
ATL I, Acts as a SIRT6 Activator to Alleviate Hepatic Steatosis in Mice via Suppression of NLRP3 Inflammasome Formation
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
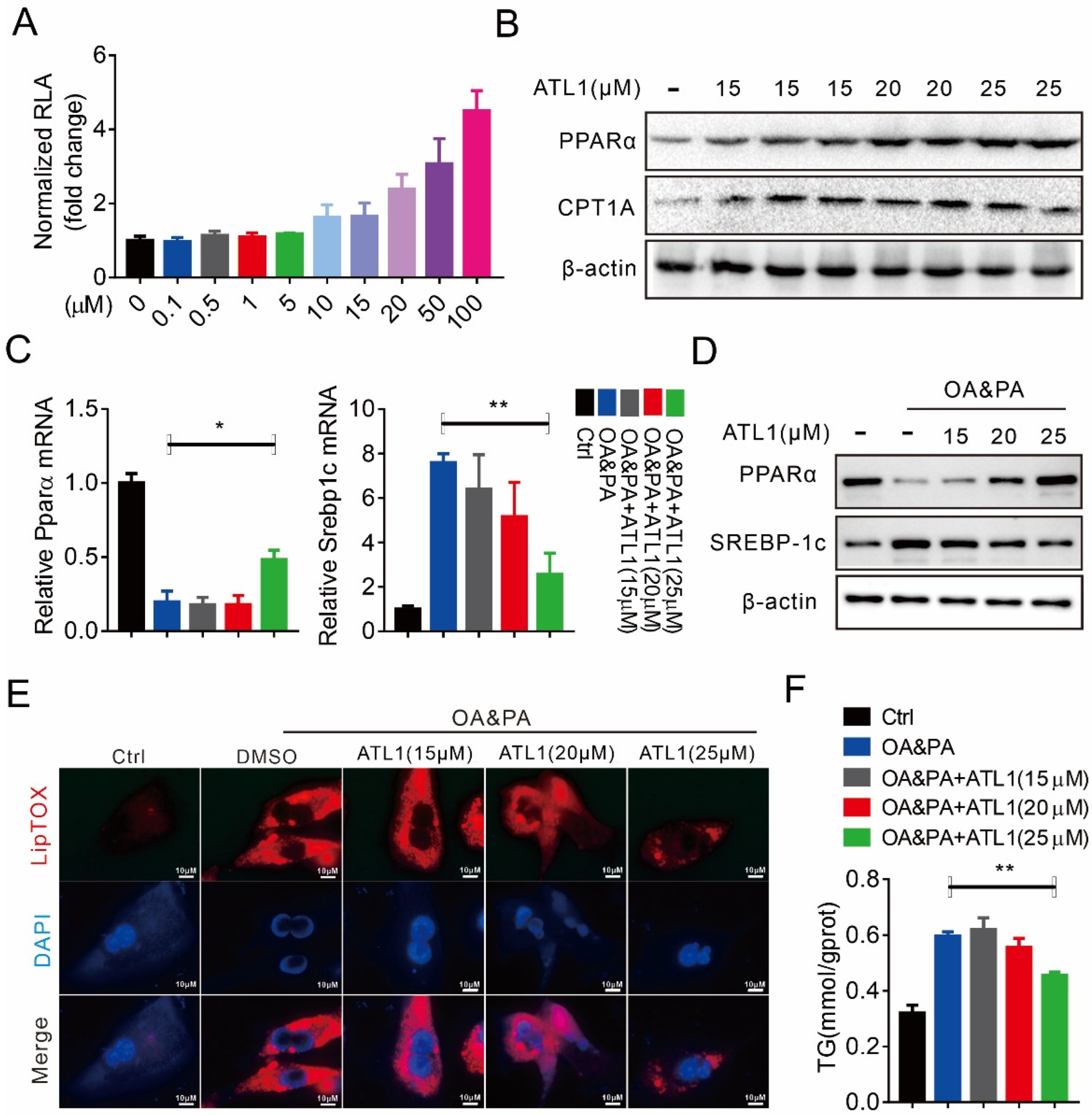
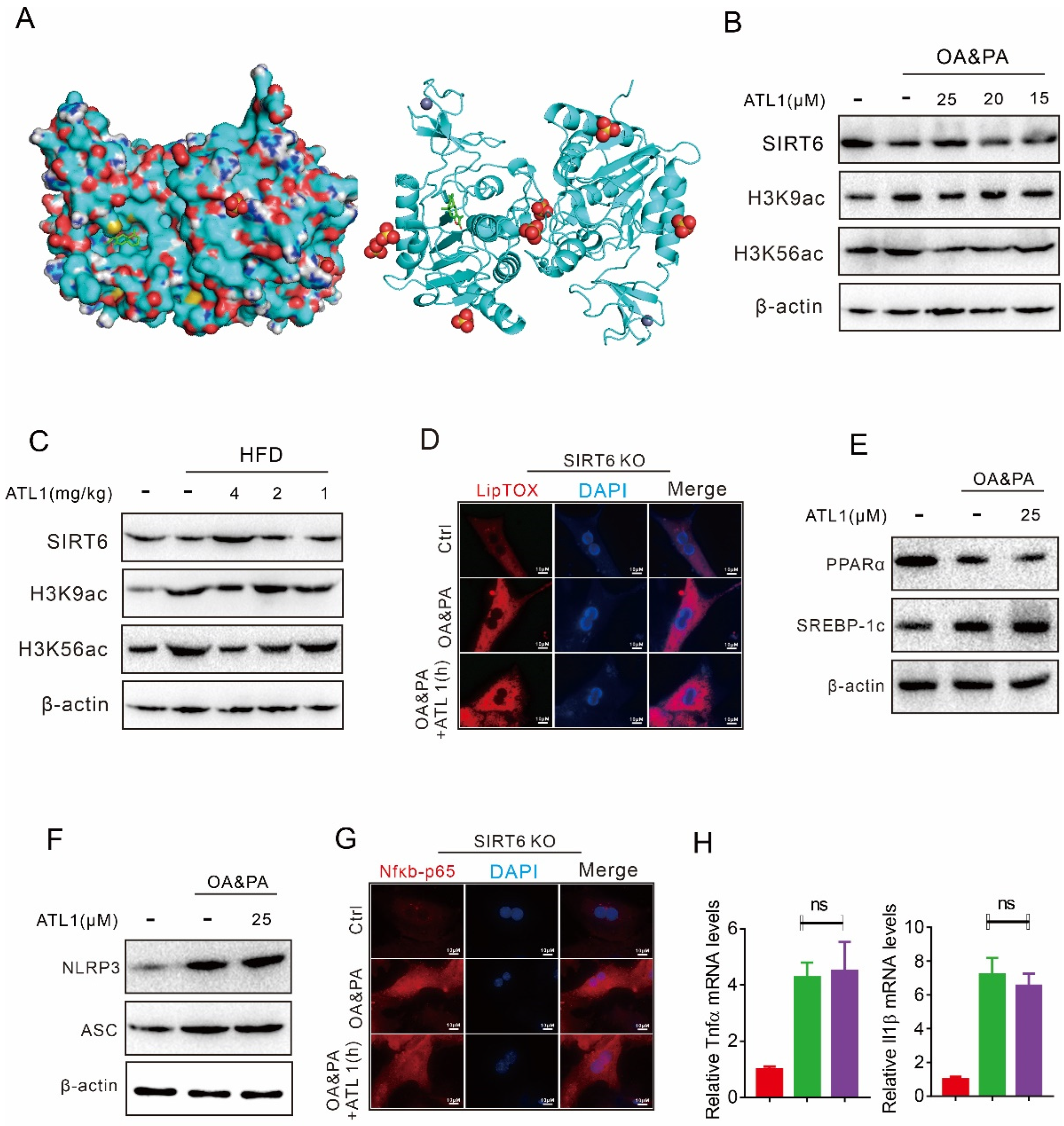
2.1. ATL I Treatment Alleviates Lipid Accumulation In Vitro
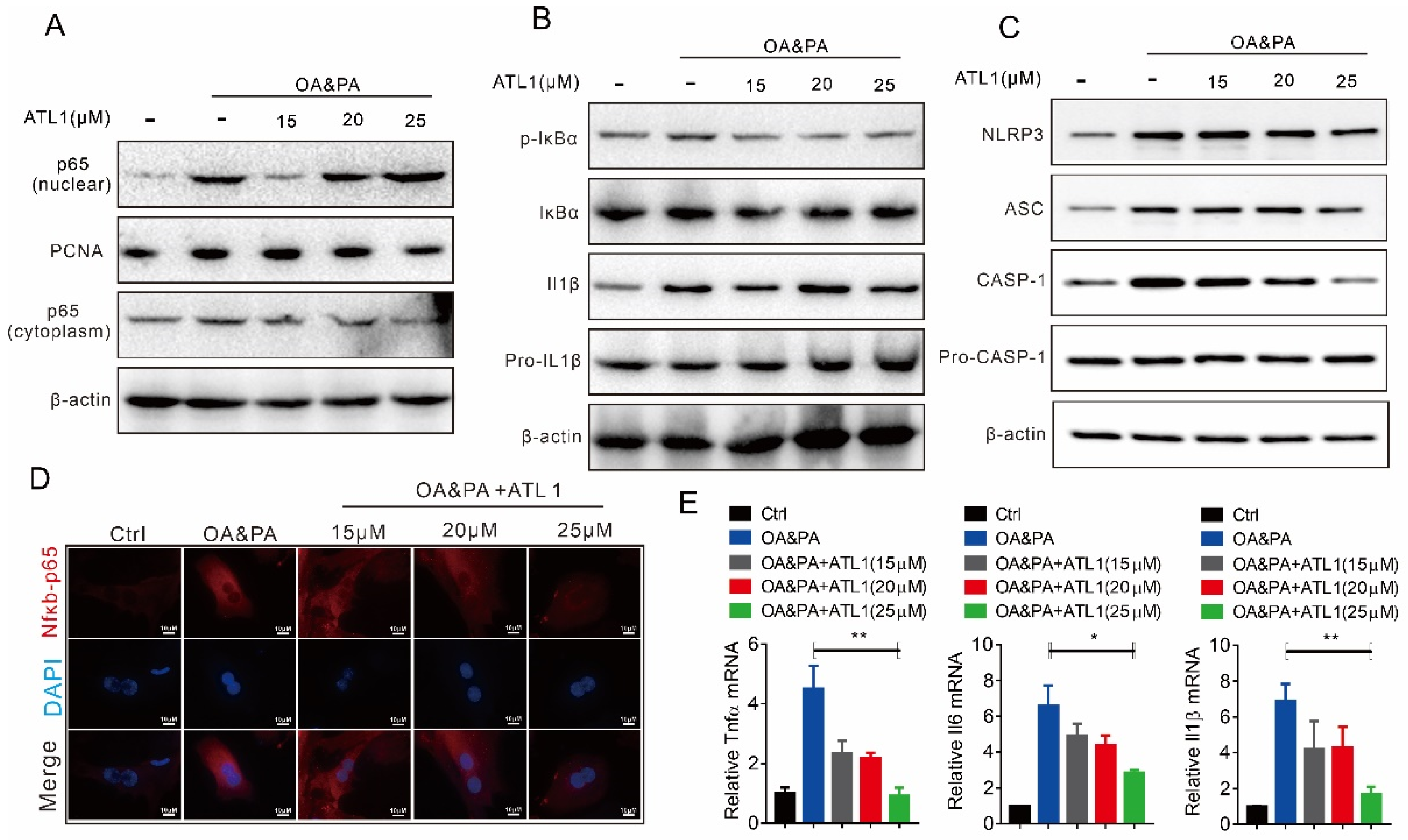
2.2. ATL I Treatment Suppressed OA&PA-Induced NLRP3 Inflammasome Activation and the Inflammatory Response In Vitro
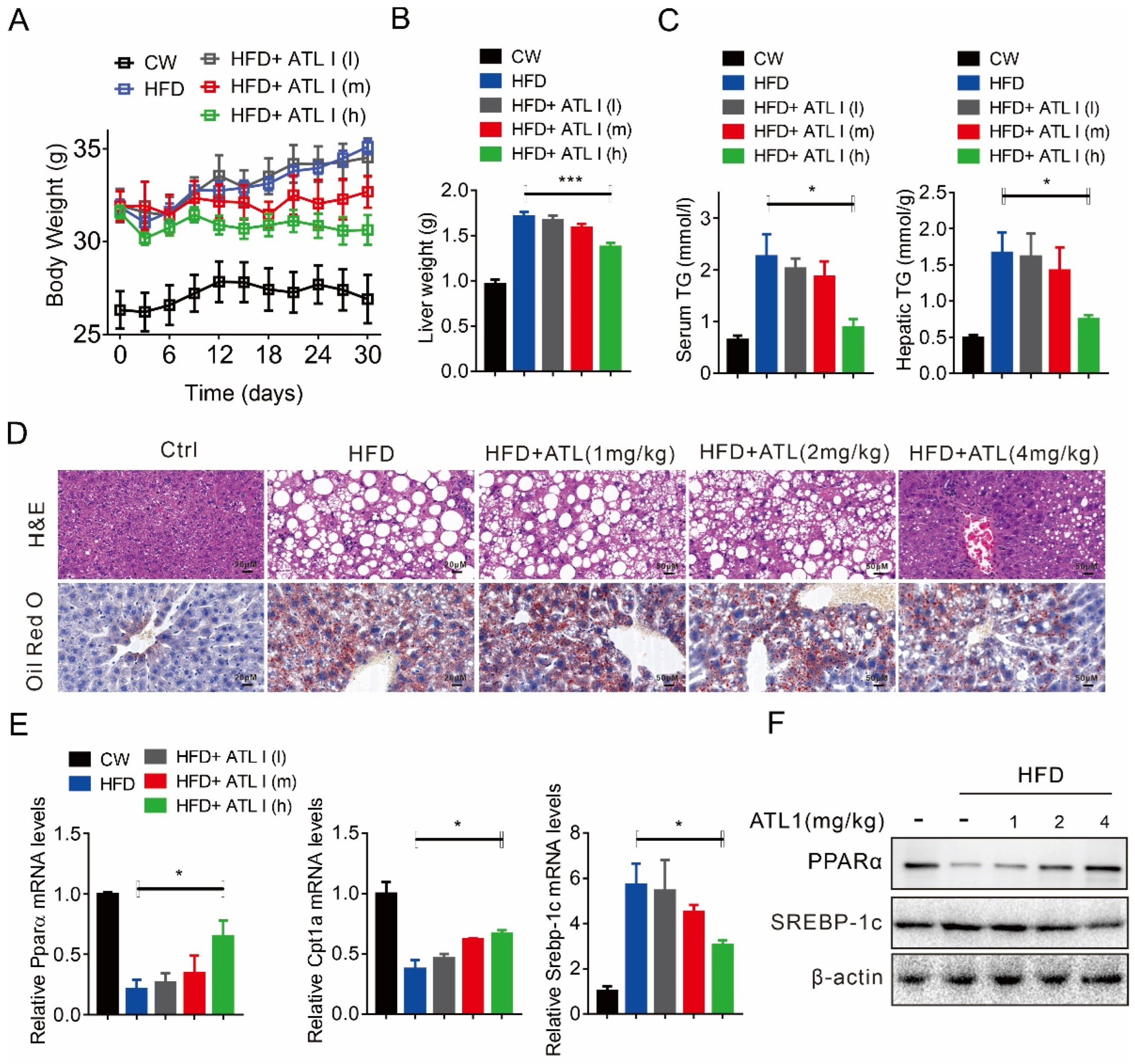
2.3. ATL I Treatment Alleviates HFD-Induced Fatty Liver in Mice
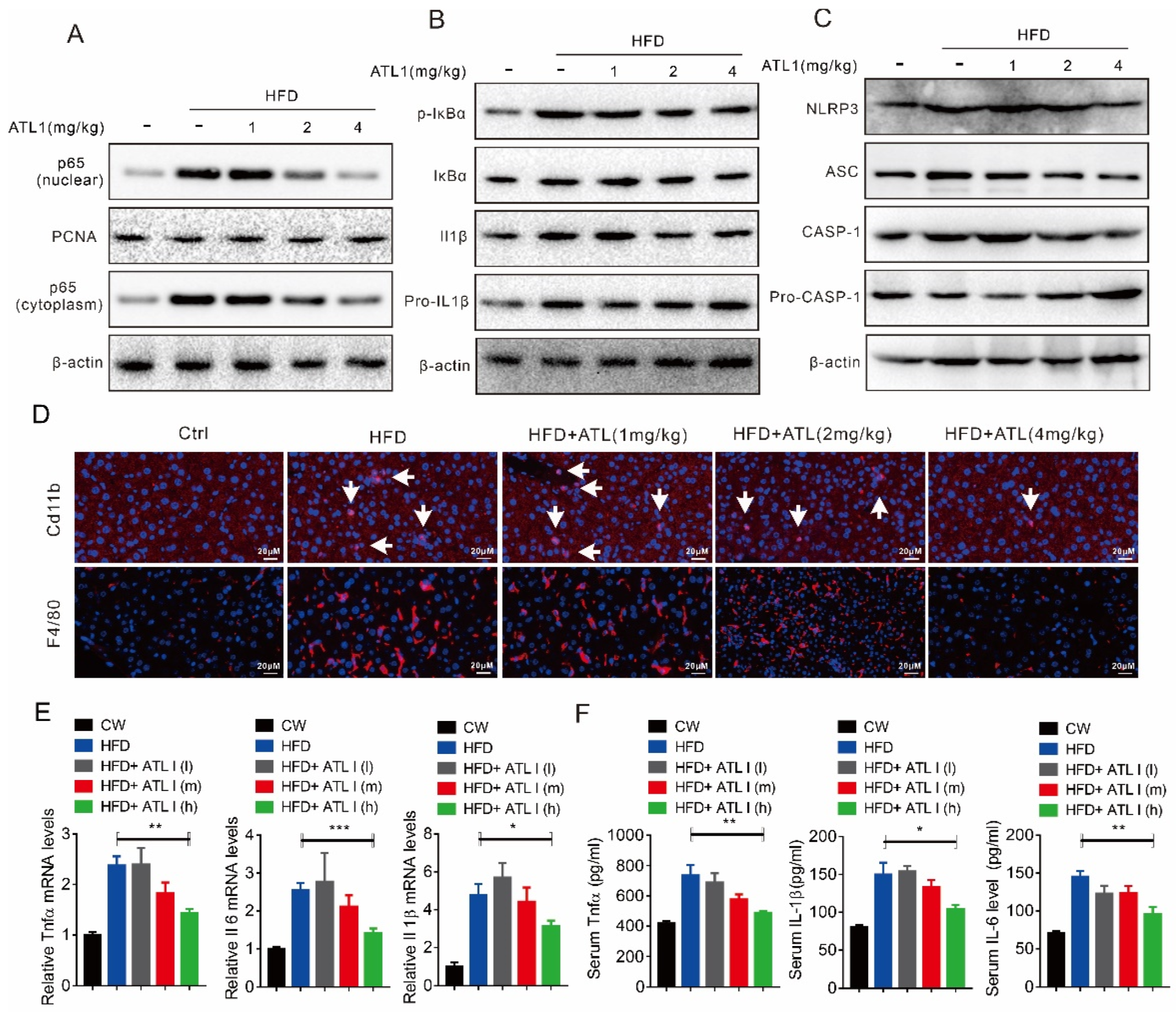
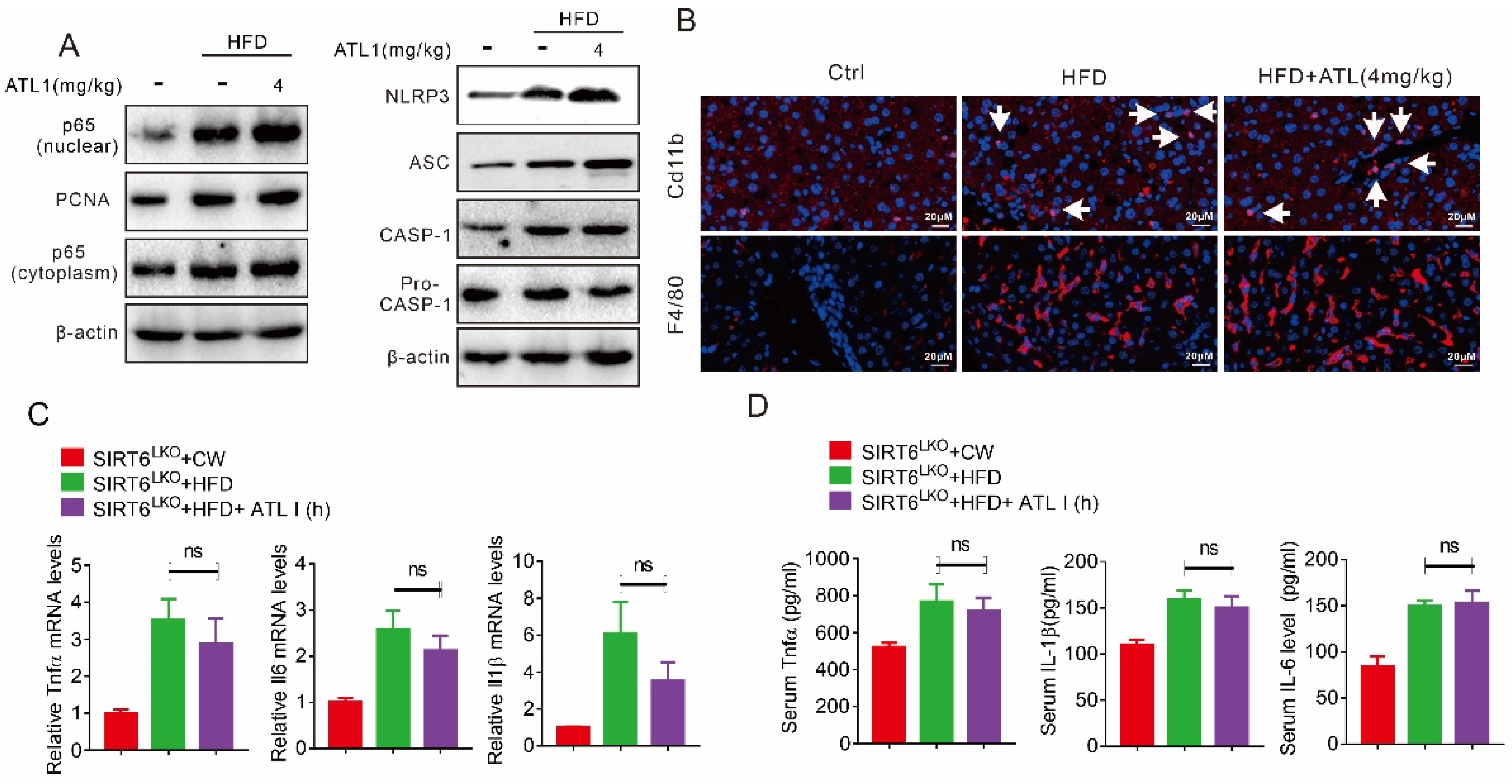
2.4. ATL I Treatment Suppressed NLRP3 Inflammasome and Inflammatory Response in HFD-Induced Obese Mice
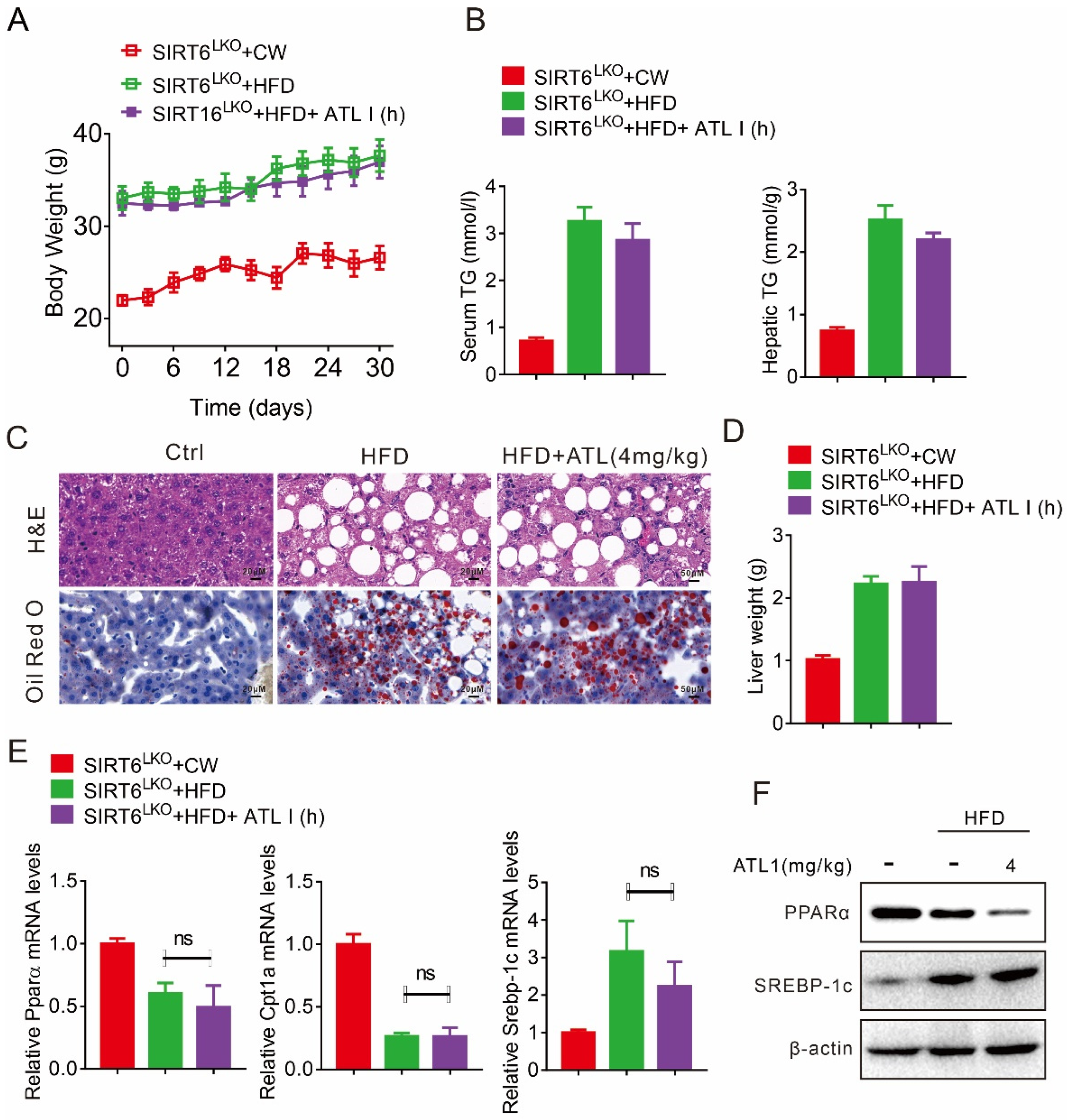
2.5. Hepatic SIRT6 Mediates the Protective Effects of ATL I Treatment against Lipid Disorder
2.6. ATL I Treatment Failed to Alter HFD-Induced Fatty Liver in Hepatic SIRT6 Knockout Mice
2.7. Hepatic SIRT6 Deletion Diminished the Protective Effects of ATL I
3. Discussion
4. Materials and Methods
4.1. Animal Treatment
4.2. Isolation of Mouse Primary Hepatocytes (MPHs)
4.3. Molecular Docking
4.4. Western Blotting
4.5. Luciferase Reporter Assays
4.6. Real Time PCR
4.7. Immunofluorescence
4.8. TG Content Test
4.9. Lipid TOX Assays
4.10. Nuclear Protein Extraction
4.11. HE and Oil Red O Staining
4.12. Statistical Analysis
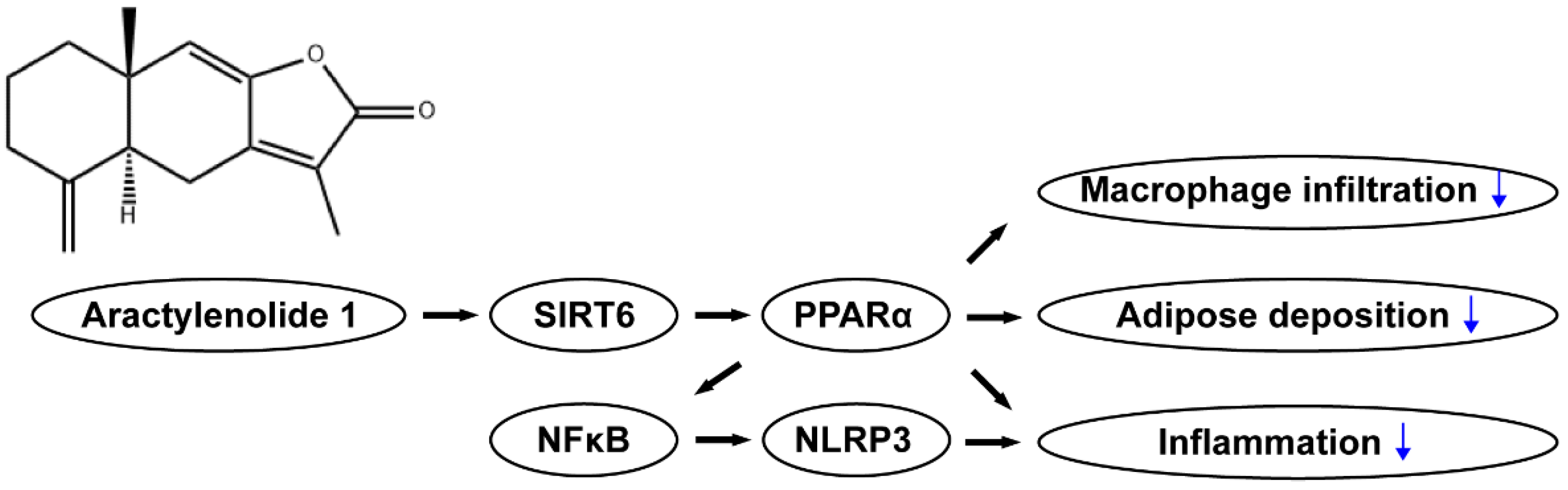
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Qayyum, A.; Nystrom, M.; Noworolski, S.M.; Chu, P.; Mohanty, A.; Merriman, R. MRI steatosis grading: Development and initial validation of a color mapping system. Am. J. Roentgenol. 2012, 198, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat. not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver. steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Uehara, K.; Sostre-Colón, J.; Gavin, M.; Santoleri, D.; Leonard, K.A.; Jacobs, R.L.; Titchenell, P.M. Activation of Liver mTORC1 Protects Against NASH via Dual Regulation of VLDL-TAG Secretion and De Novo Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1625–1647. [Google Scholar] [CrossRef]
- Carvalho-Gontijo, R.; Han, C.; Zhang, L.; Zhang, V.; Hosseini, M.; Mekeel, K.; Schnabl, B.; Loomba, R.; Karin, M.; Brenner, D.A.; et al. Metabolic Injury of Hepatocytes Promotes Progression of NAFLD and AALD. Semin. Liver Dis. 2022, 42, 233–249. [Google Scholar] [CrossRef]
- Bao, X.; Ma, X.; Huang, R.; Chen, J.; Xin, H.; Zhou, M.; Li, L.; Tong, S.; Zhang, Q.; Shui, G.; et al. Knockdown of hepatocyte Perilipin-3 mitigates hepatic steatosis and steatohepatitis caused by hepatocyte CGI-58 deletion in mice. J. Mol. Cell Biol. 2022, mjac055. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Chait, A. Hypertriglyceridemia. Endocrinol. Metab. Clin. N. Am. 2022, 51, 539–555. [Google Scholar] [CrossRef]
- Toth, P.P. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc. Health Risk Manag. 2016, 12, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Mooli, R.G.R.; Ramakrishnan, S.K. Liver Steatosis is a Driving Factor of Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1267–1270. [Google Scholar] [CrossRef]
- Maitiabula, G.; Tian, F.; Wang, P.; Zhang, L.; Gao, X.; Wan, S.; Sun, H.; Yang, J.; Zhang, Y.; Gao, T.; et al. Liver PP2A-Cα Protects from Parenteral Nutrition-associated Hepatic Steatosis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 669–692. [Google Scholar] [CrossRef]
- Guthrie, G. Parenteral Nutrition Associated Hepatic Steatosis and NAFLD Intersect at AMPK. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 724–725. [Google Scholar] [CrossRef]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. Loss of hepatic PPARα promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Krüppel-like factor 16 (KLF16) targets PPARα to improve steatohepatitis and insulin resistance. Gut 2021, 70, 2183–2195. [Google Scholar] [CrossRef]
- Huang, Q.; Xin, X.; Sun, Q.; An, Z.; Gou, X.; Feng, Q. Plant-derived bioactive compounds regulate the NLRP3 inflammasome to treat NAFLD. Front. Pharmacol. 2022, 13, 896899. [Google Scholar] [CrossRef]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Kaufmann, B.; Kui, L.; Reca, A.; Leszczynska, A.; Kim, A.D.; Booshehri, L.M.; Wree, A.; Friess, H.; Hartmann, D.; Broderick, L.; et al. Cell-specific Deletion of NLRP3 Inflammasome Identifies Myeloid Cells as Key Drivers of Liver Inflammation and Fibrosis in Murine Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 751–767. [Google Scholar] [CrossRef]
- Liu, C.; Pan, Z.; Wu, Z.; Tang, K.; Zhong, Y.; Chen, Y.; Xiao, X.; Guo, J.; Duan, S.; Cui, T.; et al. Hepatic SIRT6 Modulates Transcriptional Activities of FXR to Alleviate Acetaminophen-induced Hepatotoxicity. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Huang, M.; Kim, H.G.; Chowdhury, K.; Gao, J.; Liu, S.; Wan, J.; Wei, L.; Dong, X.C. SIRT6 controls hepatic lipogenesis by suppressing LXR. ChREBP, and SREBP1. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166249. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Huang, M.; Kim, H.G.; Zhang, Y.; Chowdhury, K.; Cai, W.; Saxena, R.; Schwabe, R.F.; Liangpunsakul, S.; Dong, X.C. SIRT6 Protects Against Liver Fibrosis by Deacetylation and Suppression of SMAD3 in Hepatic Stellate Cells. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 341–364. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chiang, H.H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Huang, G.; Wei, T.; Gao, J.; Huang, C.; Sun, M.; Zhu, L.; Shen, W. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 764–777. [Google Scholar] [CrossRef]
- Yang, T.; Chen, Y.; Xu, J.; Li, J.; Liu, H.; Liu, N. Bioinformatics screening the novel and promising targets of curcumin in hepatocellular carcinoma chemotherapy and prognosis. BMC Complement. Med. Ther. 2022, 22, 21. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Wei, L.; Yang, X.; Zhang, Y.; Cheng, B.; Yang, Z.; Gao, W.; Song, C.; Miao, W.; et al. The Protective Effects of Imperatorin on Acetaminophen Overdose-Induced Acute Liver Injury. Oxid. Med. Cell Longev. 2020, 2020, 8026838. [Google Scholar] [CrossRef]
- Li, Q.; Tan, J.X.; He, Y.; Bai, F.; Li, S.W.; Hou, Y.W.; Ji, L.S.; Gao, Y.T.; Zhang, X.; Zhou, Z.H.; et al. Atractylenolide III ameliorates Non-Alcoholic Fatty Liver Disease by activating Hepatic Adiponectin Receptor 1-Mediated AMPK Pathway. Int. J. Biol. Sci. 2022, 18, 1594–1611. [Google Scholar] [CrossRef]
- Li, L.; Zhu, G.; Fu, G.; Zha, W.; Li, H. Metabolic Syndrome Ameliorated by 4-Methylesculetin by Reducing Hepatic Lipid Accumulation. Int. J. Mol. Sci. 2022, 23, 10465. [Google Scholar] [CrossRef]
- Aibaidula, Y.; Aimaiti, M.; Tan, H.; Chen, B.; Yang, J.; Ma, X. Lactucin & Lactucopicrin ameliorates FFA-induced steatosis in HepG2 cells via modulating lipid metabolism. J. Pharmacol. Sci. 2022, 150, 110–122. [Google Scholar]
- Xu, R.; Liu, X.; Tian, M.; Chen, D. Atractylodes-I overcomes the oxidative stress-induced colonic mucosal epithelial cells dysfunction to prevent irritable bowel syndrome via modulating the miR-34a-5p-LDHA signaling pathway. Curr. Mol. Med. 2022. [Google Scholar] [CrossRef]
- Fan, M.; Gu, X.; Zhang, W.; Shen, Q.; Zhang, R.; Fang, Q.; Wang, Y.; Guo, X.; Zhang, X.; Liu, X. Atractylenolide I ameliorates cancer cachexia through inhibiting biogenesis of IL-6 and tumour-derived extracellular vesicles. J. Cachexia Sarcopenia Muscle 2022. [Google Scholar] [CrossRef]
- Du, Z.; Ma, Z.; Lai, S.; Ding, Q.; Hu, Z.; Yang, W.; Qian, Q.; Zhu, L.; Dou, X.; Li, S. Atractylenolide I Ameliorates Acetaminophen-Induced Acute Liver Injury via the TLR4/MAPKs/NF-κB Signaling Pathways. Front. Pharmacol. 2022, 13, 797499. [Google Scholar] [CrossRef]
- Bailly, C. Atractylenolides. essential components of Atractylodes-based traditional herbal medicines: Antioxidant, anti-inflammatory and anticancer properties. Eur. J. Pharmacol. 2021, 891, 173735. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, Y.; Lee, G.; Doh, E.J.; Hong, S. Quantitative Interrelation between Atractylenolide I. II, and III in Atractylodes japonica Koidzumi Rhizomes, and Evaluation of Their Oxidative Transformation Using a Biomimetic Kinetic Model. ACS Omega 2018, 3, 14833–14840. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Geng, Q.; Wang, Y. Protective effect of atractylenolide I on immunological liver injury. Zhongguo Zhong Yao Za Zhi 2012, 37, 1809–1813. [Google Scholar]
- Cignarella, F.; Cantoni, C.; Ghezzi, L.; Salter, A.; Dorsett, Y.; Chen, L.; Phillips, D.; Weinstock, G.M.; Fontana, L.; Cross, A.H.; et al. Intermittent Fasting Confers Protection in CNS Autoimmunity by Altering the Gut Microbiota. Cell Metab. 2018, 27, 1222–1235.e6. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Li, W.; Shi, G.; Huang, Y.; Sun, X.; Sun, N.; Jiang, D. Atractylenolide III Improves Mitochondrial Function and Protects Against Ulcerative Colitis by Activating AMPK/SIRT1/PGC-1α. Mediat. Inflamm. 2022, 2022, 9129984. [Google Scholar] [CrossRef]
- Song, M.Y.; Jung, H.W.; Kang, S.Y.; Park, Y.K. Atractylenolide III Enhances Energy Metabolism by Increasing the SIRT-1 and PGC1α Expression with AMPK Phosphorylation in C2C12 Mouse Skeletal Muscle Cells. Biol. Pharm. Bull. 2017, 40, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.H.; Li, W.; Go, Y.; Oh, Y.C. Atractylodis Rhizoma Alba Attenuates Neuroinflammation in BV2 Microglia upon LPS Stimulation by Inducing HO-1 Activity and Inhibiting NF-κB and MAPK. Int. J. Mol. Sci. 2019, 20, 4015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.M.; Yan-Qin, H.E.; Gu-Ran, Y.U. The Effect of Atractylenolide-I on Oxidative Stress-Mediated Injury of bEnd.3 Cells Induced by Hydrogen Peroxide. Guid. J. Tradit. Chin. Med. Pharm. 2019, 25, 35–38. [Google Scholar]
- Song, M.Y.; Yi, F.; Xiao, H.; Yin, J.; Huang, Q.; Xia, J.; Yin, X.M.; Wen, Y.B.; Zhang, L.; Liu, Y.H.; et al. Energy restriction induced SIRT6 inhibits microglia activation and promotes angiogenesis in cerebral ischemia via transcriptional inhibition of TXNIP. Cell Death Dis. 2022, 13, 449. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Shang, J.; Gao, C.; Guan, X.; Chen, Y.; Zhu, L.; Zhang, L.; Zhang, C.; Zhang, J.; Pang, T. A novel SIRT6 activator ameliorates neuroinflammation and ischemic brain injury via EZH2/FOXC1 axis. Acta Pharm. Sin. B 2021, 11, 708–726. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wu, Y.; Zhao, Z.; Wu, B.; Sun, K.; Wang, H.; Qin, L.; Bai, F.; Leng, Y.; Tang, W. A new mechanism of obeticholic acid on NASH treatment by inhibiting NLRP3 inflammasome activation in macrophage. Metab. Clin. Exp. 2021, 120, 154797. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, W.; Zhang, L.; Shi, C.; Zhou, J.; Wang, P.; Wang, H.; Shi, X.; Wei, S.; Wang, Q.; et al. TGR5 Regulates Macrophage Inflammation in Nonalcoholic Steatohepatitis by Modulating NLRP3 Inflammasome Activation. Front. Immunol. 2021, 11, 609060. [Google Scholar] [CrossRef]
- Xiang, X.; Ohshiro, K.; Zaidi, S.; Yang, X.; Bhowmick, K.; Vegesna, A.K.; Bernstein, D.; Crawford, J.M.; Mishra, B.; Latham, P.S.; et al. Impaired reciprocal regulation between SIRT6 and TGF-β signaling in fatty liver. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22335. [Google Scholar] [CrossRef]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: The PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Jia, Y.; Fu, T.; Viswakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARα by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 628–638. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Y.; Wang, Y.; Qiu, Y.; Mou, H.; Deng, Y.; Yao, J.; Xia, Z.; Zhang, W.; Zhu, D.; et al. mTORC2 Facilitates Liver Regeneration Through the Sphingolipid-Induced PPAR-α-Fatty Acid Oxidation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1311–1331. [Google Scholar] [CrossRef]
- Choi, Y.; Song, M.J.; Jung, W.J.; Jeong, H.; Park, S.; Yang, B.; Lee, E.C.; Joo, J.S.; Choi, D.; Koo, S.H.; et al. Liver-Specific Deletion of Mouse CTCF Leads to Hepatic Steatosis via Augmented PPARγ Signaling. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1761–1787. [Google Scholar] [CrossRef]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e8. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, Z.; Chu, S.; Ma, X.; Wang, X.; Jiang, M.; Bai, G. Atractylenolide-I covalently binds to CYP11B2, selectively inhibits aldosterone synthesis, and improves hyperaldosteronism. Acta Pharm. Sin. B 2022, 12, 135–148. [Google Scholar] [CrossRef]
- Qin, Y.; Yu, Y.; Yang, C.; Wang, Z.; Yang, Y.; Wang, C.; Zheng, Q.; Li, D.; Xu, W. Atractylenolide I Inhibits NLRP3 Inflammasome Activation in Colitis-Associated Colorectal Cancer via Suppressing Drp1-Mediated Mitochondrial Fission. Front. Pharmacol. 2021, 12, 674340. [Google Scholar] [CrossRef]
- Han, Y.; Bai, C.; He, X.M.; Ren, Q.L. P2X7 receptor involved in antitumor activity of atractylenolide I in human cervical cancer cells. Purinergic Signal. 2022. [Google Scholar] [CrossRef]
- Wang, M.; Li, X.Z.; Zhang, M.X.; Ye, Q.Y.; Chen, Y.X.; Chang, X. Atractylenolide-I Sensitizes Triple-Negative Breast Cancer Cells to Paclitaxel by Blocking CTGF Expression and Fibroblast Activation. Front. Oncol. 2021, 11, 738534. [Google Scholar] [CrossRef]
- Cheng, B.; Gao, W.; Wu, X.; Zheng, M.; Yu, Y.; Song, C.; Miao, W.; Yang, Z.; He, Y.; Liu, C.; et al. Ginsenoside Rg2 Ameliorates High-Fat Diet-Induced Metabolic Disease through SIRT1. J. Agric. Food Chem. 2020, 68, 4215–4226. [Google Scholar] [CrossRef]
- Pan, Z.; Guo, J.; Tang, K.; Chen, Y.; Gong, X.; Chen, Y.; Zhong, Y.; Xiao, X.; Duan, S.; Cui, T.; et al. Ginsenoside Rc Modulates SIRT6-NRF2 Interaction to Alleviate Alcoholic Liver Disease. J. Agric. Food Chem. 2022, 70, 14220–14234. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, D.; Mai, Z.; Chen, Y.; Luo, L.; Liu, H.; Zhao, L.; Huang, R.; Wang, S.; Chen, R.; Zhou, H.; et al. ATL I, Acts as a SIRT6 Activator to Alleviate Hepatic Steatosis in Mice via Suppression of NLRP3 Inflammasome Formation. Pharmaceuticals 2022, 15, 1526. https://doi.org/10.3390/ph15121526
Kong D, Mai Z, Chen Y, Luo L, Liu H, Zhao L, Huang R, Wang S, Chen R, Zhou H, et al. ATL I, Acts as a SIRT6 Activator to Alleviate Hepatic Steatosis in Mice via Suppression of NLRP3 Inflammasome Formation. Pharmaceuticals. 2022; 15(12):1526. https://doi.org/10.3390/ph15121526
Chicago/Turabian StyleKong, Danli, Zhenhua Mai, Yongze Chen, Ling Luo, Hao Liu, Le Zhao, Ruixian Huang, Shuang Wang, Rong Chen, Hao Zhou, and et al. 2022. "ATL I, Acts as a SIRT6 Activator to Alleviate Hepatic Steatosis in Mice via Suppression of NLRP3 Inflammasome Formation" Pharmaceuticals 15, no. 12: 1526. https://doi.org/10.3390/ph15121526