
Therapeutic Targeting of TLR4 for Inflammation, Infection, and Cancer: A Perspective for Disaccharide Lipid A Mimetics

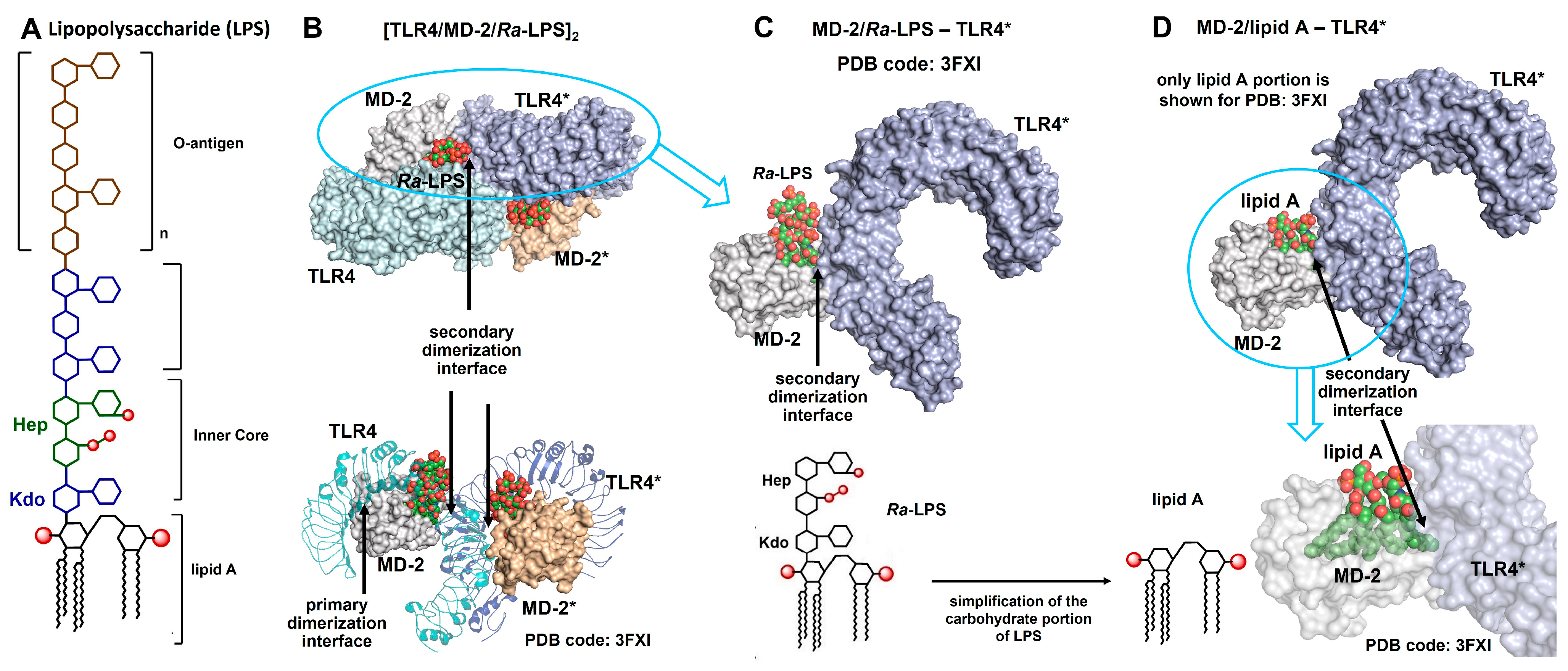{kind=link}
{kind=link}
{kind=link}
{kind=link}
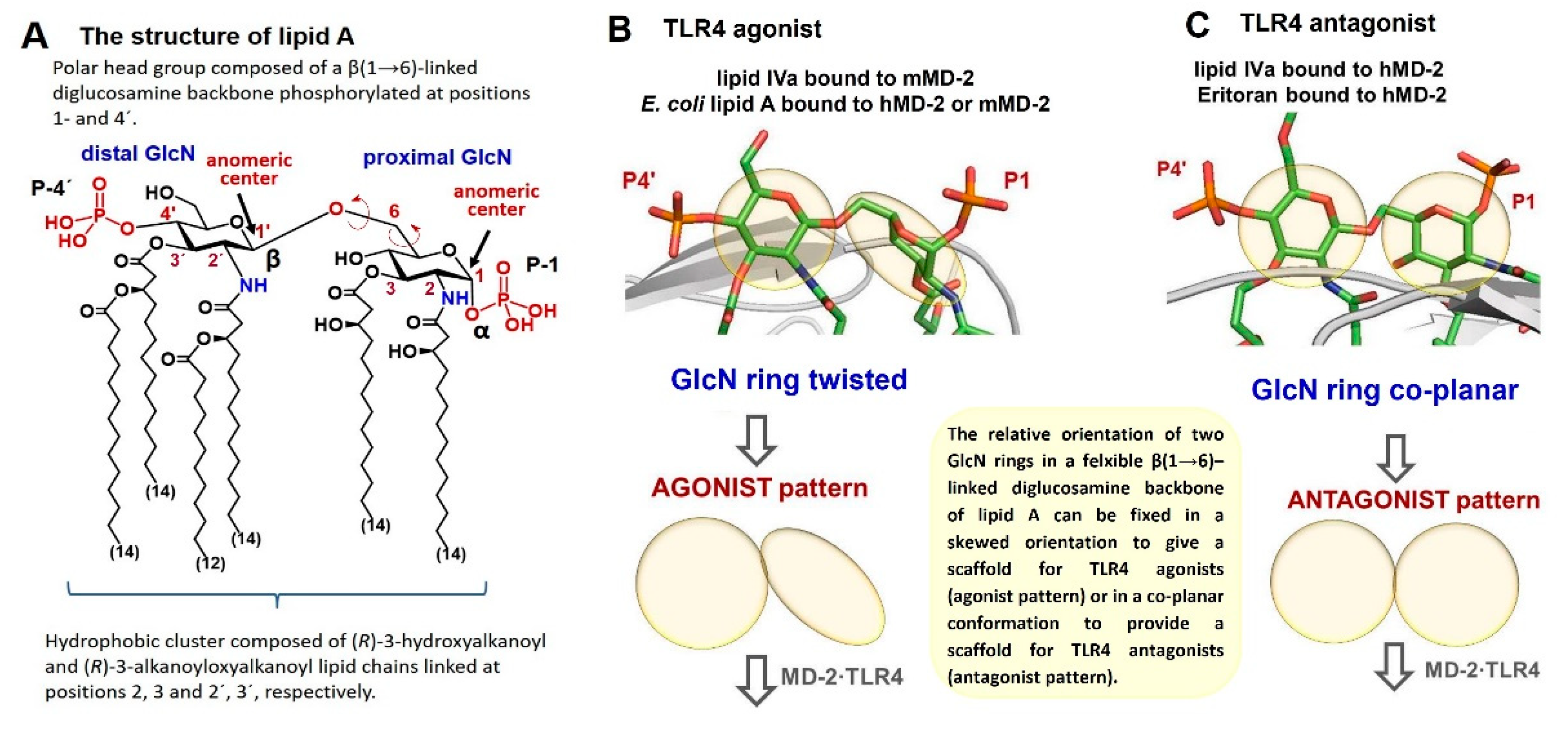{kind=link}
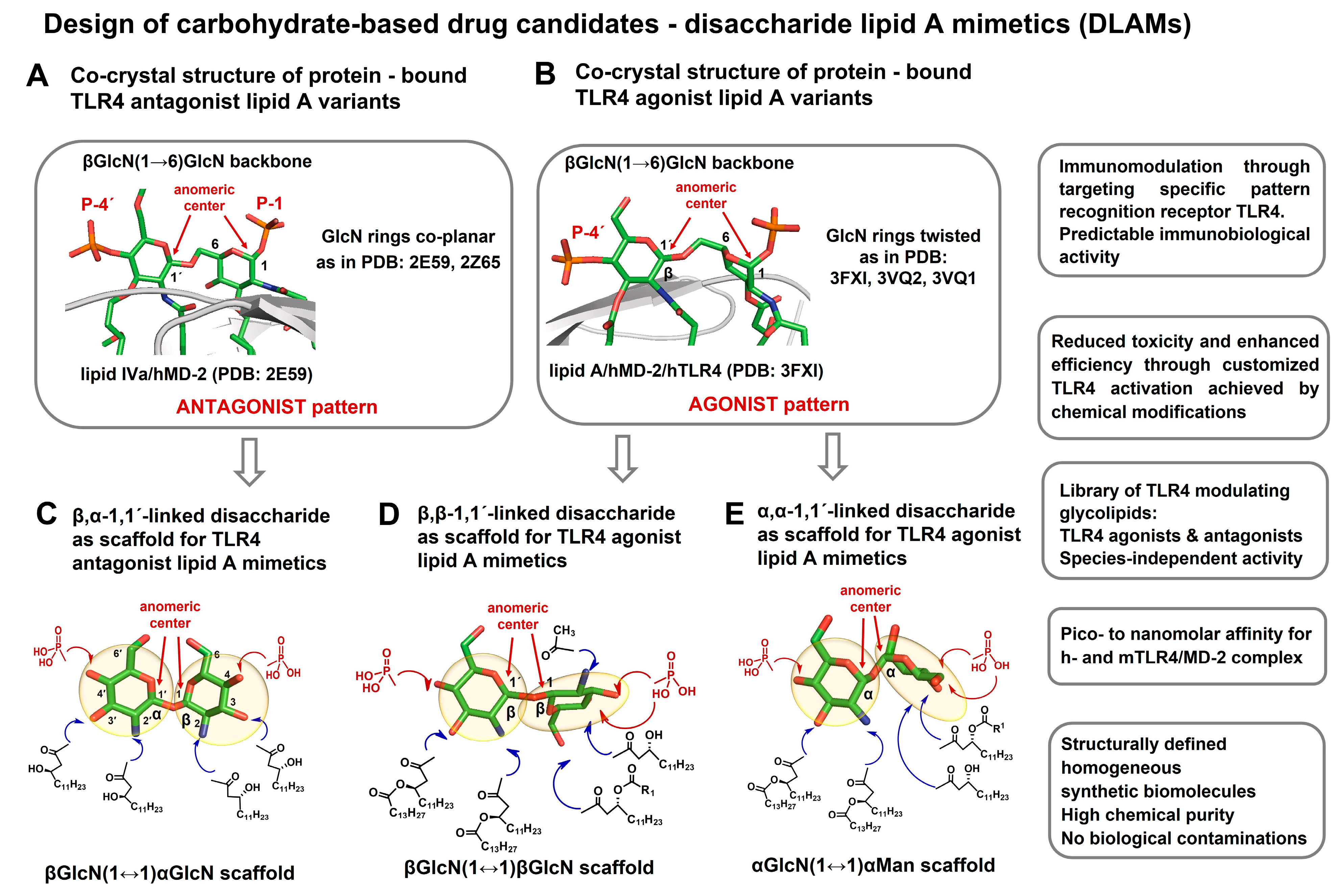{kind=link}
{kind=link}
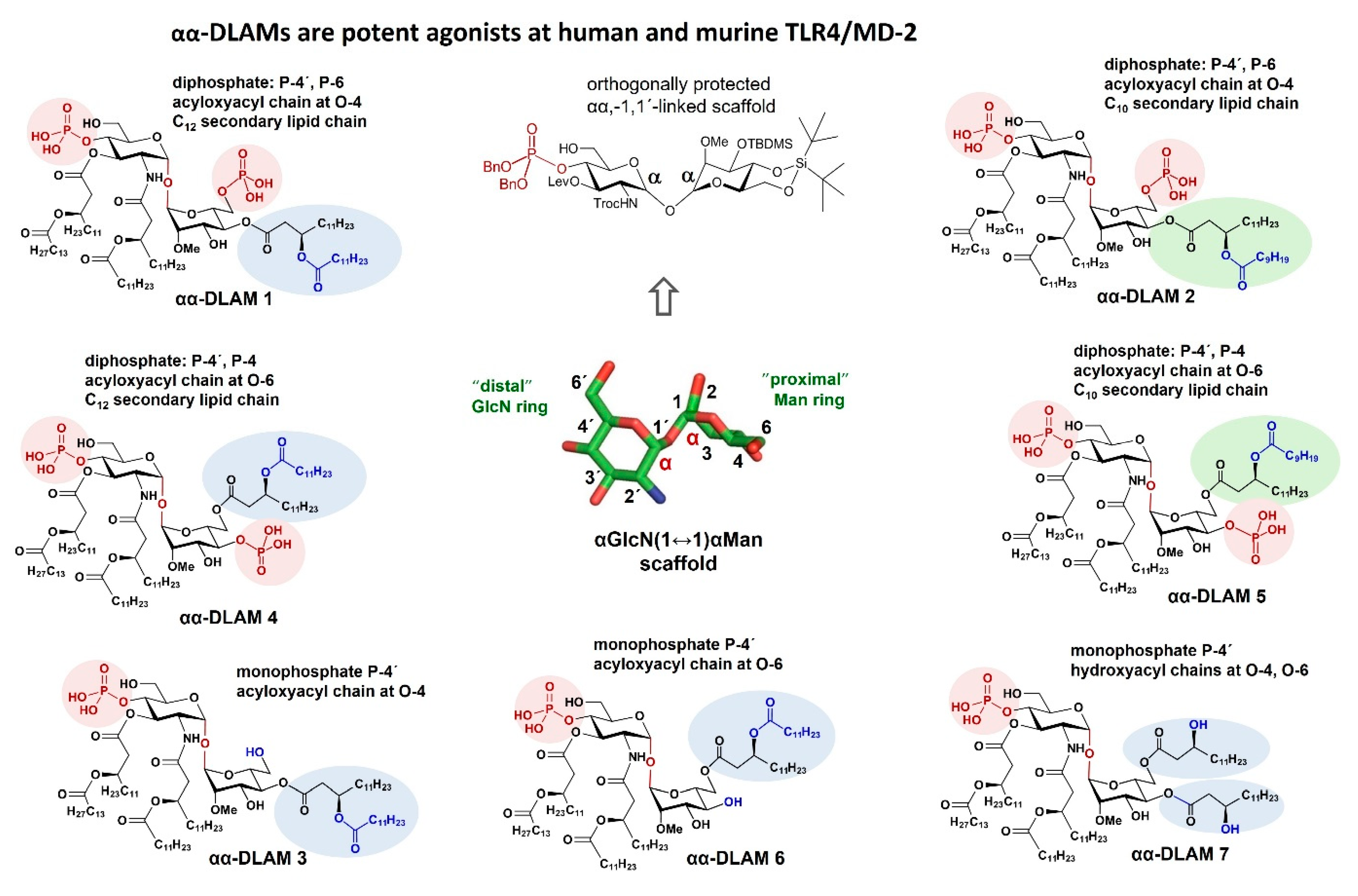{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Species-Specific Activity of LPS
3. Therapeutic Potential of TLR4 Antagonists
4. Therapeutic Potential of TLR4 Agonists
5. Challenges in Development of Synthetic TLR4 Ligands
6. Co-crystal-Structure-Based Design of Disaccharide Lipid A Mimetics (DLAMs)
7. TLR4 Antagonists Based on a βGlcN(1↔1)αGlcN Scaffold: Nanomolar Potency and Species-Independent Activity of βα-DLAMs
8. TLR4 Agonists Based on αGlcN(1↔1)αMan and βGlcN(1↔1)βGlcN Scaffolds: Tailored Modulation of TLR4-Mediated Pro-inflammatory Signaling by αα-DLAMs and ββ-DLAMs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALI | acute lung injury |
| BEAS-2B | human bronchial epithelial cell line |
| Calu-3 | human lung adenocarcinoma cell line |
| CD14 | cluster of differentiation 14 |
| COPD | chronic obstructive pulmonary disease |
| DAMP | danger-associated molecular pattern |
| DCs | dendritic cells |
| DLAM | disaccharide lipid A mimetic |
| GPI | glycosyl phosphatidyl inositol |
| HEK293 | human embryonic kidney 293 cells |
| HMGB1 | high-mobility group box 1 protein |
| IFN-β | interferon-β |
| IL-1β | interleukin-1β |
| IRF3 | interferon regulatory factor 3 |
| Kdo | 3-deoxy-D-manno-octulosonic acid |
| LBP | LPS-binding protein |
| LPS | lipopolysaccharide |
| LRR | leucine-rich repeat |
| MCP-1 | monocyte chemoattractant protein-1 |
| MD-2 | myeloid differentiation factor 2 |
| MNC | mononuclear cells |
| MPLA | monophosphoryl lipid A |
| MPL® | clinical-grade MPL® adjuvant (GlaxoSmithKline) |
| MyD88 | myeloid differentiation primary response 88 |
| NF-ĸB | nuclear factor kappa B |
| PAMP | pathogen-associated molecular pattern |
| PDB | Protein Data Bank |
| PRR | pattern recognition receptor |
| SEAP | secreted embryonic alkaline phosphatase |
| SIRS | severe inflammatory response syndrome |
| THP-1 | human acute monocytic leukemia cells |
| TIR | Toll-interleukin-1 receptor |
| TIRAP | TIR-domain-containing adaptor protein |
| TLR4 | Toll-like receptor 4 |
| TNF-α | tumor necrosis factor-α |
| TRAF | TNF-receptor-associated factor |
| TRAM | TRIF-related adaptor molecule |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
References
- Pfeiffer, R. Untersuchungen über das Choleragift. Z. Für Hyg. Und Infekt. 1892, 11, 393–412. [Google Scholar] [CrossRef]
- Beutler, B.; Rietschel, E.T. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F. Bacterial endotoxin: Molecular relationships of structure to activity and function. FASEB J. 1994, 8, 217–225. [Google Scholar] [CrossRef]
- Seydel, U.; Schromm, A.B.; Blunck, R.; Brandenburg, K. Chemical structure, molecular conformation, and bioactivity of endotoxins. Chem. Immunol. 2000, 74, 5–24. [Google Scholar]
- Holst, O. Structure of the Lipopolysaccharide Core Region. In Bacterial Lipopolysaccharides; Knirel, Y.A., Valvano, M.A., Eds.; Springer: Vienna, Austria, 2011; pp. 21–39. [Google Scholar]
- Knirel, Y. Structure of O-Antigens. In Bacterial Lipopolysaccharides; Knirel, Y.A., Valvano, M.A., Eds.; Springer: Vienna, Austria, 2011; pp. 41–115. [Google Scholar]
- Galanos, C.; Lüderitz, O.; Rietschel, E.T.; Westphal, O.; Brade, H.; Brade, L.; Freudenberg, M.A.; Schade, U.F.; Imoto, M.; Yoshimura, H.; et al. Synthetic and Escheria coli free lipid A express identical endotoxic activities. Eur. J. Biochem. 1985, 148, 1–5. [Google Scholar] [CrossRef]
- Schletter, J.; Heine, H.; Ulmer, A.J.; Rietschel, E.T. Molecular mechanisms of endotoxin activity. Arch. Microbiol. 1995, 164, 383–389. [Google Scholar] [CrossRef]
- Trent, M.S.; Stead, C.M.; Tran, A.X.; Hankins, J.V. Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 2006, 12, 205–223. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Kirikae, T.; Schade, F.U.; Zähringer, U.; Kirikae, F.; Brade, H.; Kusumoto, S.; Kusama, T.; Rietschel, E.T. The significance of the hydrophilic backbone and the hydrophobic fatty acid regions of lipid a for macrophage binding and cytokine induction. FEMS Immunol. Med. Microbiol. 1994, 8, 13–26. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kobayashi, M.; Saitoh, S.I.; Tanimura, N.; Takahashi, K.; Kawasaki, K.; Nishijima, M.; Fujimoto, Y.; Fukase, K.; Akashi-Takamura, S.; Miyake, K. Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J. Immunol. 2006, 176, 6211–6218. [Google Scholar] [CrossRef]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14 and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Delude, R.L.; Savedra, R.; Zhao, H.; Thieringer, R.; Yamamoto, S.; Fenton, M.J.; Golenbock, D.T. CD14 enhances cellular responses to endotoxin without imparting ligand-specific recognition. Proc. Natl. Acad. Sci. USA 1995, 92, 9288–9292. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef] [Green Version]
- Resman, N.; Vasl, J.; Oblak, A.; Pristovsek, P.; Gioannini, T.L.; Weiss, J.P.; Jerala, R. Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 2009, 284, 15052–15060. [Google Scholar] [CrossRef] [Green Version]
- Krüger, C.L.; Zeuner, M.-T.; Cottrell, G.S.; Widera, D.; Heilemann, M. Quantitative single-molecule imaging of TLR4 reveals ligand-specific receptor dimerization. Sci. Signal. 2017, 10, eaan1308. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Muzio, M.; Ni, J.; Feng, P.; Dixit, V.M. IRAK (Pelle) Family Member IRAK-2 and MyD88 as Proximal Mediators of IL-1 Signaling. Science 1997, 278, 1612–1615. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Seong, S.-Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.C.; Wang, H.; et al. Identification of oxidative stress and Toll-like Receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundbäck, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1–dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.R.; Yamasaki, K.; Radek, K.A.; Nardo, A.D.; Goodarzi, H.; Golenbock, D.; Beutler, B.; Gallo, R.L. Recognition of Hyaluronan Released in Sterile Injury Involves a Unique Receptor Complex Dependent on Toll-like Receptor 4, CD44 and MD-2. J. Biol. Chem. 2007, 282, 18265–18275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, Y.; Watari, M.; Jerud, E.S.; Young, D.W.; Ishizaka, S.T.; Rose, J.; Chow, J.C.; Strauss, J.F. The Extra Domain A of Fibronectin Activates Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 10229–10233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Lee, B.L.; Stowe, I.B.; Gupta, A.; Kornfeld, O.S.; Roose-Girma, M.; Anderson, K.; Warming, S.; Zhang, J.; Lee, W.P.; Kayagaki, N. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J. Exp. Med. 2018, 215, 2279–2288. [Google Scholar] [CrossRef]
- Casson, C.N.; Yu, J.; Reyes, V.M.; Taschuk, F.O.; Yadav, A.; Copenhaver, A.M.; Nguyen, H.T.; Collman, R.G.; Shin, S. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 2015, 112, 6688–6693. [Google Scholar] [CrossRef] [Green Version]
- Zamyatina, A.; Heine, H. Lipopolysaccharide Recognition in the Crossroads of TLR4 and Caspase-4/11 Mediated Inflammatory Pathways. Front. Immunol. 2020, 11, 585146. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszýnski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, S.; Nagai, Y.; Ogata, H.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Kawasaki, K.; Nishijima, M.; Hayashi, S.; Kimoto, M.; et al. Human MD-2 confers on mouse Toll-like receptor 4 species-specific lipopolysaccharide recognition. Int. Immunol. 2001, 13, 1595–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Drolet, J.R.; Monks, B.G.; Golenbock, D.T. MD-2 residues Tyrosine 42, Arginine 69, Aspartic acid 122, and Leucine 125 provide species specificity for lipid IVa. J. Biol. Chem. 2010, 285, 27935–27943. [Google Scholar] [CrossRef] [Green Version]
- Galanos, C.; Lehmann, V.; Lüderitz, O.; Rietschel, E.T.; Westphal, O.; Brade, H.; Brade, L.; Freudenberg, M.A.; Hansen-Hagge, T.; Lüderitz, T.; et al. Endotoxic properties of chemically synthesized lipid A part structures. Comparison of synthetic lipid A precursor and synthetic analogues with biosynthetic lipid A precursor and free lipid A. Eur. J. Biochem. 1984, 140, 221–227. [Google Scholar] [CrossRef]
- Feist, W.; Ulmer, A.J.; Musehold, J.; Brade, H.; Kusumoto, S.; Flad, H.D. Induction of tumor necrosis factor-alpha release by lipopolysaccharide and defined lipopolysaccharide partial structures. Immunobiology 1989, 179, 293–307. [Google Scholar] [CrossRef]
- Loppnow, H.; Brade, L.; Brade, H.; Rietschel, E.T.; Kusumoto, S.; Shiba, T.; Flad, H.D. Induction of human interleukin 1 by bacterial and synthetic lipid A. Eur. J. Immunol. 1986, 16, 1263–1267. [Google Scholar] [CrossRef]
- Kovach, N.L.; Yee, E.; Munford, R.S.; Raetz, C.R.; Harlan, J.M. Lipid IVA inhibits synthesis and release of tumor necrosis factor induced by lipopolysaccharide in human whole blood ex vivo. J. Exp. Med. 1990, 172, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Ohto, U.; Fukase, K.; Miyake, K.; Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 316, 1632–1634. [Google Scholar] [CrossRef]
- Steeghs, L.; Keestra, A.M.; van Mourik, A.; Uronen-Hansson, H.; van der Ley, P.; Callard, R.; Klein, N.; van Putten, J.P.M. Differential Activation of Human and Mouse Toll-Like Receptor 4 by the Adjuvant Candidate LpxL1 of Neisseria meningitidis. Infect. Immun. 2008, 76, 3801–3807. [Google Scholar] [CrossRef]
- Lien, E.; Means, T.K.; Heine, H.; Yoshimura, A.; Kusumoto, S.; Fukase, K.; Fenton, M.J.; Oikawa, M.; Qureshi, N.; Monks, B.; et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Investig. 2000, 105, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Irvine, K.L.; Gangloff, M.; Walsh, C.M.; Spring, D.R.; Gay, N.J.; Bryant, C.E. Identification of key residues that confer Rhodobacter sphaeroides LPS activity at horse TLR4/MD-2. PLoS ONE 2014, 9, e98776. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, K.L.; Vandenplas, M.L.; Barton, M.H.; Bryant, C.E.; Moore, J.N. The equine TLR4/MD-2 complex mediates recognition of lipopolysaccharide from Rhodobacter sphaeroides as an agonist. J. Endotoxin Res. 2007, 13, 235–242. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M.; Laterre, P.-F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.-P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of Eritoran, an Antagonist of MD2-TLR4, on Mortality in Patients With Severe Sepsis: The ACCESS Randomized Trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Bridges, C.B.; Cox, N.J.; Fukuda, K. Influenza-Associated Hospitalizations in the United States. JAMA 2004, 292, 1333–1340. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Shirey, K.A.; Blanco, J.C.G.; Vogel, S.N. Targeting TLR4 Signaling to Blunt Viral-Mediated Acute Lung Injury. Front. Immunol. 2021, 12, 705080. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory Syncytial Virus Fusion Protein-Induced Toll-Like Receptor 4 (TLR4) Signaling Is Inhibited by the TLR4 Antagonists Rhodobacter sphaeroides Lipopolysaccharide and Eritoran (E5564) and Requires Direct Interaction with MD-2. mBio 2012, 3, e00218-12. [Google Scholar] [CrossRef] [Green Version]
- Sabouri, A.H.; Marcondes, M.C.G.; Flynn, C.; Berger, M.; Xiao, N.; Fox, H.S.; Sarvetnick, N.E. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 2014, 1574, 84–95. [Google Scholar] [CrossRef]
- Younan, P.; Ramanathan, P.; Graber, J.; Gusovsky, F.; Bukreyev, A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice from Lethal Filovirus Challenge. mBio 2017, 8, e00226-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olejnik, J.; Hume, A.J.; Mühlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 2018, 14, e1007390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinya, K.; Ito, M.; Makino, A.; Tanaka, M.; Miyake, K.; Eisfeld, A.J.; Kawaoka, Y. The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J. Virol. 2012, 86, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35, 1146183. [Google Scholar] [CrossRef]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Stromdahl, A.C.; Cerps, S.; Uller, L.; Kjellstrom, S.; Bond, P.J.; Schmidtchen, A.A. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 2021, 12, 916–932. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021, 2021, 818–820. [Google Scholar] [CrossRef]
- Brandão, S.C.S.; Ramos, J.d.O.X.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2021, 58, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Kruglikov, I.L.; Scherer, P.E. Preexisting and inducible endotoxemia as crucial contributors to the severity of COVID-19 outcomes. PLoS Pathog. 2021, 17, e1009306. [Google Scholar] [CrossRef]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced Cytokine Production in Human Monocytes and Macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef]
- Wang, H.; Reddy, S.T.; Fogelman, A.M. The role of gut-derived oxidized lipids and bacterial lipopolysaccharide in systemic inflammation and atherosclerosis. Curr. Opin. Lipidol. 2022, 33, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.S.; Doherty, T.M.; Shah, P.K.; Arditi, M. TLR Signaling: An Emerging Bridge from Innate Immunity to Atherogenesis. J. Immunol. 2004, 173, 5901–5907. [Google Scholar] [CrossRef] [Green Version]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [Green Version]
- Doherty, T.M.; Shah, P.K.; Arditi, M.; Stoll, L.L.; Denning, G.M.; Weintraub, N.L. Lipopolysaccharide, Toll-Like Receptors, and the Immune Contribution to Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, e38–e39. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, E.S.; Wondrak, T.G. TLR4-directed Molecular Strategies Targeting Skin Photodamage and Carcinogenesis. Curr. Med. Chem. 2018, 25, 5487–5502. [Google Scholar] [CrossRef]
- Blohm-Mangone, K.; Burkett, N.B.; Tahsin, S.; Myrdal, P.B.; Aodah, A.; Ho, B.; Janda, J.; McComas, M.; Saboda, K.; Roe, D.J.; et al. Pharmacological TLR4 Antagonism Using Topical Resatorvid Blocks Solar UV-Induced Skin Tumorigenesis in SKH-1 Mice. Cancer Prev. Res. (Phila.) 2018, 11, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, W.; Gao, R.; Su, Y.; Umehara, H.; Dong, L.; Gong, F. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology 2013, 52, 1739–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, I.; Behl, T.; Bungau, S.; Kumar, A.; Mehta, V.; Setia, D.; Uddin, M.S.; Zengin, G.; Aleya, L.; Arora, S. Exploring the therapeutic promise of targeting HMGB1 in rheumatoid arthritis. Life Sci. 2020, 258, 118164. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Joosten, L.A.B.; Roelofs, M.F.; Radstake, T.R.D.J.; Matera, G.; Popa, C.; Van der Meer, J.W.M.; Netea, M.G.; van den Berg, W.B. Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Villalvilla, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G. TLR4 signalling in osteoarthritis—Finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 2015, 11, 159–170. [Google Scholar] [CrossRef]
- Zuo, L.; Lucas, K.; Fortuna, C.A.; Chuang, C.C.; Best, T.M. Molecular Regulation of Toll-like Receptors in Asthma and COPD. Front. Physiol. 2015, 6, 312. [Google Scholar] [CrossRef] [Green Version]
- Bolourani, S.; Brenner, M.; Wang, P. The interplay of DAMPs, TLR4, and proinflammatory cytokines in pulmonary fibrosis. J. Mol. Med. 2021, 99, 1373–1384. [Google Scholar] [CrossRef]
- Bezemer, G.F.G.; Sagar, S.; van Bergenhenegouwen, J.; Georgiou, N.A.; Garssen, J.; Kraneveld, A.D.; Folkerts, G. Dual Role of Toll-Like Receptors in Asthma and Chronic Obstructive Pulmonary Disease. Pharmacol. Rev. 2012, 64, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Speletas, M.; Merentiti, V.; Kostikas, K.; Liadaki, K.; Minas, M.; Gourgoulianis, K.; Germenis, A.E. Association of TLR4-T399I Polymorphism with Chronic Obstructive Pulmonary Disease in Smokers. Clin. Develop. Immunol. 2009, 2009, 260286. [Google Scholar] [CrossRef] [Green Version]
- Sabroe, I.; Whyte, M.K.; Wilson, A.G.; Dower, S.K.; Hubbard, R.; Hall, I. Toll-like receptor (TLR) 4 polymorphisms and COPD. Thorax 2004, 59, 81. [Google Scholar]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016, 7, e2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, L.; Sluijter, J.P.G.; Keulen, J.K.v.; Hoefer, I.E.; Nederhoff, M.G.J.; Goumans, M.-J.; Doevendans, P.A.; Echteld, C.J.A.v.; Joles, J.A.; Quax, P.H.; et al. Toll-Like Receptor 4 Mediates Maladaptive Left Ventricular Remodeling and Impairs Cardiac Function After Myocardial Infarction. Circ. Res. 2008, 102, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, C.; Gao, M.; Cao, X.; Ha, T.; Kalbfleisch, J.H.; Williams, D.L.; Li, C.; Kao, R.L. Toll-Like Receptor 4 plays a central role in cardiac dysfunction during trauma hemorrhage shock. Shock 2014, 42, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun-Fahrländer, C.; Riedler, J.; Herz, U.; Eder, W.; Waser, M.; Grize, L.; Maisch, S.; Carr, D.; Gerlach, F.; Bufe, A.; et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 2002, 347, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedler, J.; Braun-Fahrländer, C.; Eder, W.; Schreuer, M.; Waser, M.; Maisch, S.; Carr, D.; Schierl, R.; Nowak, D.; von Mutius, E. Exposure to farming in early life and development of asthma and allergy: A cross-sectional survey. Lancet 2001, 358, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P. Hay fever, hygiene, and household size. Brit. Med. J. 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [Green Version]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-enhanced, Toll-like Receptor 4–dependent T Helper Cell Type 2 Responses to Inhaled Antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef] [Green Version]
- Thorne, P.S. Environmental endotoxin exposure and asthma. J. Allergy Clin. Immunol. 2021, 148, 61–63. [Google Scholar] [CrossRef]
- Trompette, A.; Divanovic, S.; Visintin, A.; Blanchard, C.; Hegde, R.S.; Madan, R.; Thorne, P.S.; Wills-Karp, M.; Gioannini, T.L.; Weiss, J.P.; et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 2009, 457, 585–588. [Google Scholar] [CrossRef] [Green Version]
- Thomas, W.R.; Hales, B.J.; Smith, W.-A. Structural biology of allergens. Curr. Allergy Asthma Rep. 2005, 5, 388–393. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.N.; Yang, L.F.; Yan, Y.; Cai, L.M.; Li, Y.T.; Qiao, Y.K.; Chen, Z.G. TLR4 antagonist suppresses airway remodeling in asthma by inhibiting the T-helper 2 response. Exp. Ther. Med. 2017, 14, 2911–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalaby, K.H.; Al Heialy, S.; Tsuchiya, K.; Farahnak, S.; McGovern, T.K.; Risse, P.-A.; Suh, W.-K.; Qureshi, S.T.; Martin, J.G. The TLR4–TRIF pathway can protect against the development of experimental allergic asthma. Immunology 2017, 152, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Grönlund, H.; Brooks, H.; Hopkins, L.; Waggoner, L.; Murton, B.; Gangloff, M.; Opaleye, O.; Chilvers, E.R.; Fitzgerald, K.; et al. Allergens as Immunomodulatory Proteins: The Cat Dander Protein Fel d 1 Enhances TLR Activation by Lipid Ligands. J. Immunol. 2013, 191, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; García-Rodríguez, C.; Villalobos, C.; Núñez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front. Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Grimaldi, M.P.; Chiappelli, M.; Licastro, F.; Castiglia, L.; Listì, F.; Vasto, S.; Lio, D.; Caruso, C.; Candore, G. Association between the polymorphisms of TLR4 and CD14 genes and Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 2672–2677. [Google Scholar] [CrossRef]
- Michaud, J.P.; Hallé, M.; Lampron, A.; Thériault, P.; Préfontaine, P.; Filali, M.; Tribout-Jover, P.; Lanteigne, A.M.; Jodoin, R.; Cluff, C.; et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. USA 2013, 110, 1941–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Liu, Y.; Hao, W.; Decker, Y.; Tomic, I.; Menger, M.D.; Liu, C.; Fassbender, K. Stimulation of TLR4 Attenuates Alzheimer’s Disease–Related Symptoms and Pathology in Tau-Transgenic Mice. J. Immunol. 2016, 197, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Morefield, G.L.; Hawkins, L.D.; Ishizaka, S.T.; Kissner, T.L.; Ulrich, R.G. Synthetic Toll-Like Receptor-4 Agonist Enhances Vaccine Efficacy in an Experimental Model of Toxic-Shock Syndrome. Clin. Vaccine Immunol. 2007, 14, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Bazin-Lee, H.; Evans, J.T.; Casella, C.R.; Mitchell, T.C. MPL Adjuvant Contains Competitive Antagonists of Human TLR4. Front. Immunol. 2020, 11, 577823. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Segovia, J.A.; Chang, T.H.; Morris, I.R.; Berton, M.T.; Tessier, P.A.; Tardif, M.R.; Cesaro, A.; Bose, S. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: Role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 2014, 10, e1003848. [Google Scholar] [CrossRef] [PubMed]
- Shinya, K.; Okamura, T.; Sueta, S.; Kasai, N.; Tanaka, M.; Ginting, T.E.; Makino, A.; Eisfeld, A.J.; Kawaoka, Y. Toll-like receptor pre-stimulation protects mice against lethal infection with highly pathogenic influenza viruses. Virol. J. 2011, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeuner, M.-T.; Krüger, C.L.; Volk, K.; Bieback, K.; Cottrell, G.S.; Heilemann, M.; Widera, D. Biased signalling is an essential feature of TLR4 in glioma cells. Biochim. Biophys. Acta (BBA) 2016, 1863, 3084–3095. [Google Scholar] [CrossRef]
- Ou, T.; Lilly, M.; Jiang, W. The Pathologic Role of Toll-Like Receptor 4 in Prostate Cancer. Front. Immunol. 2018, 9, 1188. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, H.; Fu, Y.; Zhu, Y.; Kong, L.; Chen, L.; Zhao, F.; Yu, L.; Chen, X. TLR4/MyD88 signaling determines the metastatic potential of breast cancer cells. Mol. Med. Rep. 2018, 18, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Kelsh, R.M.; McKeown-Longo, P.J. Topographical changes in extracellular matrix: Activation of TLR4 signaling and solid tumor progression. Trends Cancer Res. 2013, 9, 1–13. [Google Scholar]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-Like Receptor 4 Prompts Human Breast Cancer Cells Invasiveness via Lipopolysaccharide Stimulation and Is Overexpressed in Patients with Lymph Node Metastasis. PLoS ONE 2014, 9, e109980. [Google Scholar] [CrossRef] [PubMed]
- Haricharan, S.; Brown, P. TLR4 has a TP53-dependent dual role in regulating breast cancer cell growth. Proc. Natl. Acad. Sci. USA 2015, 112, E3216–E3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahiro, K.; Matsumoto, Y.; Yamada, H.; Endo, M.; Setsu, N.; Fujiwara, T.; Nakagawa, M.; Kimura, A.; Shimada, E.; Okada, S.; et al. Activation of TLR4 signaling inhibits progression of osteosarcoma by stimulating CD8-positive cytotoxic lymphocytes. Cancer Immunol. Immunother. 2020, 69, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Ang, B.; Xu, X.; Huang, X.; Wu, Y.; Sun, Y.; Wang, W.; Li, N.; Cao, X.; Wan, T. TLR4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by DAMPs released from chemically stressed cancer cells. Cell. Mol. Immunol. 2014, 11, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Mu, R.; Wang, Z.; Xing, P.; Zhang, J.; Dong, L.; Wang, C. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat. Commun. 2019, 10, 2272. [Google Scholar] [CrossRef] [Green Version]
- Matzner, P.; Sorski, L.; Shaashua, L.; Elbaz, E.; Lavon, H.; Melamed, R.; Rosenne, E.; Gotlieb, N.; Benbenishty, A.; Reed, S.G.; et al. Perioperative treatment with the new synthetic TLR4 agonist GLA-SE reduces cancer metastasis without adverse effects. Int. J. Cancer 2016, 138, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Shetab Boushehri, M.A.; Lamprecht, A. TLR4-Based Immunotherapeutics in Cancer: A Review of the Achievements and Shortcomings. Mol. Pharm. 2018, 15, 4777–4800. [Google Scholar] [CrossRef]
- Reisser, D.; Jeannin, J.-F. Lipid A in cancer therapies preclinical results. In Lipid A in Cancer Therapy; Jeannin, J.-F., Ed.; Springer New York: New York, NY, USA, 2010; pp. 101–110. [Google Scholar] [CrossRef]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. OncoImmunology 2018, 7, e1526250. [Google Scholar] [CrossRef]
- Andreani, V.; Gatti, G.; Simonella, L.; Rivero, V.; Maccioni, M. Activation of TLR4 on Tumor Cells In vitro Inhibits Subsequent Tumor Growth In vivo. Cancer Res. 2007, 67, 10519–10527. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C.; Glover, D.W. Spontaneous regression: A hidden treasure buried in time. Med. Hypotheses 2002, 58, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Starnes, C.O. Coley’s toxins in perspective. Nature 1992, 357, 11–12. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Nauts, H.C.; McLaren, J.R. Coley Toxins—The First Century. In Consensus on Hyperthermia for the 1990s: Clinical Practice in Cancer Treatment; Bicher, H.I., McLaren, J.R., Pigliucci, G.M., Eds.; Springer USA: Boston, MA, USA, 1990; pp. 483–500. [Google Scholar]
- Huang, L.; Xu, H.; Peng, G. TLR-mediated metabolic reprogramming in the tumor microenvironment: Potential novel strategies for cancer immunotherapy. Cell Mol. Immunol. 2018, 15, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Cen, X.; Liu, S.; Cheng, K. The Role of Toll-Like Receptor in Inflammation and Tumor Immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [Green Version]
- Pandey, N.; Chauhan, A.; Jain, N. TLR4 Polymorphisms and Expression in Solid Cancers. Mol. Diagn. Ther. 2018, 22, 683–702. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Lenz, L.L.; Harris, R.A. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016, 76, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Øynebråten, I.; Corthay, A. Toll-Like Receptor Ligands and Interferon-γ Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Betancur, A.; Chen, M.; Ter Meulen, J.H. Toll-Like Receptor 4 Expression on Lymphoma Cells Is Critical for Therapeutic Activity of Intratumoral Therapy With Synthetic TLR4 Agonist Glucopyranosyl Lipid, A. Front. Oncol. 2020, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, C.; Qin, X.; Xia, J.; Wu, A. LPS alters the immuno-phenotype of glioma and glioma stem-like cells and induces in vivo antitumor immunity via TLR4. J. Exp. Clin. Cancer Res. 2017, 36, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albershardt, T.C.; Leleux, J.; Parsons, A.J.; Krull, J.E.; Berglund, P.; ter Meulen, J. Intratumoral immune activation with TLR4 agonist synergizes with effector T cells to eradicate established murine tumors. NPJ Vaccines 2020, 5, 50. [Google Scholar] [CrossRef]
- Roscigno, G.; Cirella, A.; Affinito, A.; Quintavalle, C.; Scognamiglio, I.; Palma, F.; Ingenito, F.; Nuzzo, S.; De Micco, F.; Cuccuru, A.; et al. miR-216a Acts as a Negative Regulator of Breast Cancer by Modulating Stemness Properties and Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 2313. [Google Scholar] [CrossRef] [Green Version]
- Rajamanickam, V.; Yan, T.; Xu, S.; Hui, J.; Xu, X.; Ren, L.; Liu, Z.; Liang, G.; Wang, O.; Wang, Y. Selective targeting of the TLR4 co-receptor, MD2, prevents colon cancer growth and lung metastasis. Int. J. Biol. Sci. 2020, 16, 1288–1301. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J.; Yan, A. Kdo2-lipid A: Structural diversity and impact on immunopharmacology. Biol. Rev. 2015, 90, 408–427. [Google Scholar] [CrossRef] [Green Version]
- Needham, B.D.; Carroll, S.M.; Giles, D.K.; Georgiou, G.; Whiteley, M.; Trent, M.S. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc. Natl. Acad. Sci. USA 2013, 110, 1464–1469. [Google Scholar] [CrossRef] [Green Version]
- Paramo, T.; Piggot, T.J.; Bryant, C.E.; Bond, P.J. The structural basis for Endotoxin-induced allosteric regulation of the Toll-Like Receptor 4 (TLR4) innate immune receptor. J. Biol. Chem. 2013, 288, 36215–36225. [Google Scholar] [CrossRef] [Green Version]
- Christ, W.J.; McGuinness, P.D.; Asano, O.; Wang, Y.; Mullarkey, M.A.; Perez, M.; Hawkins, L.D.; Blythe, T.A.; Dubuc, G.R.; Robidoux, A.L. Total synthesis of the proposed structure of Rhodobacter sphaeroides lipid A resulting in the synthesis of new potent lipopolysaccharide antagonists. J. Am. Chem. Soc. Mass. Spec. 1994, 116, 3637–3638. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Teghanemt, A.; Re, F.; Prohinar, P.; Widstrom, R.; Gioannini, T.L.; Weiss, J.P. Novel roles in human MD-2 of Phenylalanines 121 and 126 and Tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J. Biol. Chem. 2008, 283, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Gioannini, T.; Teghanemt, A.; Zhang, D.; Esparza, G.; Yu, L.; Weiss, J. Purified monomeric ligand.MD-2 complexes reveal molecular and structural requirements for activation and antagonism of TLR4 by Gram-negative bacterial endotoxins. Immunol. Res. 2014, 59, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Gong, M.; Björkbacka, H.; Golenbock, D.T. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J. Immunol. 2011, 187, 3683–3693. [Google Scholar] [CrossRef] [Green Version]
- Casella, C.R.; Mitchell, T.C. Inefficient TLR4/MD-2 heterotetramerization by monophosphoryl lipid A. PLoS ONE 2013, 8, e62622. [Google Scholar] [CrossRef]
- Bohannon, J.K.; Hernandez, A.; Enkhbaatar, P.; Adams, W.L.; Sherwood, E.R. The immunobiology of TLR4 agonists: From endotoxin tolerance to immunoadjuvants. Shock 2013, 40, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Sass, H.-J.; Zähringer, U.; Grzesiek, S. Structure and dynamics of 13C,15N-labeled lipopolysaccharides in a membrane mimetic. Angew. Chem. Int. Ed. 2008, 47, 9870–9874. [Google Scholar] [CrossRef]
- Gutsmann, T.; Schromm, A.B.; Brandenburg, K. The physicochemistry of endotoxins in relation to bioactivity. Int. J. Med. Microbiol. 2007, 297, 341–352. [Google Scholar] [CrossRef]
- Artner, D.; Oblak, A.; Ittig, S.; Garate, J.A.; Horvat, S.; Arrieumerlou, C.; Hofinger, A.; Oostenbrink, C.; Jerala, R.; Kosma, P.; et al. Conformationally constrained Lipid A mimetics for exploration of structural basis of TLR4/MD-2 activation by lipopolysaccharide. ACS Chem. Biol. 2013, 8, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Adanitsch, F.; Ittig, S.; Stöckl, J.; Oblak, A.; Haegman, M.; Jerala, R.; Beyaert, R.; Kosma, P.; Zamyatina, A. Development of αGlcN (1 ↔ 1) αMan-based Lipid A mimetics as a novel class of potent Toll-like Receptor 4 agonists. J. Med. Chem. 2014, 57, 8056–8071. [Google Scholar] [CrossRef] [PubMed]
- Adanitsch, F.; Shi, J.; Shao, F.; Beyaert, R.; Heine, H.; Zamyatina, A. Synthetic glycan-based TLR4 agonists targeting caspase-4/11 for the development of adjuvants and immunotherapeutics. Chem. Sci. 2018, 9, 3957–3963. [Google Scholar] [CrossRef] [PubMed]
- Heine, H.; Adanitsch, F.; Peternelj, T.T.; Haegman, M.; Kasper, C.; Ittig, S.; Beyaert, R.; Jerala, R.; Zamyatina, A. Tailored Modulation of Cellular Pro-inflammatory Responses With Disaccharide Lipid A Mimetics. Front. Immunol. 2021, 12, 631797. [Google Scholar] [CrossRef] [PubMed]
- Garate, J.A.; Stöckl, J.; del Carmen Fernández-Alonso, M.; Artner, D.; Haegman, M.; Oostenbrink, C.; Jimènez-Barbero, J.; Beyaert, R.; Heine, H.; Kosma, P.; et al. Anti-endotoxic activity and structural basis for human MD-2·TLR4 antagonism of tetraacylated lipid A mimetics based on βGlcN (1 ↔ 1) αGlcN scaffold. Innate Immun. 2015, 21, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, J.R.; Cluff, C.W.; Evans, J.T.; Lacy, M.J.; Stephens, J.R.; Brookshire, V.G.; Rong, W.; Ward, J.R.; Yorgensen, Y.M.; Persing, D.H.; et al. Immunostimulatory activity of aminoalkyl glucosaminide 4-phosphates (AGPs): Induction of protective innate immune responses by RC-524 and RC-529. J. Endotoxin Res. 2002, 8, 453–458. [Google Scholar] [CrossRef]
- Ishizaka, S.T.; Hawkins, L.D. E6020: A synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev. Vaccines 2007, 6, 773–784. [Google Scholar] [CrossRef]
- Akamatsu, M.; Fujimoto, Y.; Kataoka, M.; Suda, Y.; Kusumoto, S.; Fukase, K. Synthesis of lipid A monosaccharide analogues containing acidic amino acid: Exploring the structural basis for the endotoxic and antagonistic activities. Bioorg. Med. Chem. 2006, 14, 6759–6777. [Google Scholar] [CrossRef]
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Whitby, L.R.; Surakattula, M.M.R.P.; Huang, H.; Shi, H.; Choi, J.H.; Wang, K.-w.; et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [Google Scholar] [CrossRef] [Green Version]
- Strobl, S.; Hofbauer, K.; Heine, H.; Zamyatina, A. Lipid A Mimetics Based on Unnatural Disaccharide Scaffold as Potent TLR4 Agonists for Prospective Immunotherapeutics and Adjuvants. Chem. Eur. J. 2022, 28, e202200547. [Google Scholar] [CrossRef]
- Chebrolu, C.; Artner, D.; Sigmund, A.M.; Buer, J.; Zamyatina, A.; Kirschning, C.J. Species and mediator specific TLR4 antagonism in primary human and murine immune cells by βGlcN (1 ↔ 1) αGlcN based lipid A mimetics. Mol. Immunol. 2015, 67, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Borio, A.; Holgado, A.; Garate, J.A.; Beyaert, R.; Heine, H.; Zamyatina, A. Disaccharide-Based Anionic Amphiphiles as Potent Inhibitors of Lipopolysaccharide-Induced Inflammation. ChemMedChem 2018, 13, 2317–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Opal, S.; Calandra, T. Sepsis studies need new direction. Lancet Infect. Dis. 2012, 12, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Opal, S.M. Immunotherapy for Sepsis: A new approach against an ancient foe. N. Engl. J. Med. 2010, 363, 87–89. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, J.G.; Rossignol, D.; Francis, G.A.; Chan, T.; Lynn, M.; Wasan, K.M. Deactivation of the lipopolysaccharide antagonist eritoran (E5564) by high-density lipoprotein-associated apolipoproteins. Innate Immun. 2012, 18, 171–178. [Google Scholar] [CrossRef]
- Rao, V.S.R.; Qasba, P.K.; Balaji, P.V.; Chandrasekaran, R. Conformation of Disaccharides. In Conformation of Carbohydrates; OPA: Overseas Publishers Association, Ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998; pp. 91–130. [Google Scholar]
- Dowd, M.K.; Reilly, P.J.; French, A.D. Conformational analysis of trehalose disaccharides and analogues using MM3. J. Comput. Chem. 1992, 13, 102–114. [Google Scholar] [CrossRef]
- French, A.D.; Johnson, G.P.; Kelterer, A.M.; Dowd, M.K.; Cramer, C.J. Quantum mechanics studies of the intrinsic conformation of trehalose. J. Phys. Chem. A 2002, 106, 4988–4997. [Google Scholar] [CrossRef]
- Nunes, S.C.C.; Jesus, A.J.L.; Moreno, M.J.; Eusebio, M.E. Conformational preferences of α,α-trehalose in gas phase and aqueous solution. Carbohydr. Res. 2010, 345, 2048–2059. [Google Scholar] [CrossRef]
- Färnbäck, M.; Eriksson, L.; Widmalm, G. Octa-O-acetyl-α,α-trehalose ethanol disolvate. Acta Crystallogr. Sect. E 2004, 60, 1483–1485. [Google Scholar] [CrossRef] [Green Version]
- Bock, K.; Defaye, J.; Driguez, H.; Bar-Guilloux, E. Conformations in solution of α,α-trehalose, α-D-glucopyranosyl α-D-mannopyranoside, and their 1-thioglycosyl analogs, and a tentative correlation of their behaviour with respect to the enzyme trehalase. Eur. J. Biochem. 1983, 132, 595–600. [Google Scholar] [CrossRef]
- Lee, C.K.; Koh, L.L. Structure of β,β-trehalose. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1993, 49, 621–624. [Google Scholar] [CrossRef] [Green Version]
- Farnback, M.; Eriksson, L.; Widmalm, G. Octa-O-acetyl-β,β-thiotrehalose. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2000, 56, 700–701. [Google Scholar] [CrossRef]
- Stortz, C.A.; French, A.D. Disaccharide conformational maps: Adiabaticity in analogues with variable ring shapes. Mol. Simul. 2008, 34, 373–389. [Google Scholar] [CrossRef]
- Peric-Hassler, L.; Hansen, H.S.; Baron, R.; Hünenberger, P.H. Conformational properties of glucose-based disaccharides investigated using molecular dynamics simulations with local elevation umbrella sampling. Carbohydr. Res. 2010, 345, 1781–1801. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Berglund, N.A.; Kargas, V.; Marzinek, J.K.; Holdbrook, D.A.; Khalid, S.; Piggot, T.J.; Schmidtchen, A.; Bond, P.J. A Thermodynamic Funnel Drives Bacterial Lipopolysaccharide Transfer in the TLR4 Pathway. Structure 2018, 26, 1151–1161.e4. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heine, H.; Zamyatina, A. Therapeutic Targeting of TLR4 for Inflammation, Infection, and Cancer: A Perspective for Disaccharide Lipid A Mimetics. Pharmaceuticals 2023, 16, 23. https://doi.org/10.3390/ph16010023
Heine H, Zamyatina A. Therapeutic Targeting of TLR4 for Inflammation, Infection, and Cancer: A Perspective for Disaccharide Lipid A Mimetics. Pharmaceuticals. 2023; 16(1):23. https://doi.org/10.3390/ph16010023
Chicago/Turabian StyleHeine, Holger, and Alla Zamyatina. 2023. "Therapeutic Targeting of TLR4 for Inflammation, Infection, and Cancer: A Perspective for Disaccharide Lipid A Mimetics" Pharmaceuticals 16, no. 1: 23. https://doi.org/10.3390/ph16010023