


In Silico Screening of Drugs That Target Different Forms of E Protein for Potential Treatment of COVID-19

 , and
, and
Abstract
:
1. Introduction
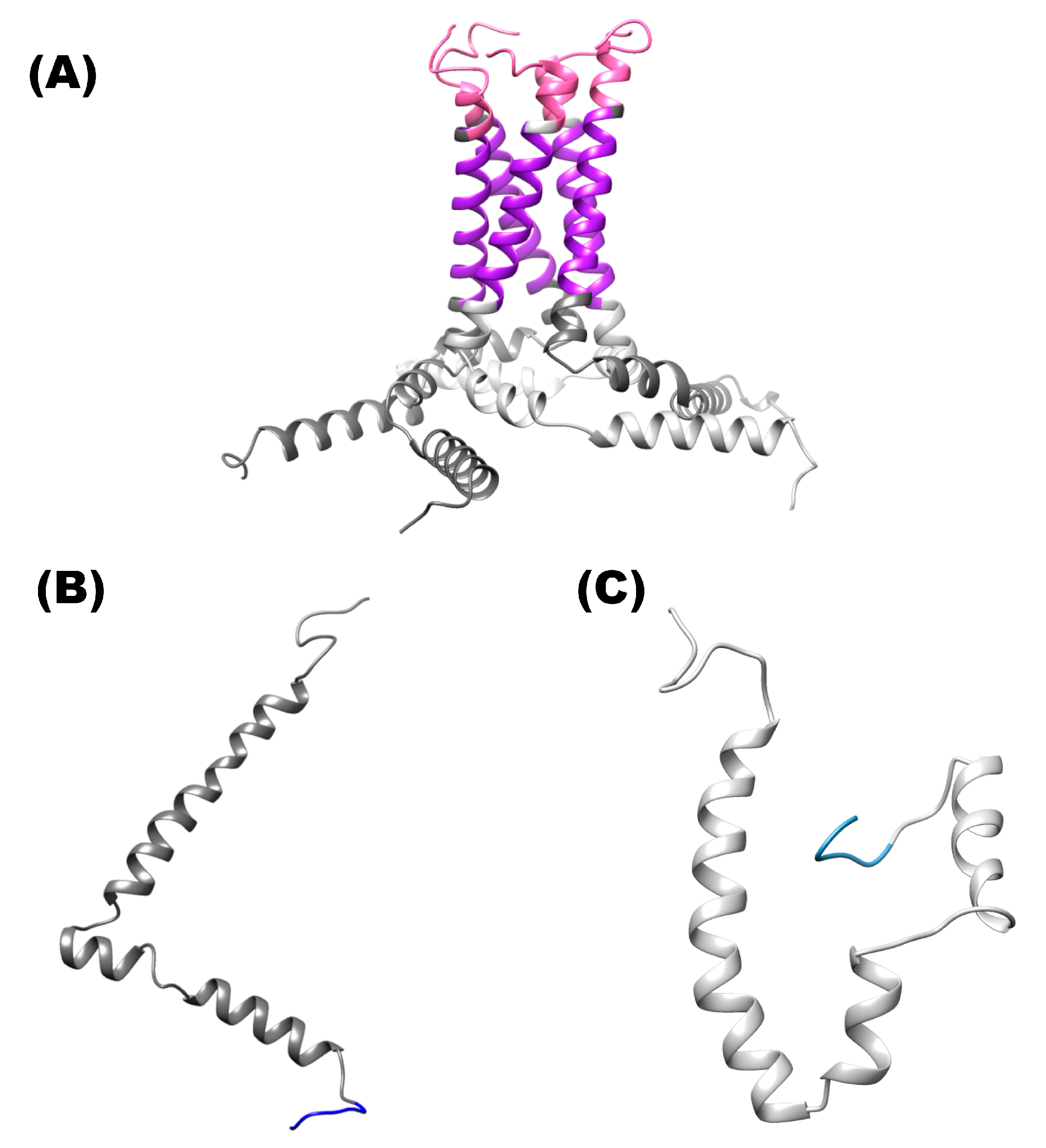
1.1. Homopentamer
1.2. Ion Channel
1.3. N-Terminal
1.4. Monomeric
2. Results
2.1. Molecular Modeling of E Protein of SARS-CoV-2
2.2. In Silico ADMET Analysis
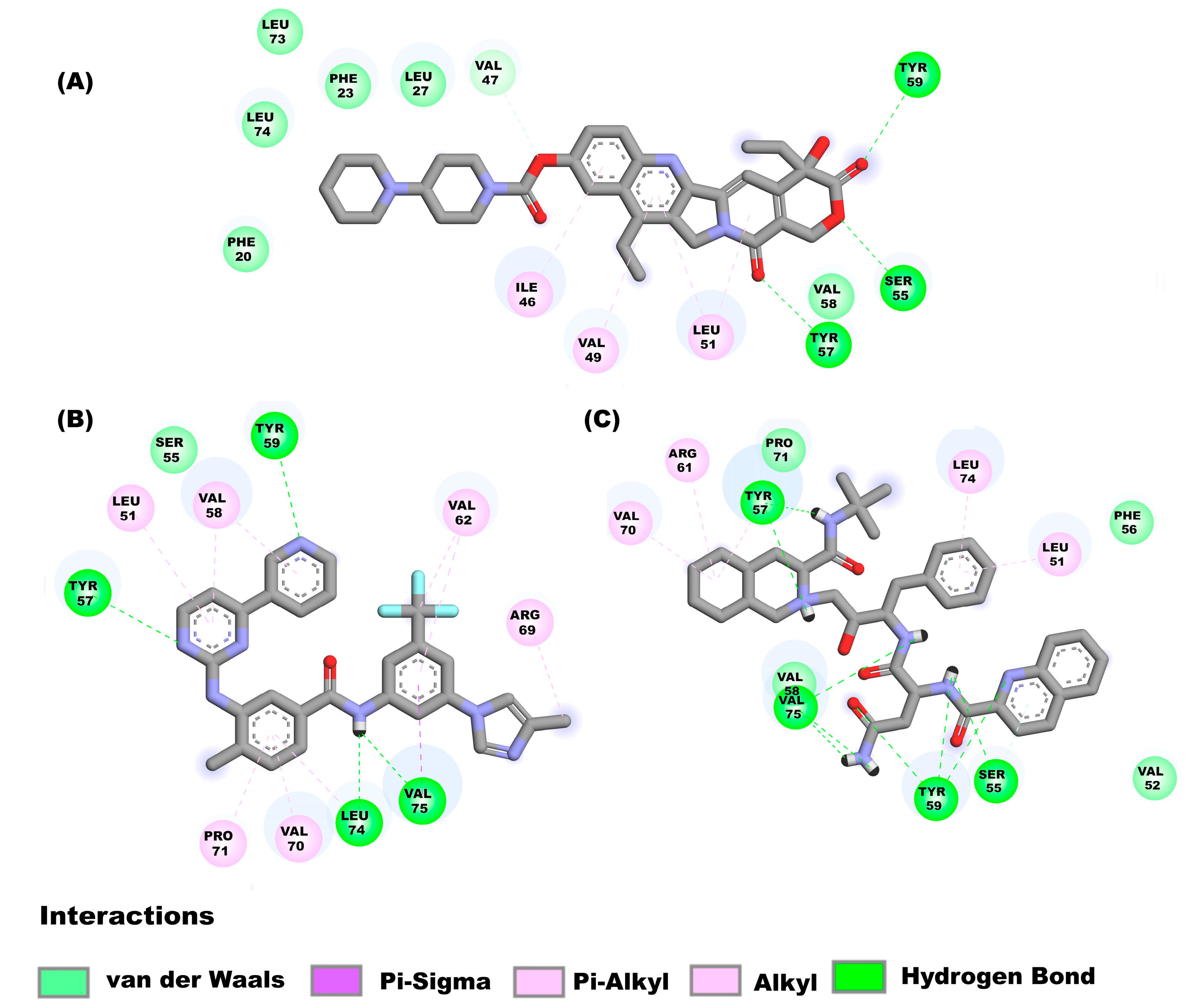
2.3. Molecular Docking Analysis on the Different Targets
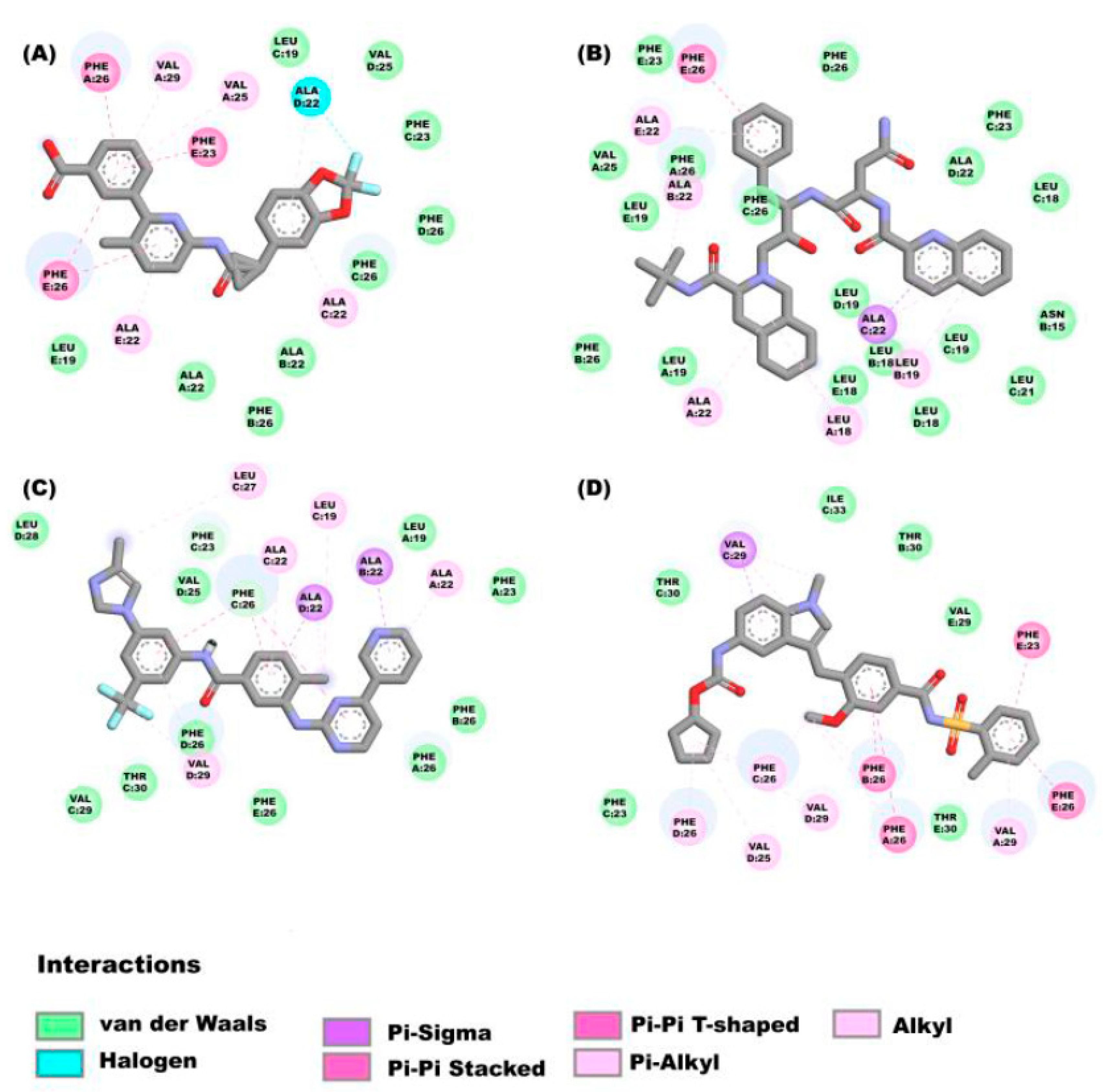
2.3.1. Molecular Docking Analysis on E Protein Form Homopentamer (Ionic Channel Region)
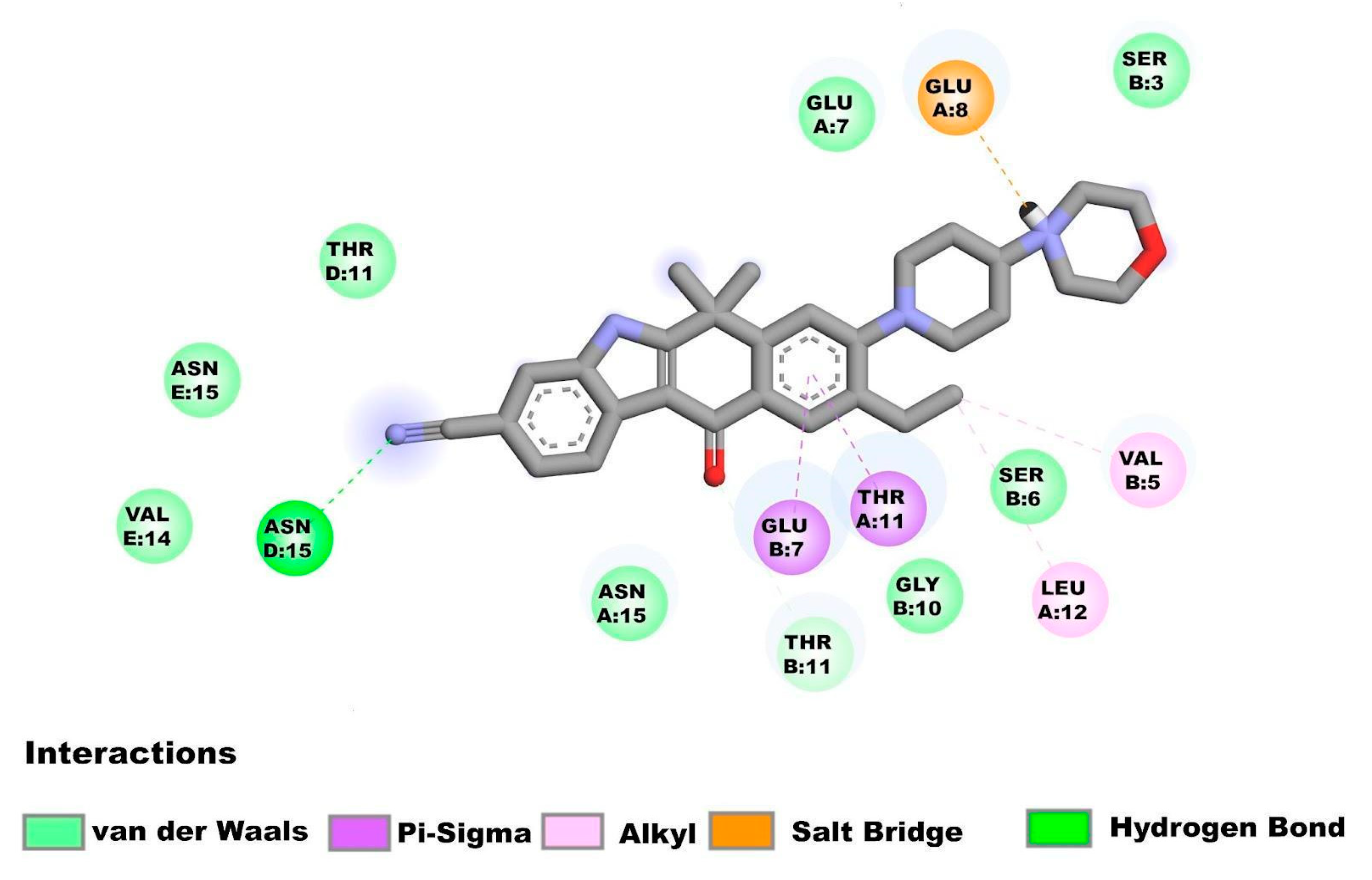
2.3.2. Molecular Docking Studies on E Protein Form Homopentamer (N-Terminal Region)
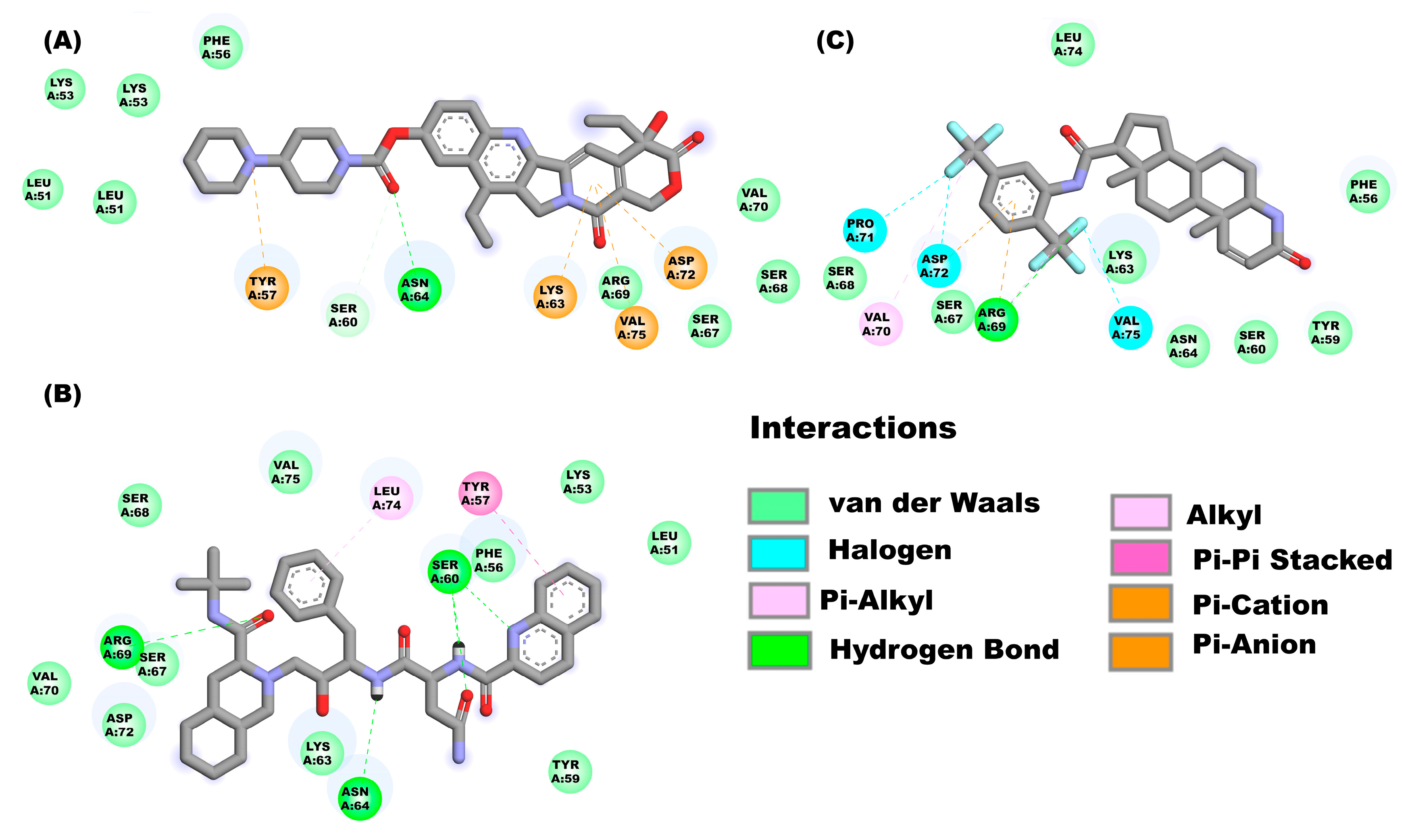
2.3.3. Molecular Docking Analysis on E Protein Form Monomer (C-Terminal Solvent Exposed)
2.3.4. Molecular Docking on E Protein Form Monomer (Harpin)
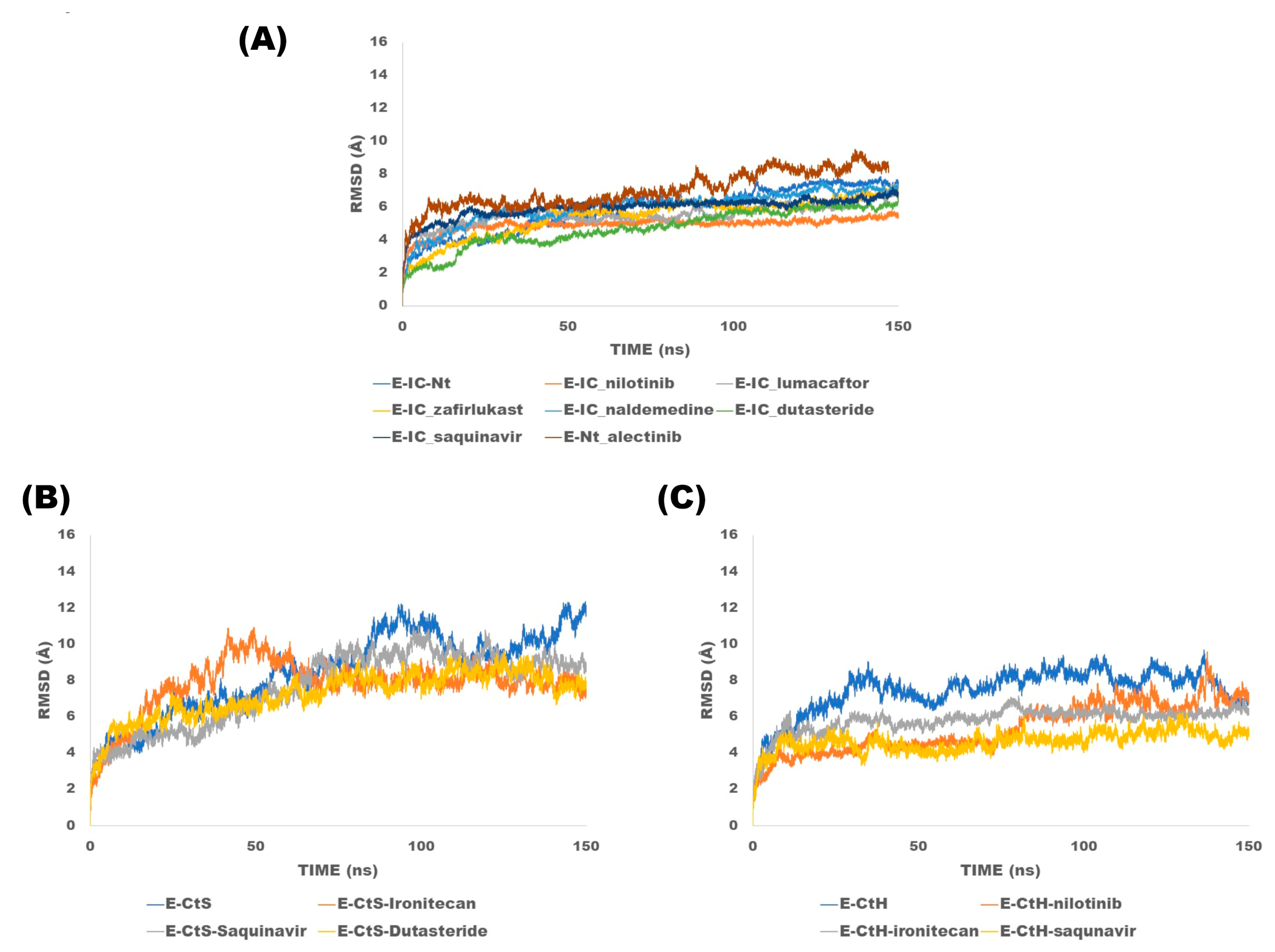
2.4. Molecular Dynamics Simulations
2.5. Binding Free Energy Calculation Using MMPBSA Approach
2.5.1. Monomeric Form
2.5.2. Homopentamer Form
3. Discussion
3.1. Monomeric
3.2. Pentameric
4. Methods
4.1. Molecular Modeling
4.2. Database Search and In Silico ADMET Analysis
4.3. Docking Analysis
4.4. Molecular Docking Studies and Visualization of the Results
4.5. Molecular Dynamics Simulations
4.6. Structural Analysis of MD Simulations
4.7. Binding Free Energy Calculations of the E Protein-Ligand Complexes
5. Conclusions
Consent for Publication
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, P.; Niyogi, S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epidemiol Infect 2020, 148, e262. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.worldometers.info/coronavirus/ (accessed on 9 February 2023).
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.S.; Choudhury, P.P.; Roy, B. SARS-CoV2 envelope protein: Non-synonymous mutations and its consequences. Genomics 2020, 112, 3890–3892. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Duart, G.; García-Murria, M.J.; Grau, B.; Acosta-Cáceres, J.M.; Martínez-Gil, L.; Mingarro, I. SARS-CoV-2 envelope protein topology in eukaryotic membranes. Open Biol. 2020, 10, 200209. [Google Scholar] [CrossRef]
- Malik, Y.A. Properties of Coronavirus and SARS-CoV-2. Malays J. Pathol. 2020, 42, 3–11. [Google Scholar]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional Flexibility in the Sequence Requirements for Coronavirus Small Envelope Protein Function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wu, S.; Wang, S.; Rubino, G.; Nickels, J.D.; Cheng, X. Refinement of SARS-CoV-2 envelope protein structure in a native-like environment by molecular dynamics simulations. Front. Mol. Biosci. 2022, 9, 1027223. [Google Scholar] [CrossRef]
- Wong, N.A.; Saier, M.H., Jr. The SARS-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef]
- Park, S.H.; Siddiqi, H.; Castro, D.V.; De Angelis, A.A.; Oom, A.L.; Stoneham, C.A.; Lewinski, M.K.; Clark, A.E.; Croker, B.A.; Carlin, A.F.; et al. Interactions of SARS-CoV-2 envelope protein with amilorides correlate with antiviral activity. PLoS Pathog 2021, 17, e1009519. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Hogue, B.G. Role of the Coronavirus E Viroporin Protein Transmembrane Domain in Virus Assembly. J. Virol. 2007, 81, 3597–3607. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. A Single Polar Residue and Distinct Membrane Topologies Impact the Function of the Infectious Bronchitis Coronavirus E Protein. PLOS Pathog. 2012, 8, e1002674. [Google Scholar] [CrossRef]
- Torres, J.; Maheswari, U.; Parthasarathy, K.; Ng, L.; Liu, D.X.; Gong, X. Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 2007, 16, 2065–2071. [Google Scholar] [CrossRef]
- De Diego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeno, J.M.J.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLOS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Islam, I.; Mishu, I.D.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Mutational insights into the envelope protein of SARS-CoV-2. Gene. Rep. 2021, 22, 100997. [Google Scholar] [CrossRef] [PubMed]
- Sariol, A.; Perlman, S. SARS-CoV-2 takes its Toll. Nat. Immunol. 2021, 22, 801–802. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Rémi, P.; Jean-Baptiste, B.; Sofiane, T.; Lbachir, B.; Elmostafa, B. SARS-CoV-2 Envelope protein (E) binds and activates TLR2: A novel target for COVID-19 interventions. bioRxiv 2021. bioRxiv:11.10.468173. [Google Scholar]
- Caillet-Saguy, C.; Durbesson, F.; Rezelj, V.V.; Gogl, G.; Tran, Q.D.; Twizere, J.C.; Vignuzzi, M.; Vincentelli, R.; Wolff, N. Host PDZ-containing proteins targeted by SARS-CoV-2. FEBS J. 2021, 288, 5148–5162. [Google Scholar] [CrossRef]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLOS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef]
- Li, Y.; Surya, W.; Claudine, S.; Torres, J. Structure of a Conserved Golgi Complex-targeting Signal in Coronavirus Envelope Proteins. J. Biol. Chem. 2014, 289, 12535–12549. [Google Scholar] [CrossRef]
- De Maio, F.; Lo Cascio, E.; Babini, G.; Sali, M.; Della Longa, S.; Tilocca, B.; Roncada, P.; Arcovito, A.; Sanguinetti, M.; Scambia, G.; et al. Improved binding of SARS-CoV-2 Envelope protein to tight junction-associated PALS1 could play a key role in COVID-19 pathogenesis. Microbes Infect 2020, 22, 592–597. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Sagum, C.A.; Oliva, I.; Rybakovsky, E.; DiGuilio, K.; Liang, J.; Bedford, M.T.; Cassel, J.; Sudol, M.; Mullin, J.M.; et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PLoS ONE 2021, 16, e0251955. [Google Scholar] [CrossRef]
- Toto, A.; Ma, S.; Malagrinò, F.; Visconti, L.; Pagano, L.; Stromgaard, K.; Gianni, S. Comparing the binding properties of peptides mimicking the Envelope protein of SARS-CoV and SARS-CoV-2 to the PDZ domain of the tight junction-associated PALS1 protein. Protein Sci. 2020, 29, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Javorsky, A.; Humbert, P.O.; Kvansakul, M. Structural basis of coronavirus E protein interactions with human PALS1 PDZ domain. Commun. Biol. 2021, 4, 724. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Lara-Ureña, N.; García-Domínguez, M. Relevance of BET Family Proteins in SARS-CoV-2 Infection. Biomolecules 2021, 11, 1126. [Google Scholar] [CrossRef]
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.J.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Rudraraju, R.; et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 2021, 184, 2167–2182. [Google Scholar] [CrossRef]
- Corse, E.; Machamer, C.E. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 2003, 312, 25–34. [Google Scholar] [CrossRef]
- de Haan, C.A.; Vennema, H.; Rottier, P.J. Assembly of the coronavirus envelope: Homotypic interactions between the M proteins. J. Virol. 2000, 74, 4967–4978. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2020, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Kuzmin, A.; Orekhov, P.; Astashkin, R.; Gordeliy, V.; Gushchin, I. Structure and dynamics of the SARS-CoV-2 envelope protein monomer. Proteins 2022, 90, 1102–1114. [Google Scholar] [CrossRef]
- Surya, W.; Li, Y.; Torres, J. Structural model of the SARS coronavirus E channel in LMPG micelles. Biochim. Biophys. Acta (BBA) 2018, 1860, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shen, L.; Gao, P.; Huang, L.; Liu, G.; Zhou, L.; Peng, L. Discovery of Potential Therapeutic Drugs for COVID-19 Through Logistic Matrix Factorization with Kernel Diffusion. Front. Microbiol. 2022, 13, 740382. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.P.; Liu, D.X. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 2001, 276, 17515–17523. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. The Coronavirus E Protein: Assembly and Beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus Pathogenesis and the Emerging Pathogen Severe Acute Respiratory Syndrome Coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef]
- Koepke, L.; Hirschenberger, M.; Hayn, M.; Kirchhoff, F.; Sparrer, K.M. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy 2021, 17, 2659–2661. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Alam, I.; Kamau, A.A.; Kulmanov, M.; Jaremko, Ł.; Arold, S.T.; Pain, A.; Gojobori, T.; Duarte, C.M. Functional Pangenome Analysis Shows Key Features of E Protein Are Preserved in SARS and SARS-CoV-2. Front. Cell Infect Microbiol. 2020, 10, 405. [Google Scholar] [CrossRef]
- Duart, G.; García-Murria, M.J.; Mingarro, I. The SARS-CoV-2 envelope (E) protein has evolved towards membrane topology robustness. Biochim. Biophys. Acta 2021, 1863, 183608. [Google Scholar] [CrossRef]
- Mukherjee, S.; Harikishore, A.; Bhunia, A. Targeting C-terminal Helical bundle of NCOVID19 Envelope (E) protein. Int. J. Biol. Macromol. 2021, 175, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Troyano-Hernáez, P.; Reinosa, R.; Holguín, Á. Evolution of SARS-CoV-2 Envelope, Membrane, Nucleocapsid, and Spike Structural Proteins from the Beginning of the Pandemic to September 2020: A Global and Regional Approach by Epidemiological Week. Viruses 2021, 13, 243. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Salinas, G.L.; Martínez-Archundia, M.; Correa-Basurto, J.; García-Machorro, J. Repositioning of Ligands That Target the Spike Glycoprotein as Potential Drugs for SARS-CoV-2 in an In Silico Study. Molecules 2020, 25, 5615. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.C.; Hsu, M.S.; Chen, Y.T.; Yang, J.M.; Tsay, Y.G.; Huang, H.C.; Huang, C. Repurposing existing drugs: Identification of SARS-CoV-2 3C-like protease inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Saha, S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PLoS ONE 2020, 15, e0237300. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Borkotoky, S.; Banerjee, M. In silico identification of Tretinoin as a SARS-CoV-2 envelope (E) protein ion channel inhibitor. Comput. Biol. Med. 2020, 127, 104063. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2021, 39, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Nath Jha, A. A Comprehensive Review of Drug Repurposing Strategies against Known Drug Targets of COVID-19. COVID 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Indari, O.; Singh, A.K.; Tiwari, D.; Jha, H.C.; Jha, A.N. Deciphering antiviral efficacy of malaria box compounds against malaria exacerbating viral pathogens-Epstein Barr virus and SARS-CoV-2, an in silico study. Med. Drug Discov. 2022, 16, 100146. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.; Sali, A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007, 16, 2412–2426. [Google Scholar] [CrossRef] [PubMed]
- Eramian, D.; Shen, M.-Y.; Devos, D.; Melo, F.; Sali, A.; Marti-Renom, M.A. A composite score for predicting errors in protein structure models. Protein Sci. 2006, 15, 1653–1666. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Domínguez-Villa, F.X.; Durán-Iturbide, N.A.; Ávila-Zárraga, J.G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl)indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorg. Chem. 2021, 106, 104497. [Google Scholar] [CrossRef]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In Silico ADME/Tox Profiling of Natural Products: A Focus on biofacquim. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, G.E.A.; Aguilar, M.E.H.; Covarrubias, D.H.; Duran, F.R. Amantadine as a drug to mitigate the effects of COVID-19. Med. Hypotheses 2020, 140, 109755. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Visualizer Software, Version 4.0; BIOVIA: San Diego, CA, USA, 2012.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. Charmm-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Tan, H.; Zhao, Y.; Zhao, W.; Xie, H.; Chen, Y.; Tong, Q.; Yang, J. Dynamics properties of membrane proteins in native cell membranes revealed by solid-state NMR spectroscopy. Biochim. et Biophys. Acta (BBA) 2022, 1864, 183791. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef]
- Liu, H.; Hou, T. CaFE: A tool for binding affinity prediction using end-point free energy methods. Bioinformatics 2016, 32, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Hsin, J.; Arkhipov, A.; Yin, Y.; Stone, J.E.; Schulten, K. Using VMD: An Introductory Tutorial. Curr. Protoc. Bioinform. 2008, 24, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. VMD Store-A VMD Plugin to Browse, Discover, and Install VMD Extensions. J. Chem. Inf. Model. 2019, 59, 4519–4523. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Affinities of Some Compounds and Considering Different Forms | ||||
|---|---|---|---|---|
| Homopentamer | Monomer | |||
| Binding Site | ||||
| Drugs | Ion Channel | N-terminal | C-terminal solvent-exposed | C-terminal Harpin |
| ΔG (kcal/mol) | ||||
| lumacaftor | −10.9 | −9.1 | −7.6 * | −7.6 |
| nilotinib | −10.8 | −9.8 | −7.5 * | −8.2 |
| dutasteride | −10.6 | −8.9 | −7.7 | −8.1 |
| naldemedine | −10.5 | −8.7 | −7.4 | −7.7 |
| zafirlukast | −10.2 | −9.1 | −7.0 | −7.3 |
| irinotecan | −10.1 | −9.0 | −8.0 | −8.2 |
| saquinavir | −9.9 | −9.3 | −7.9 | −7.7 |
| alectinib | −9.4 | −9.6 | −7.0 | −7.0 |
| Binding Free Energy MMPBSA for the E-Homopentamer | ||||
|---|---|---|---|---|
| Site Bind: Ion Channel | ||||
| Drugs | Complex Total (kcal/mol) | Receptor (kcal/mol) | Ligand (kcal/mol) |
ΔG
(kcal/mol) |
| Lumacaftor | −7885.9200 | −7178.3400 | −125.6700 | −13.1500 |
| Nilotinib | −7965.2763 | −7537.2451 | −404.9531 | −23.0781 |
| Dutasteride | −7392.0455 | −7351.4972 | −23.6330 | −16.9153 |
| Naldemedine | −7364.0697 | −7341.7927 | −15.1966 | −7.0804 |
| Zafirlukast | −7603.0986 | −7358.9720 | −241.1082 | −3.0184 |
| Saquinavir | −7582.0157 | −7511.3579 | −45.7430 | −24.9147 |
| Site bind: N-terminal | ||||
| Alectinib | −4203.9200 | −4307.8700 | −65.5600 | −116.6700 |
| Binding Free Energy MMPBSA for the E-Monomer | ||||
|---|---|---|---|---|
| Binding Site: Solvent Exposed C-Terminal | ||||
| Drugs | Complex Total (kcal/mol) | Receptor (kcal/mol) | Ligand (kcal/mol) |
ΔG
(kcal/mol) |
| Dutasteride | −2175.4155 | −2058.9008 | −105.8080 | −10.7068 |
| Irinotecan | −2111.4271 | −2062.2341 | −44.8761 | −4.3169 |
| Saquinavir | −2206.3300 | −2245.6700 | −50.1300 | −2.1400 |
| Binding site: Harpin | ||||
| Nilotinib | −2238.1565 | −2071.3184 | −163.2539 | −3.5842 |
| Irinotecan | −2114.7274 | −2059.0421 | −45.8114 | −9.8740 |
| Saquinavir | −2122.5089 | −2073.9448 | −36.6094 | −11.9547 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez Salinas, G.L.; López Rincón, A.; García Machorro, J.; Correa Basurto, J.; Martínez Archundia, M. In Silico Screening of Drugs That Target Different Forms of E Protein for Potential Treatment of COVID-19. Pharmaceuticals 2023, 16, 296. https://doi.org/10.3390/ph16020296
Ramírez Salinas GL, López Rincón A, García Machorro J, Correa Basurto J, Martínez Archundia M. In Silico Screening of Drugs That Target Different Forms of E Protein for Potential Treatment of COVID-19. Pharmaceuticals. 2023; 16(2):296. https://doi.org/10.3390/ph16020296
Chicago/Turabian StyleRamírez Salinas, Gema Lizbeth, Alejandro López Rincón, Jazmín García Machorro, José Correa Basurto, and Marlet Martínez Archundia. 2023. "In Silico Screening of Drugs That Target Different Forms of E Protein for Potential Treatment of COVID-19" Pharmaceuticals 16, no. 2: 296. https://doi.org/10.3390/ph16020296