ||||
| CLC7 | Lysosomal chloride channel | NS3736 | Chloride channel inhibitor | Preclinical | Osteoporosis [48] |
| NPC1 | Lysosomal cholesterol channel, intracellular receptor for Ebola virus | NPC1-AAV9 gene therapy | Correct NPC1 mutation and restore the function of NPC1 [49] | Clinical | Niemann–Pick disease, type C [49] |
| Benzylpiperazine adamantane diamide 3.0 | Inhibit NPC1 to block viral entry | Preclinical | Ebola virus infection [50] | ||
| TPCs | Na+ channel | Ned19 | NAADP antagonist, block TPC | Preclinical | Viral infection [51] |
| NAADP | Activate TPC | ||||
| PI (3,5) P2 | Activate TPC2 | ||||
| CLN7 | Lysosomal chloride channel | Gene therapy | Rescue CLN7 | Clinical | Batten Disease [52] |
| LAMP2 | Lysosomal membrane structural protein | LAMP2B-AAV9 gene therapy | Restore the function of LAMP2B [53] | Phase I | Danon Disease (NCT03882437) |
| Cystinosin | Transport cystine | ELX-02 | Binds to ribosomes and rescue cystinosin W138X mutation [54] | Phase II | Cystinosis (NCT04069260) |
| Cysteamine | Binds to lysosomal cystine and transport them out through lysosomal cationic amino acid transporter | FDA-approved drug | Cystinosis [55,56] | ||
| LIMP-2 | Lysosomal membrane structural protein | Gene therapy | Restore the function of LIMP-2 | Preclinical | Gaucher disease [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Zhu, Y.; Liu, H.; Liang, T.; Wei, Y. Advances in Drug Discovery Targeting Lysosomal Membrane Proteins. Pharmaceuticals 2023, 16, 601. https://doi.org/10.3390/ph16040601
Wang H, Zhu Y, Liu H, Liang T, Wei Y. Advances in Drug Discovery Targeting Lysosomal Membrane Proteins. Pharmaceuticals. 2023; 16(4):601. https://doi.org/10.3390/ph16040601
Chicago/Turabian StyleWang, Hongna, Yidong Zhu, Huiyan Liu, Tianxiang Liang, and Yongjie Wei. 2023. "Advances in Drug Discovery Targeting Lysosomal Membrane Proteins" Pharmaceuticals 16, no. 4: 601. https://doi.org/10.3390/ph16040601