


Structure-Based Virtual Screening of Furan-1,3,4-Oxadiazole Tethered N-phenylacetamide Derivatives as Novel Class of hTYR and hTYRP1 Inhibitors

 ,
,  ,
,  ,
,  , , and
, , and
Abstract
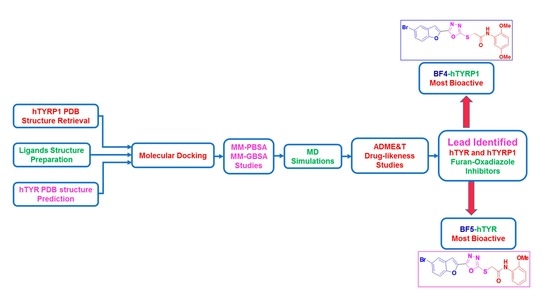
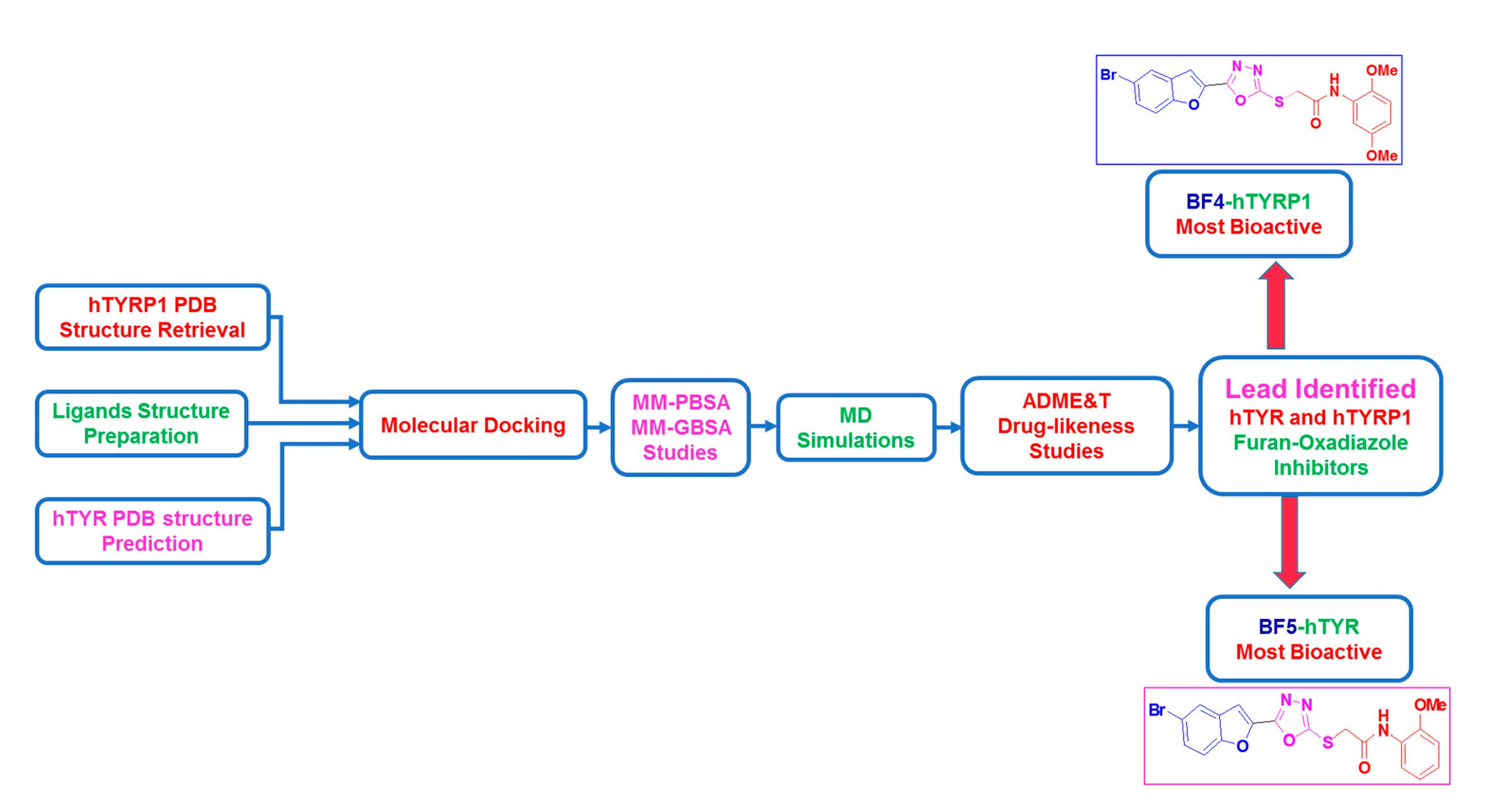
:
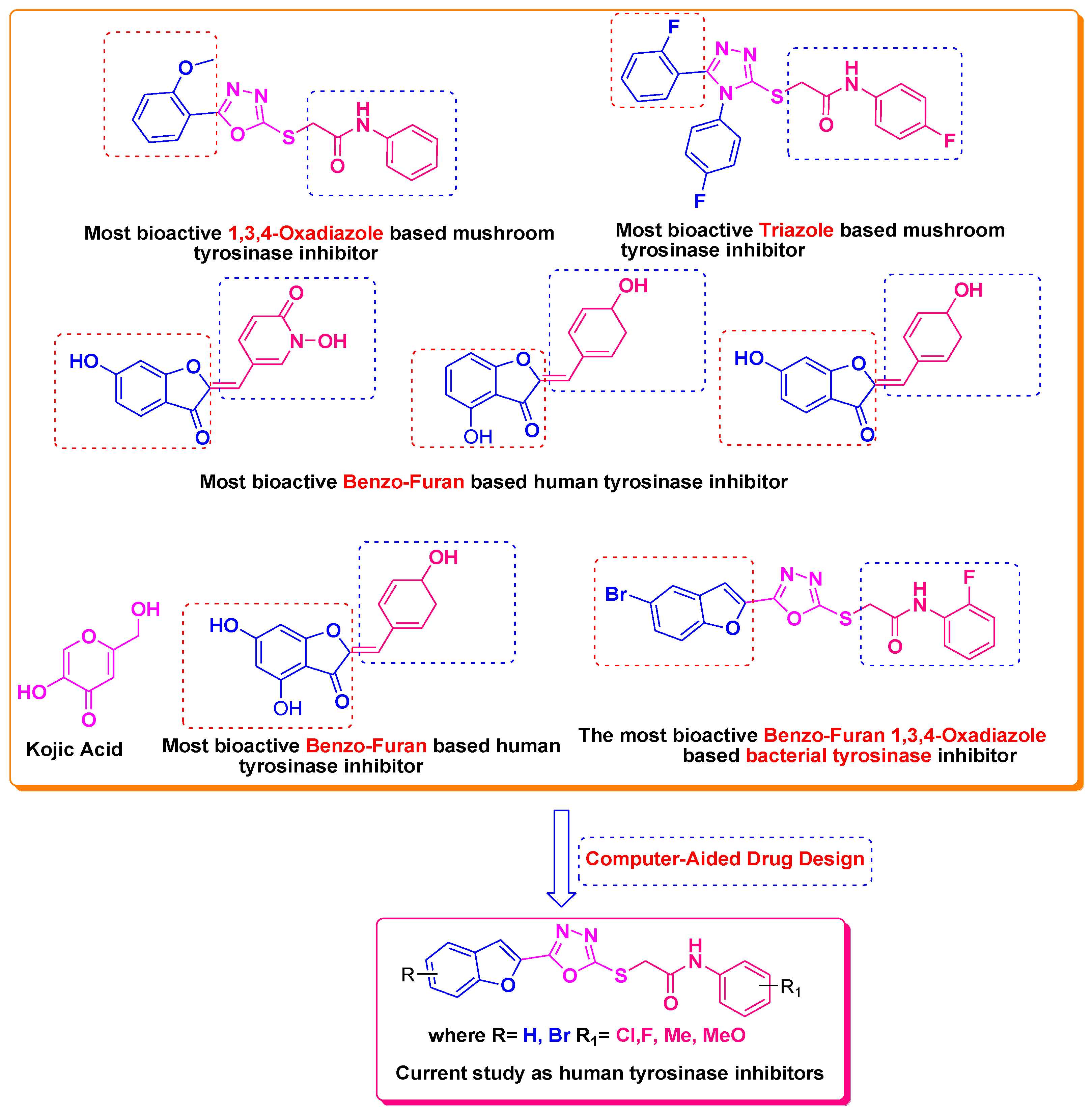
1. Introduction
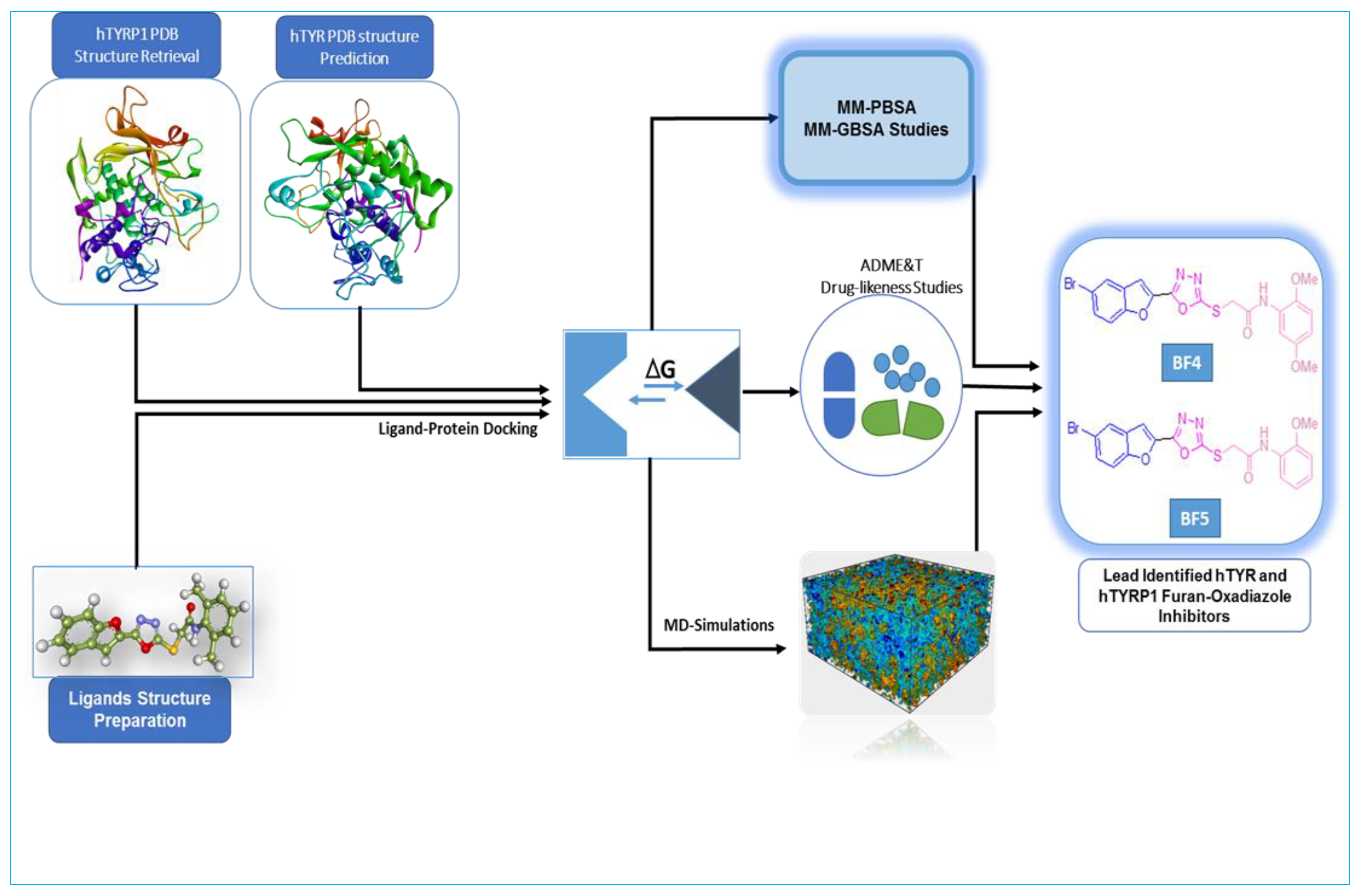
2. Results and Discussion
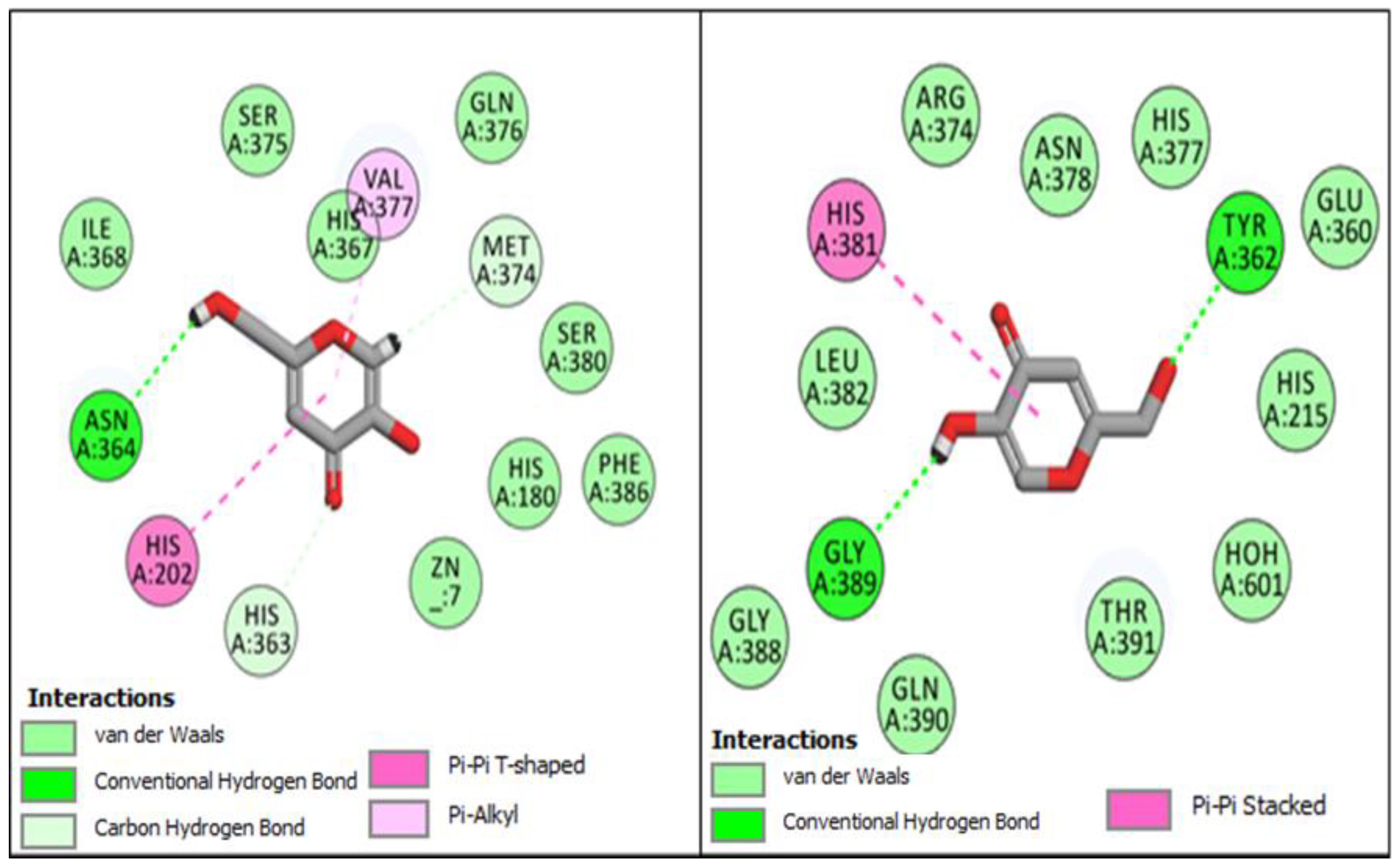
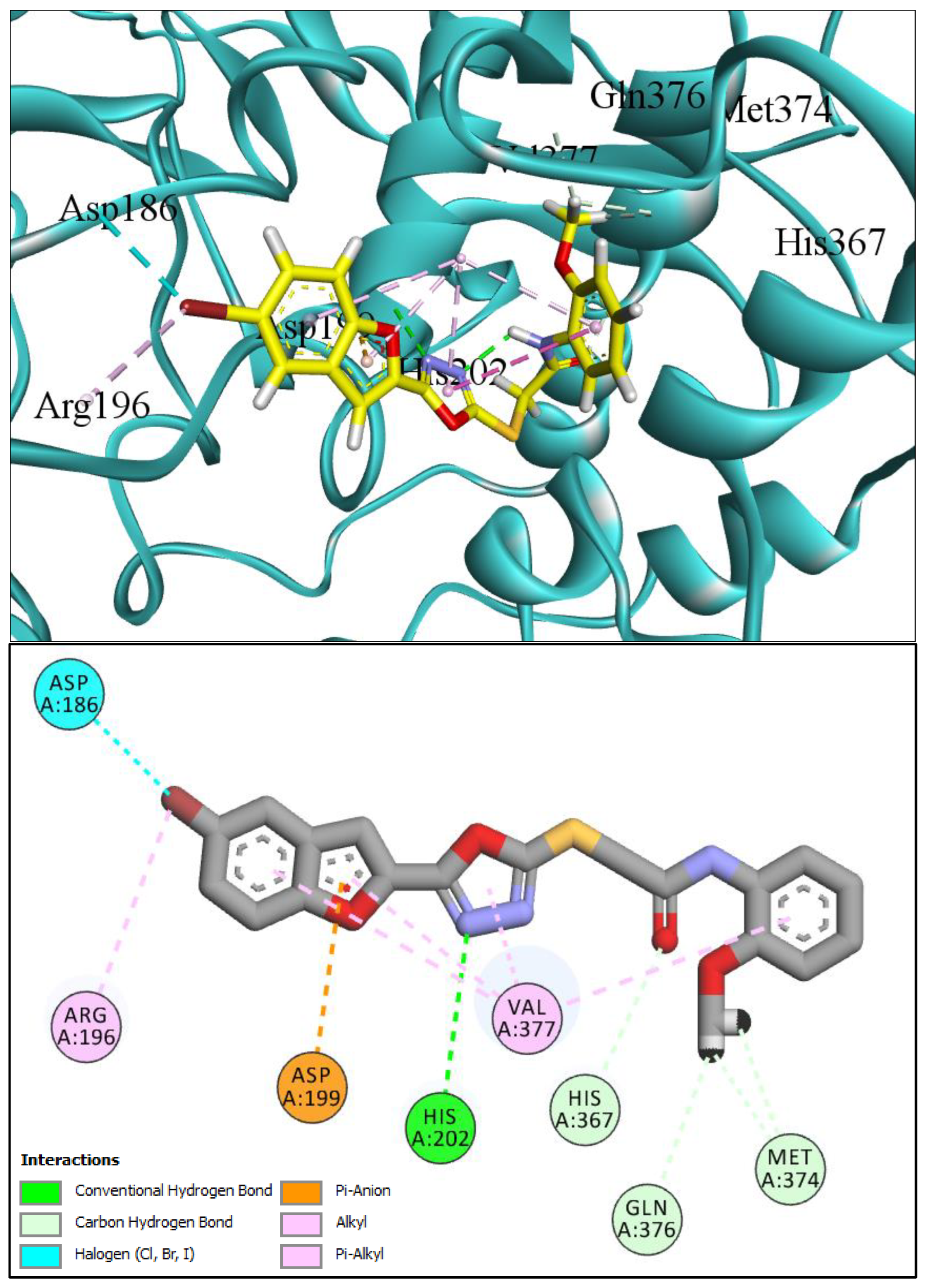
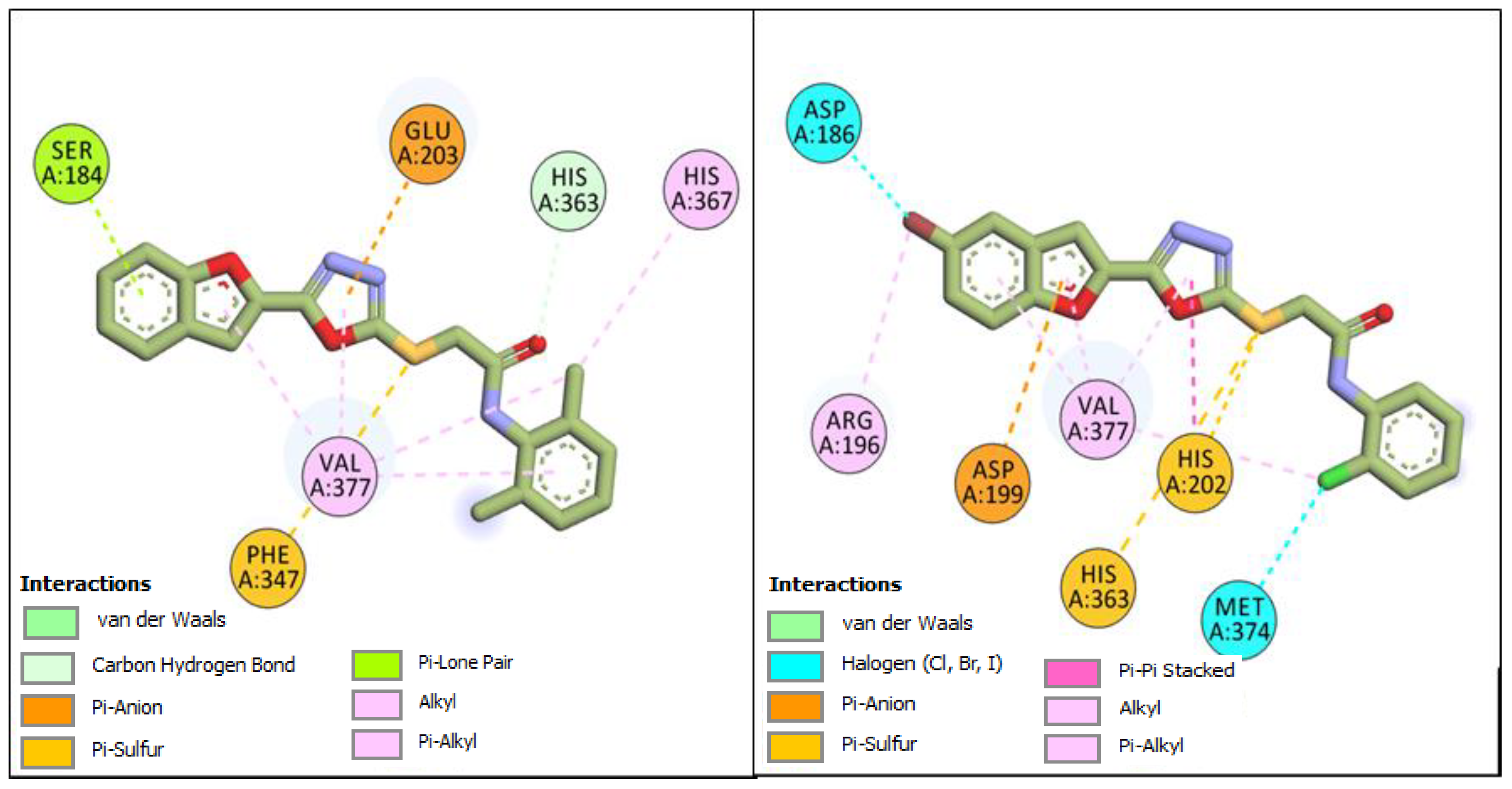
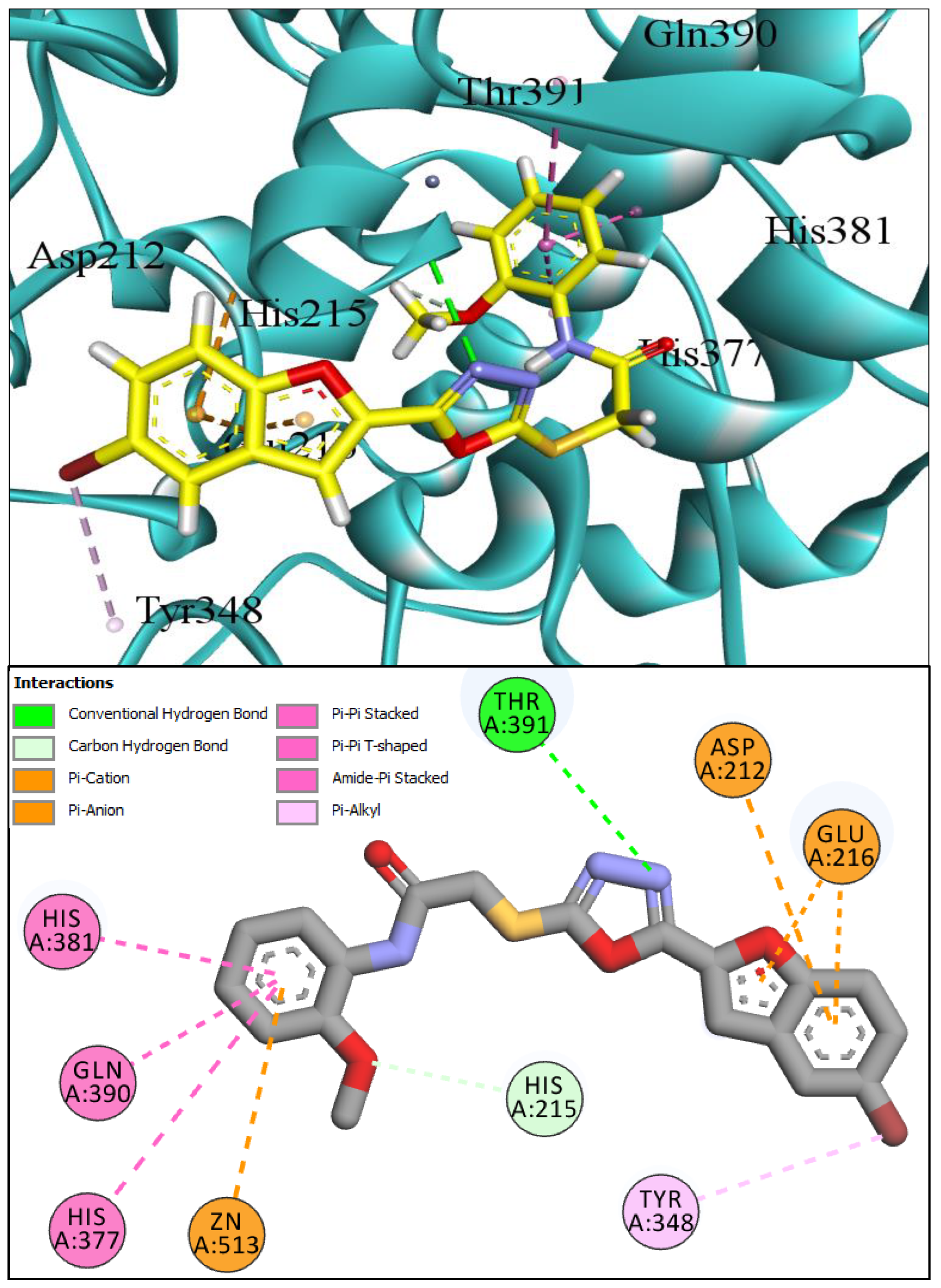
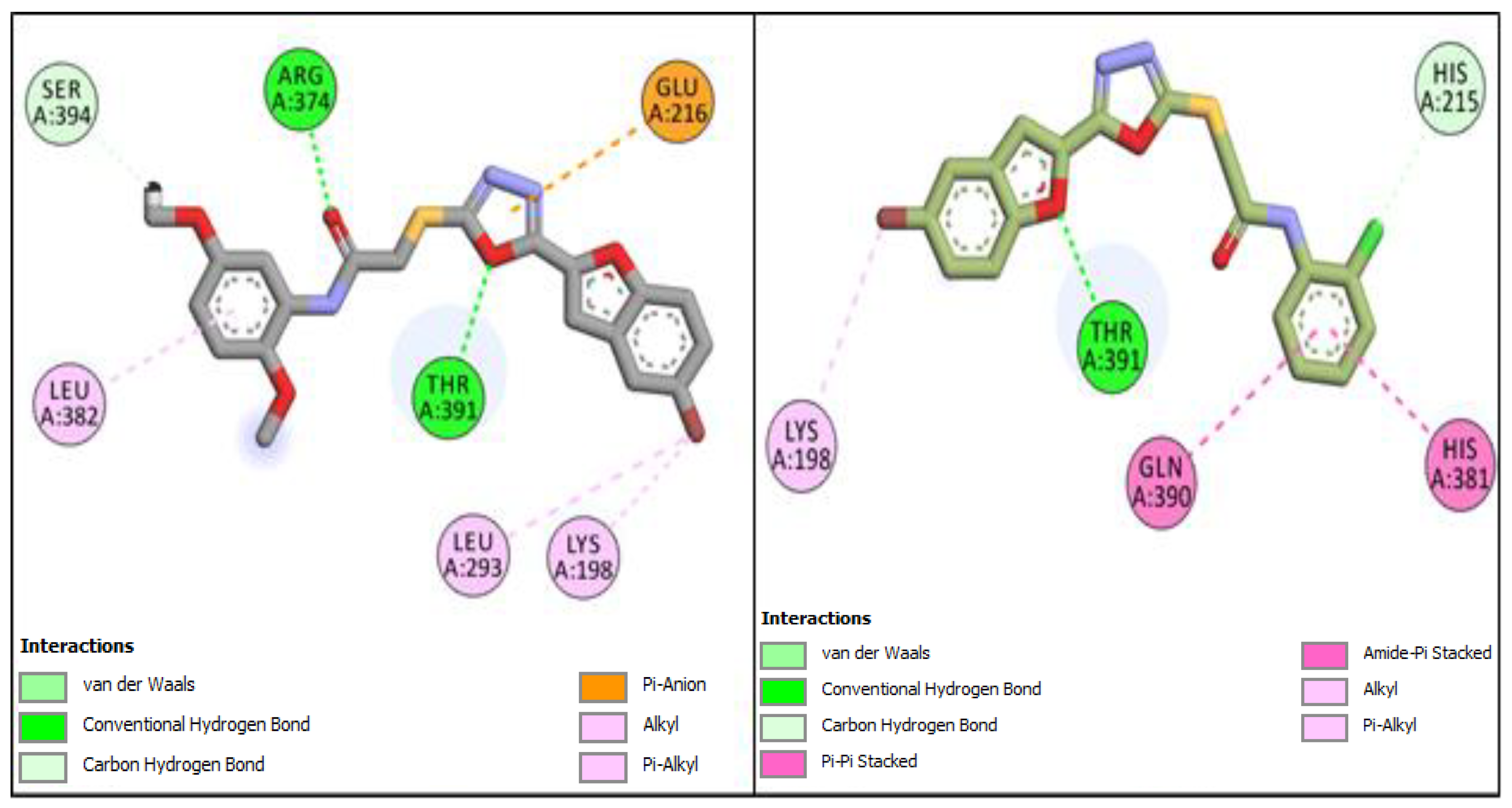
2.1. Computational Investigations of BF1–BF16 against hTYR and hTYRP1
2.2. ADMET and Drug-Likeness Predictive Studies
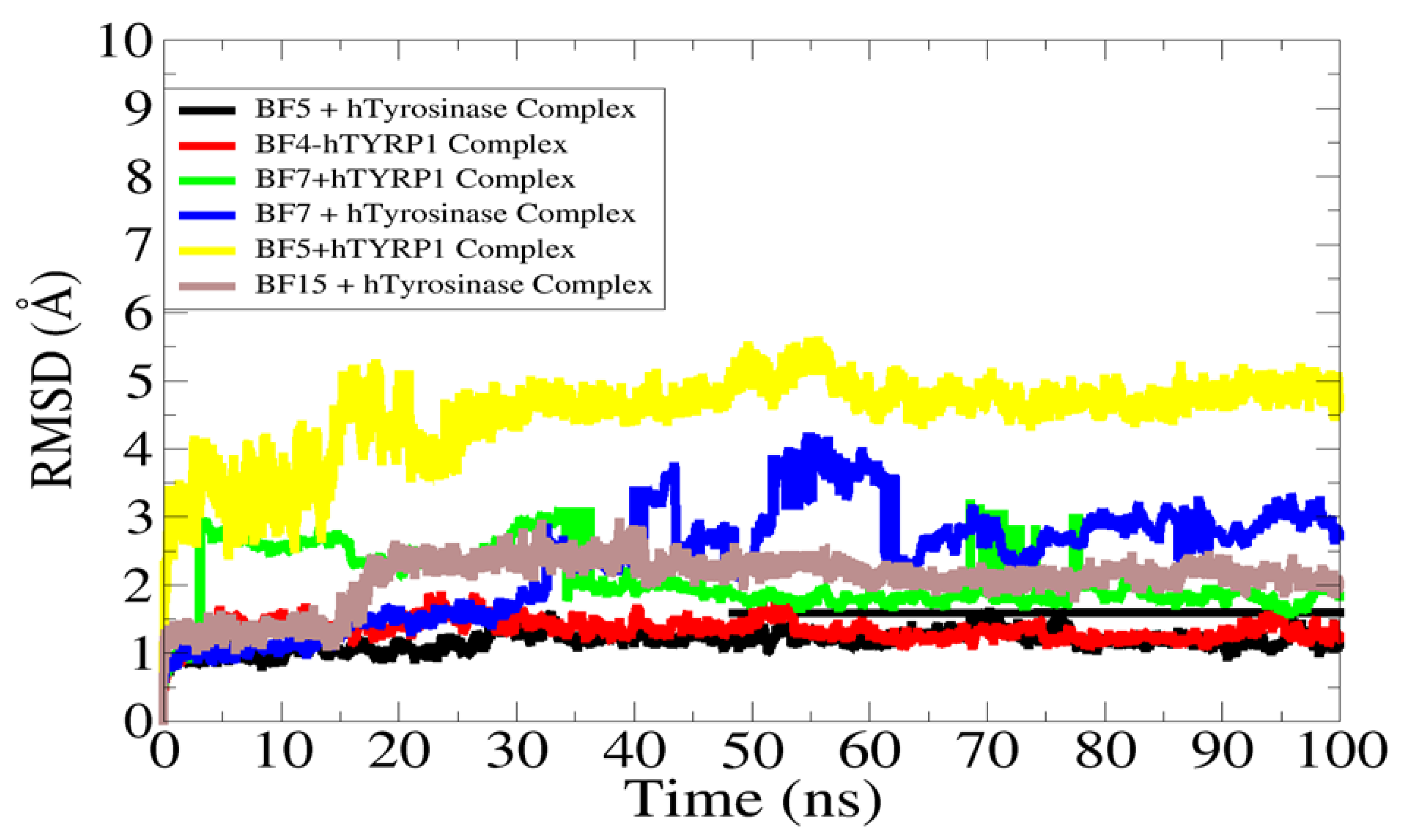
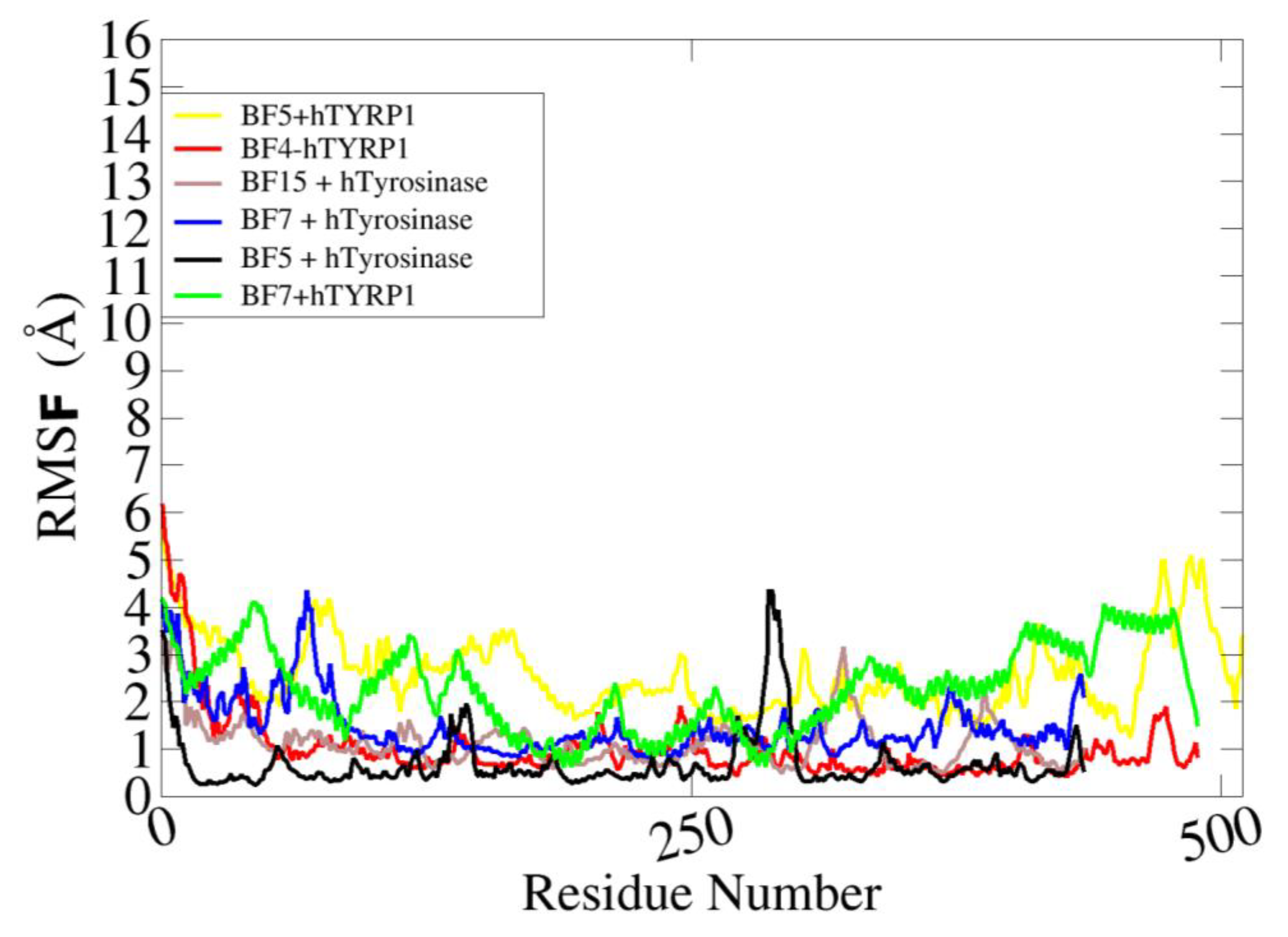
2.3. Molecular Dynamics Stability Analysis of the Ligand-Protein Complexes
2.4. MM-GBSA/MM-PBSA Binding Free Energy Analysis of Complexes
3. Material and Methods
3.1. Structures of Synthsized Furan-1,3,4-Oxadiazoles BF1–BF16
3.2. In Silico Biological Evaluation of Furan-1,3,4-Oxadiazoles BF1–BF16
3.2.1. Molecular Docking, ADME&T, Drug-Likeness, and Protein Homology Modeling Studies
3.2.2. Molecular Dynamic Simulation Studies
3.2.3. Estimation of Binding Energies and Interactions (MM-GBSA and MM-PBSA)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Zeng, W.; Jiang, A.; He, Z.; Shen, X.; Dong, X.; Feng, J.; Lu, H. Global, Regional and National Incidence, Mortality and Disability-Adjusted Life-Years of Skin Cancers and Trend Analysis from 1990 to 2019: An Analysis of the Global Burden of Disease Study 2019. Cancer Med. 2021, 10, 4905–4922. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, S.; Akhtar, N.; Khan, M.S.; Hameed, A.; Irfan, M.; Arshad, M.A.; Ali, S.; Asrar, M. Plant Derived Anticancer Agents: A Green Approach towards Skin Cancers. Biomed. Pharmacother. 2018, 103, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological Trends in Skin Cancer. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, M.C.F.; Sousa, J.J.S.; Pais, A.A.C.C. Skin Cancer and New Treatment Perspectives: A Review. Cancer Lett. 2015, 357, 8–42. [Google Scholar] [CrossRef]
- Chummun, S.; McLean, N.R. The Management of Malignant Skin Cancers. Surgery 2017, 35, 519–524. [Google Scholar]
- Testori, A.; Rutkowski, P.; Marsden, J.; Bastholt, L.; Chiarion-Sileni, V.; Hauschild, A.; Eggermont, A.M.M. Surgery and Radiotherapy in the Treatment of Cutaneous Melanoma. Ann. Oncol. 2009, 20, vi22–vi29. [Google Scholar] [CrossRef]
- Shtivelman, E.; Davies, M.A.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and Therapeutic Targets in Melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [Green Version]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma Biology and New Targeted Therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, P.J.; Elder, D.E.; Haupt, H.M.; Stern, J.B.; Multhaupt, H.A.B. Tyrosinase Expression in Malignant Melanoma, Desmoplastic Melanoma, and Peripheral Nerve Tumors. Arch. Pathol. Lab. Med. 2003, 127, 1083–1085. [Google Scholar] [CrossRef]
- Bandarchi, B.; Ma, L.; Navab, R.; Seth, A.; Rasty, G. From Melanocyte to Metastatic Malignant Melanoma. Dermatol. Res. Pract. 2010, 2010, 583748. [Google Scholar] [CrossRef]
- Kobayashi, T.; Urabe, K.; Winder, A.; Jiménez-Cervantes, C.; Imokawa, G.; Brewington, T.; Solano, F.; García-Borrön, J.C.; Hearing, V.J. Tyrosinase Related Protein 1 (TRP1) Functions as a DHICA Oxidase in Melanin Biosynthesis. EMBO J. 1994, 13, 5818–5825. [Google Scholar] [CrossRef]
- Ghanem, G.; Fabrice, J. Tyrosinase Related Protein 1 (TYRP1/Gp75) in Human Cutaneous Melanoma. Mol. Oncol. 2011, 5, 150–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, D.N.; Postow, M.A.; Ibrahim, N.; Ludwig, D.L.; Cosaert, J.; Kambhampati, S.R.P.; Tang, S.; Grebennik, D.; Kauh, J.S.W.; Lenz, H.J.; et al. An Open-Label, Dose-Escalation Phase I Study of Anti-TYRP1 Monoclonal Antibody IMC-20D7S for Patients with Relapsed or Refractory Melanoma. Clin. Cancer Res. 2016, 22, 5204–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitrago, E.; Hardré, R.; Haudecoeur, R.; Jamet, H.; Belle, C.; Boumendjel, A.; Bubacco, L.; Réglier, M. Are Human Tyrosinase and Related Proteins Suitable Targets for Melanoma Therapy? Curr. Top. Med. Chem. 2016, 16, 3033–3047. [Google Scholar] [CrossRef]
- Mroz, P.; Huang, Y.; Szokalska, A.; Zhiyentayev, T.; Janjua, S.; Nifli, A.; Sherwood, M.E.; Ruzié, C.; Borbas, K.E.; Fan, D.; et al. Stable Synthetic Bacteriochlorins Overcome the Resistance of Melanoma to Photodynamic Therapy. FASEB J. 2010, 24, 3160–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameyama, K.; Sakai, C.; Kuge, S.; Nishiyama, S.; Tomita, Y.; Ito, S.; Wakamatsu, K.; Hearing, V.J. The Expression of Tyrosinase, Tyrosinase-Related Proteins 1 and 2 (TRP1 and TRP2), the Silver Protein, and a Melanogenic Inhibitor in Human Melanoma Cells of Differing Melanogenic Activities. Pigment. Cell Res. 1995, 8, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Brozyna, A.A.; VanMiddlesworth, L.; Slominski, A.T. Inhibition of Melanogenesis as a Radiation Sensitizer for Melanoma Therapy. Int. J. Cancer 2008, 123, 1448–1456. [Google Scholar] [CrossRef]
- Sharma, K.V.; Bowers, N.; Davids, L.M. Photodynamic Therapy-Induced Killing Is Enhanced in Depigmented Metastatic Melanoma Cells. Cell Biol. Int. 2011, 35, 939–944. [Google Scholar] [CrossRef]
- Yan, J.; Tingey, C.; Lyde, R.; Gorham, T.C.; Choo, D.K.; Muthumani, A.; Myles, D.; Weiner, L.P.; Kraynyak, K.A.; Reuschel, E.L.; et al. Novel and Enhanced Anti-Melanoma DNA Vaccine Targeting the Tyrosinase Protein Inhibits Myeloid-Derived Suppressor Cells and Tumor Growth in a Syngeneic Prophylactic and Therapeutic Murine Model. Cancer Gene Ther. 2014, 21, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.J.; Sittadjody, S.; Thangasamy, T.; Mendoza, E.E.; Limesand, K.H.; Burd, R. Exploiting Tyrosinase Expression and Activity in Melanocytic Tumors: Quercetin and the Central Role of P53. Integr. Cancer Ther. 2011, 10, 328–340. [Google Scholar] [CrossRef]
- Jawaid, S.; Khan, T.H.; Osborn, H.M.I.; Williams, N.A.O. Tyrosinase Activated Melanoma Prodrugs. Anticancer Agents Med. Chem. 2012, 9, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.M.; Sargent, M.V. 3.10-Furans and their Benzo Derivatives: (i) Structure. In Comprehensive Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 1984; Volume 4, pp. 531–597. [Google Scholar] [CrossRef]
- Rymbai, E.M.; Chakraborty, A.; Choudhury, R.; Biplab De, N.V. Review on Chemistry and Therapeutic Activity of the Derivatives of Furan and Oxazole: The Oxygen Containing Heterocycles. Der Pharma Chem. 2019, 11, 20–41. [Google Scholar]
- Rodd, E.H. Chemistry of Carbon Compounds: A Modern Comprehensive Treatise; Elsevier: Amsterdam, The Netherlands, 1971. [Google Scholar]
- Limpricht, H. Ueber das Tetraphenol C4H4O. Ber. Der Dtsch. Chem. Ges. 1870, 3, 90–91. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.H.; Hu, Y.H.; Yang, J.; Liu, T.; Sun, J.; Wang, X.J. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanjare, B.D.; Choi, N.G.; Mahajan, P.G.; Raza, H.; Hassan, M.; Han, Y.; Yu, S.M.; Kim, S.J.; Seo, S.Y.; Lee, K.H. Novel 1,3,4-oxadiazole compounds inhibit the tyrosinase and melanin level: Synthesis, in-vitro, and in-silico studies. Bioorg. Med. Chem. 2021, 1, 116222. [Google Scholar] [CrossRef]
- Mann, T.; Gerwat, W.; Batzer, J.; Eggers, K.; Scherner, C.; Wenck, H.; Stäb, F.; Hearing, V.J.; Röhm, K.H.; Kolbe, L. Inhibition of Human Tyrosinase Requires Molecular Motifs Distinctively Different from Mushroom Tyrosinase. J. Investig. Dermatol. 2018, 138, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Roulier, B.; Pérès, B.; Haudecoeur, R. Advances in the Design of Genuine Human Tyrosinase Inhibitors forTargeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63, 13428–13443. [Google Scholar] [CrossRef]
- Koirala, P.; Seong, S.H.; Zhou, Y.; Shrestha, S.; Jung, H.A.; Choi, J.S. Structure–Activity Relationship of the Tyrosinase Inhibitors Kuwanon G, Mulberrofuran G, and Albanol B from Morus Species: A Kinetics and Molecular Docking Study. Molecules 2018, 23, 1413. [Google Scholar] [CrossRef] [Green Version]
- Okombi, S.; Rival, D.; Bonnet, S.; Mariotte, A.M.; Perrier, E.; Boumendjel, A. Discovery of Benzylidenebenzofuran-3(2H)-One (Aurones) as Inhibitors of Tyrosinase Derived from Human Melanocytes. J. Med. Chem. 2006, 49, 329–333. [Google Scholar] [CrossRef]
- Hu, X.; Wang, M.; Yan, G.R.; Yu, M.H.; Wang, H.Y.; Hou, A.J. 2-Arylbenzofuran and Tyrosinase Inhibitory Constituents of MorusNotabilis. J. Asian Nat. Prod. Res. 2012, 14, 1103–1108. [Google Scholar] [CrossRef]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, E.M. Design, synthesis, antimicrobial evaluation, and laccase catalysis effect of novel benzofuran–oxadiazole and benzofuran–triazole hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Hassan, M.; Ashraf, Z.; Abbas, Q.; Raza, H.; Seo, S.Y. Exploration of Novel Human Tyrosinase Inhibitors by Molecular Modeling, Docking and Simulation Studies. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Zahoor, F.A.; Rasul, A.; Mansha, A.; Ahmad, S.; Raza, Z. Synthesis, hemolytic studies, and in silico modeling of novel acefylline-1,2,4-triazole hybrids as potential anti-cancer agents against MCF-7 and A549. ACS Omega 2021, 6, 11943–11953. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Mei, H.C.; Kuo, I.C.; Lee, T.H.; Chen, Y.H.; Lee, C.K. Characterizing Tyrosinase Modulators from the Roots of Angelica Keiskei Using Tyrosinase Inhibition Assay and UPLC-MS/MS as the Combinatorial Novel Approach. Molecules 2019, 24, 3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Irfan, A.; Zahoor, A.F.; Kamal, S.; Hassan, M.; Kloczkowski, A. Ultrasonic-Assisted Synthesis of Benzofuran Appended Oxadiazole Molecules as Tyrosinase Inhibitors: Mechanistic Approach through Enzyme Inhibition, Molecular Docking, Chemoinformatics, ADMET and Drug-Likeness Studies. Int. J. Mol. Sci. 2022, 23, 10979. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the Synergistic Anticancer Potential of Benzofuran–Oxadiazoles and Triazoles: Improved Ultrasound- and Microwave-Assisted Synthesis, Molecular Docking, Hemolytic, Thrombolytic and Anticancer Evaluation of Furan-Based Molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. 2017, 129, 9944–9947. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological Macromolecular Structures Enabling Research and Education in Fundamental Biology, Biomedicine, Biotechnology and Energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Faisal, S.; Lal Badshah, S.; Kubra, B.; Sharaf, M.; Emwas, A.H.; Jaremko, M.; Abdalla, M. Computational Study of Sars-Cov-2 Rna Dependent Rna Polymerase Allosteric Site Inhibition. Molecules 2022, 27, 223. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes. BIOVIA Discovery Studio Visualizer, Release 2017; Dassault Systèmes: Vélizy-Villacoublay, France, 2023. [Google Scholar]
- Mills, N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. Commercial Price: $1910 for download, $2150 for CD-ROM; Academic Price: $710 for download, $800 for CD-ROM. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- Shinu, P.; Sharma, M.; Gupta, G.L.; Mujwar, S.; Kandeel, M.; Kumar, M.; Nair, A.B.; Goyal, M.; Singh, P.; Attimarad, M.; et al. Computational Design, Synthesis, and Pharmacological Evaluation of Naproxen-Guaiacol Chimera for Gastro-Sparing Anti-Inflammatory Response by Selective COX2 Inhibition. Molecules 2022, 27, 6905. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Sun, L.; Fidan, O. In silico evaluation of food-derived carotenoids against SARS-CoV-2 drug targets: Crocin is a promising dietary supplement candidate for COVID-19. J. Food Biochem. 2022, 46, e14219. [Google Scholar] [CrossRef] [PubMed]
- Fidan, O.; Mujwar, S.; Kciuk, M. Discovery of adapalene and dihydrotachysterol as antiviral agents for the Omicron variant of SARS-CoV-2 through computational drug repurposing. Mol. Divers. 2022, 4, 1–13. [Google Scholar] [CrossRef]
- Kciuk, M.; Mujwar, S.; Szymanowska, A.; Marciniak, B.; Bukowski, K.; Mojzych, M.; Kontek, R. Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. Int. J. Mol. Sci. 2022, 23, 5892. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comb. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber, An Accessory Software Package For Molecular Mechanical Calculations. J. Am. Chem. Soc. 2001, 222, 403. [Google Scholar]
- Saunders, W.R.; Grant, J.; Müller, E.H. Long Range Forces in a Performance Portable Molecular Dynamics Framework. Adv. Parallel Comput. 2018, 22, 37–46. [Google Scholar] [CrossRef]
- Paquet, E.; Viktor, H.L. Molecular Dynamics, Monte Carlo Simulations, and Langevin Dynamics: A Computational Review. BioMed Res. Int. 2015, 2015, 183918. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Pan, D.; Li, J.; Zhang, L.; Shao, X. Application of BerendsenBarostat in Dissipative Particle Dynamics for Nonequilibrium Dynamic Simulation. J. Chem. Phys. 2017, 146, 124108. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 5. Improved Docking Performance Using High Solute Dielectric Constant MM/GBSA and MM/PBSA Rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, Y.; Zhang, H. Computational Investigation of Structural Basis for Enhanced Binding of Isoflavone Analogues with Mitochondrial Aldehyde Dehydrogenase. ACS Omega 2022, 7, 8115–8127. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Affinity in (Kcal/mol) with hTYRP1 | Binding Affinity in (Kcal/mol) with hTYR |
|---|---|---|
| BF4 | −11.50 Kcal/mol | -- |
| BF5 | −11.55 Kcal/mol | −13.30 Kcal/mol |
| BF7 | −11.29 Kcal/mol | −11.19 Kcal/mol |
| BF15 | -- | −11.88 Kcal/mol |
| Kojic acid (Standard) | −8.90 Kcal/mol | −6.62 Kcal/mol |
| Compounds | HIA+ Values | Lipophilicity (iLogP) | CYP450 3A4 Inhibitor/ Substrate | Water Solubility | P-gp Substrate | Carcinogenicity | Renal (OCTs) |
|---|---|---|---|---|---|---|---|
| BF4 | 1.0 | 3.99 | Substrate | Moderately soluble | No | None | Non Inhibitor |
| BF5 | 1.0 | 3.75 | Substrate | Moderately soluble | No | None | Non Inhibitor |
| BF7 | 1.0 | 3.66 | Substrate | Poorly soluble | No | None | Non Inhibitor |
| BF15 | 1.0 | 3.57 | Substrate | Moderately soluble | No | None | Non Inhibitor |
| Compounds | Bioavailability Score | PAINS Alerts | Brenk Alerts | Lipinski Rule | Pfizer Rule | Golden Triangle Rule | TPSA |
|---|---|---|---|---|---|---|---|
| BF4 | 0.55 | None | None | Accepted | Accepted | Accepted | 124.92 Ų |
| BF5 | 0.55 | None | None | Accepted | Accepted | Accepted | 115.69 Ų |
| BF7 | 0.55 | None | None | Accepted | Accepted | Accepted | 106.46 Ų |
| BF15 | 0.55 | None | None | Accepted | Accepted | Accepted | 106.46 Ų |
| Energy Parameter | BF5 + hTYR (SD) | BF7 + hTYR (SD) | BF15 + hTYR (SD) | BF4-hTYRP1 (SD) | BF5 + hTYRP1 (SD) | BF7 + hTYRP1 (SD) |
|---|---|---|---|---|---|---|
| MM-GBSA | ||||||
| Van der Waals | −52.20(4.25) | −50.28 (3.68) | −52.62 (5.72) | −55.01 (5.07) | −48.62 (4.68) | −49.55 (5.21) |
| Electrostatic | −23.89(3.65) | −24.01 (3.56) | −28.21 (2.51) | −31.74 (4.68) | −22.17 (3.66) | −26.30 (4.22) |
| Delta G gas | −76.09(6.84) | −74.29 (5.38) | −80.83 (8.60) | −86.75 (6.81) | −70.79 (7.25) | −75.85 (6.38) |
| Delta G solv | 28.99 (2.38) | 27.68 (7.48) | 24.86 (3.84) | 25.08 (4.29) | 28.64 (4.21) | 23.59 (3.56) |
| Delta Total | −47.1 (5.52) | −46.61 (6.87) | −55.97 (5.69) | −61.67 (6.31) | −42.15 (6.11) | −52.26 (3.98) |
| MM-PBSA | ||||||
| Van der Waals | −52.20(4.25) | −50.28 (3.68) | −52.62 (5.72) | −55.01 (5.07) | −48.62 (4.68) | −49.55 (5.21) |
| Electrostatic | −23.89(4.25) | −24.01 (3.56) | −28.21 (2.51) | −31.74 (4.68) | −22.17 (3.66) | −26.30 (4.22) |
| Delta G gas | −76.09(6.84) | −74.29 (5.38) | −80.83 (8.60) | −86.75 (6.81) | −70.79 (7.25) | −75.85 (6.38) |
| Delta G solv | 25.60 (1.68) | 25.07 (2.58) | 28.61 (3.64) | 24.01 (3.29) | 27.64 (4.62) | 24.50 (3.33) |
| Delta Total | −50.49(4.68) | −49.22 (5.28) | −52.22 (4.22) | −62.74 (2.67) | −43.15 (3.08) | −51.35 (2.67) |
| Compounds | Structures of Furan-1,3,4-Oxadiazssoles |
|---|---|
| BF1 |  |
| BF2 |  |
| BF3 |  |
| BF4 |  |
| BF5 |  |
| BF6 |  |
| BF7 |  |
| BF8 |  |
| BF9 |  |
| BF10 |  |
| BF11 |  |
| BF12 |  |
| BF13 |  |
| BF14 |  |
| BF15 |  |
| BF16 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, A.; Faisal, S.; Ahmad, S.; Al-Hussain, S.A.; Javed, S.; Zahoor, A.F.; Parveen, B.; Zaki, M.E.A. Structure-Based Virtual Screening of Furan-1,3,4-Oxadiazole Tethered N-phenylacetamide Derivatives as Novel Class of hTYR and hTYRP1 Inhibitors. Pharmaceuticals 2023, 16, 344. https://doi.org/10.3390/ph16030344
Irfan A, Faisal S, Ahmad S, Al-Hussain SA, Javed S, Zahoor AF, Parveen B, Zaki MEA. Structure-Based Virtual Screening of Furan-1,3,4-Oxadiazole Tethered N-phenylacetamide Derivatives as Novel Class of hTYR and hTYRP1 Inhibitors. Pharmaceuticals. 2023; 16(3):344. https://doi.org/10.3390/ph16030344
Chicago/Turabian StyleIrfan, Ali, Shah Faisal, Sajjad Ahmad, Sami A. Al-Hussain, Sadia Javed, Ameer Fawad Zahoor, Bushra Parveen, and Magdi E. A. Zaki. 2023. "Structure-Based Virtual Screening of Furan-1,3,4-Oxadiazole Tethered N-phenylacetamide Derivatives as Novel Class of hTYR and hTYRP1 Inhibitors" Pharmaceuticals 16, no. 3: 344. https://doi.org/10.3390/ph16030344