
New Sulfamethoxazole Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors: Design, Synthesis, Cytotoxic Activity and Molecular Modeling
 ,
,  , , , , , , , , , ,
, , , , , , , , , ,
Abstract
:1. Introduction
2. Results and Discussion
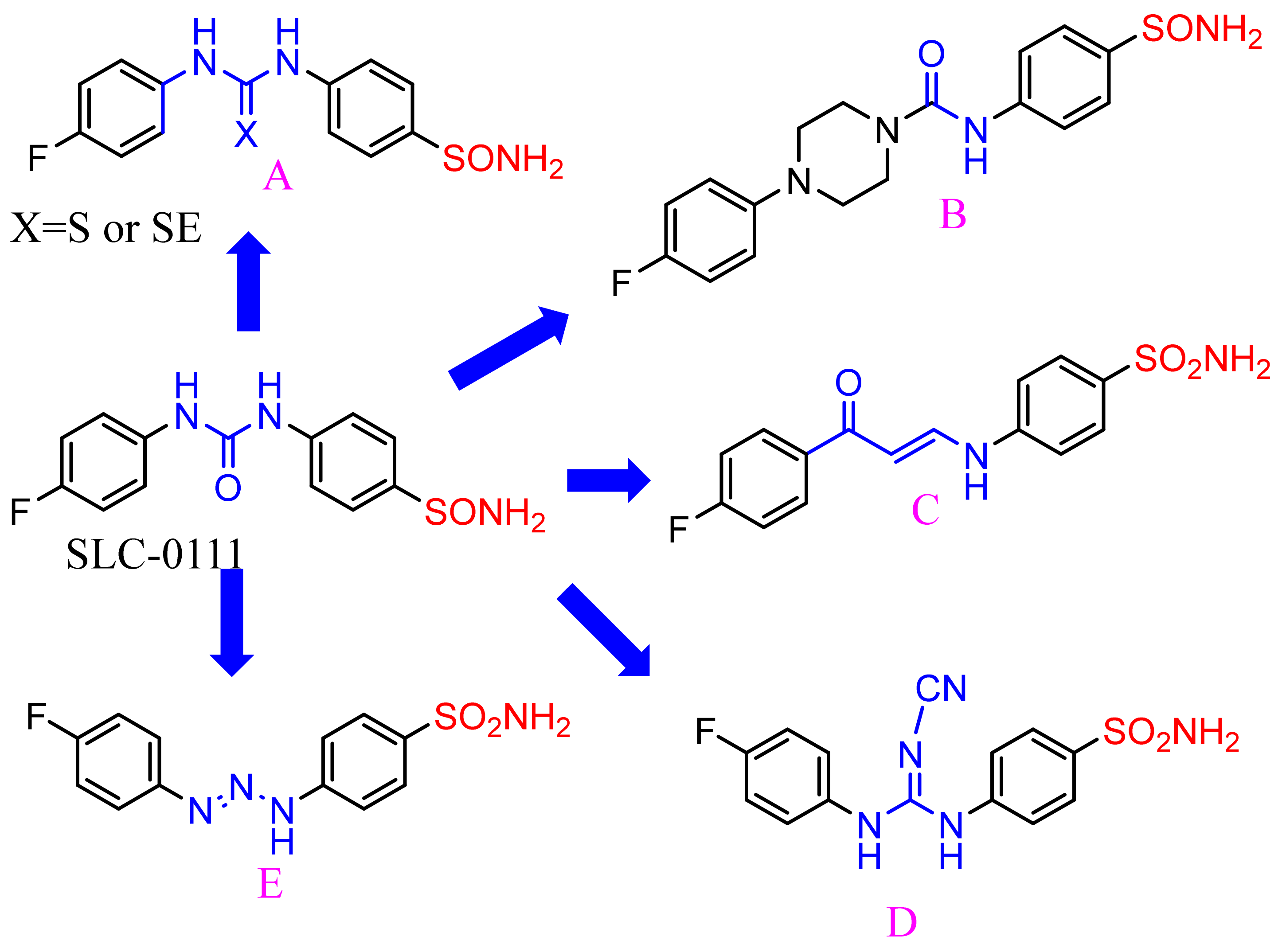
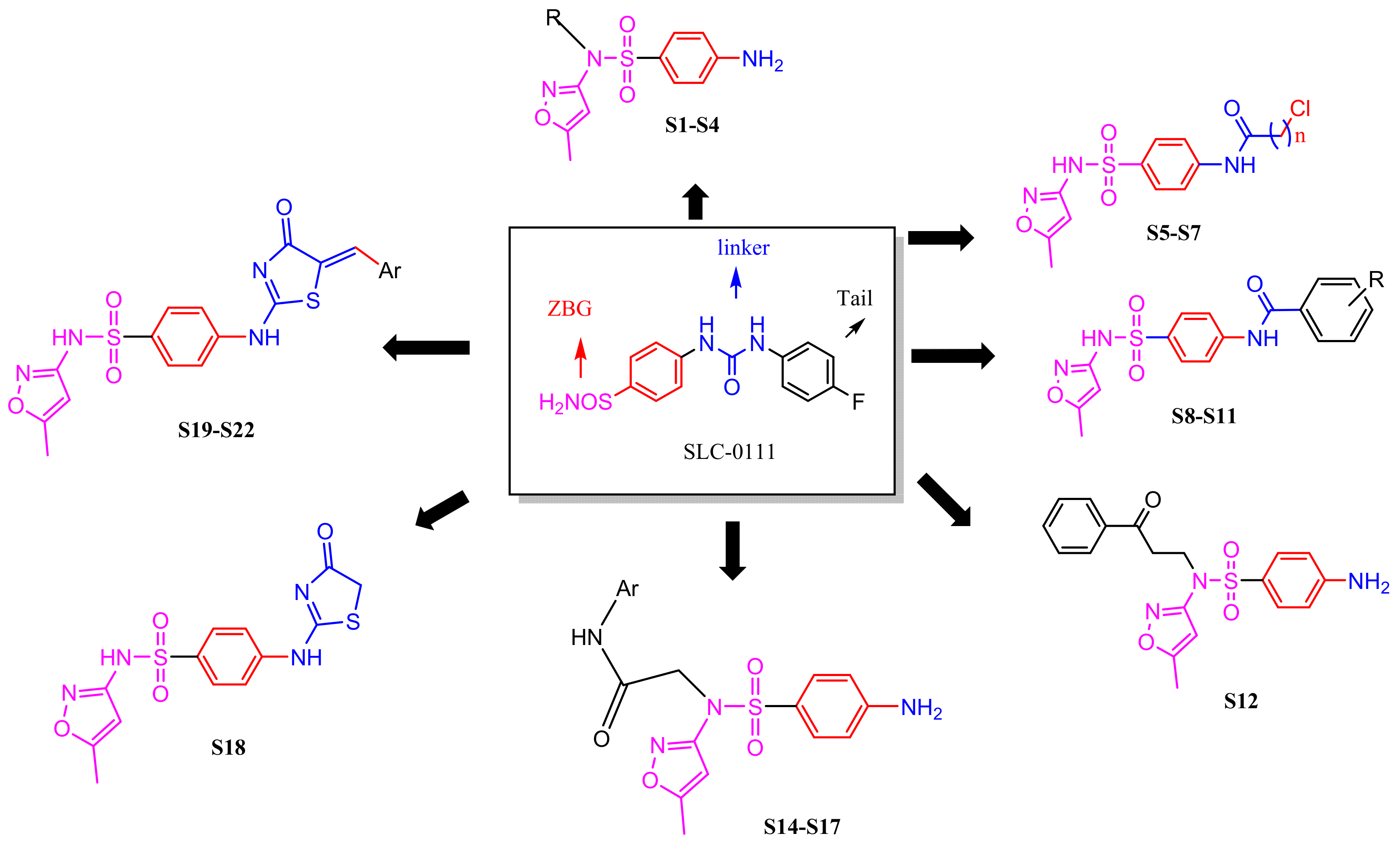
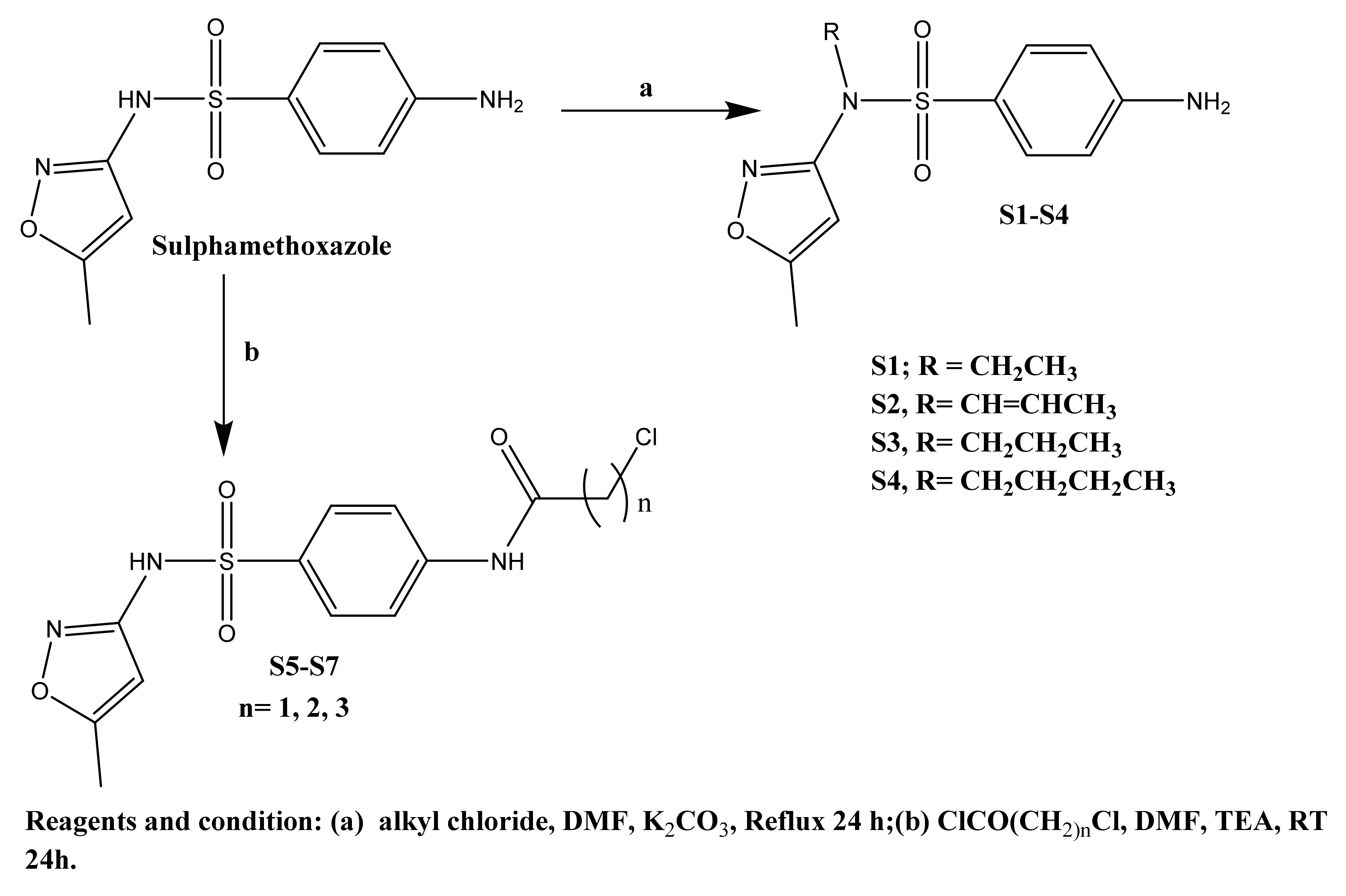
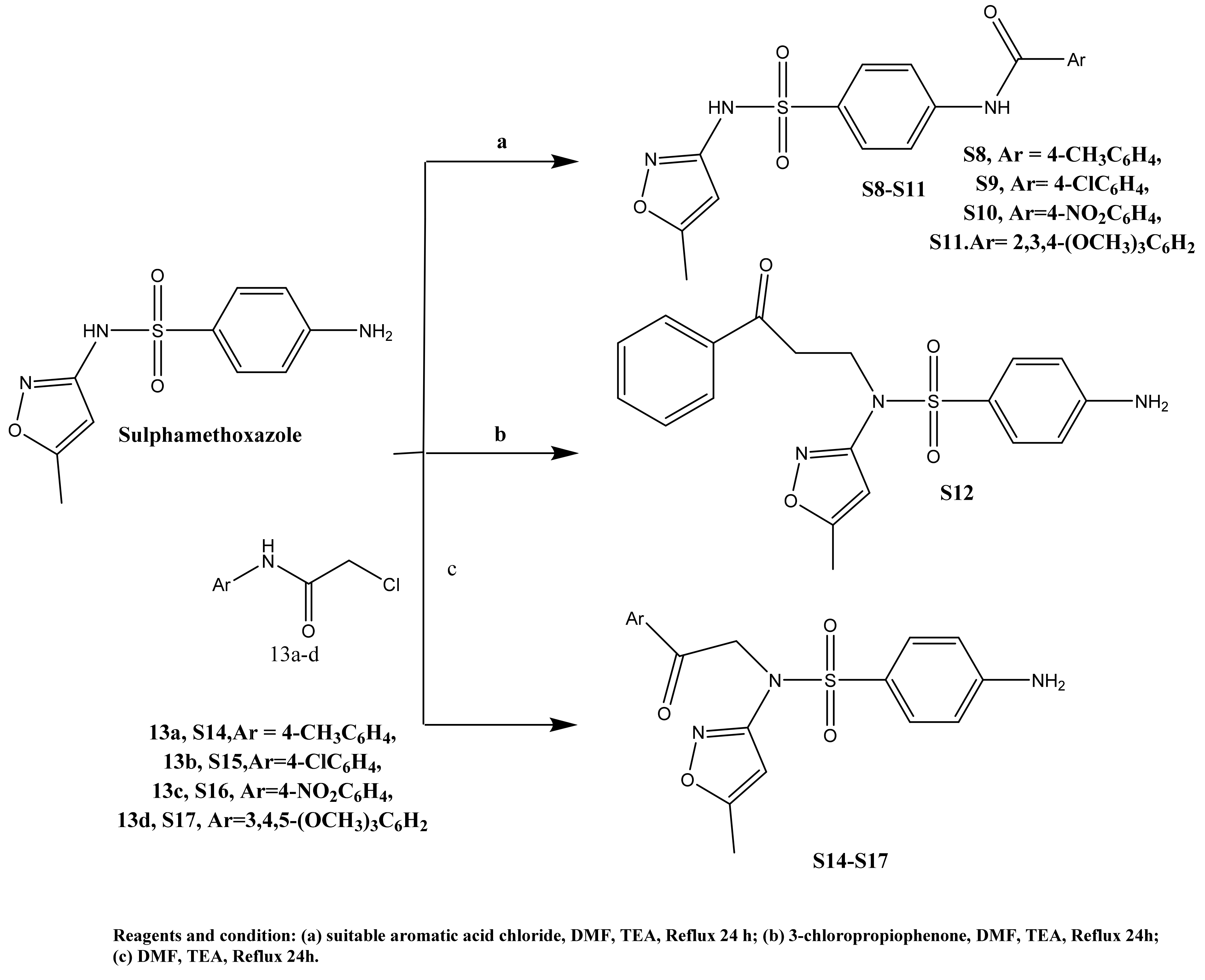
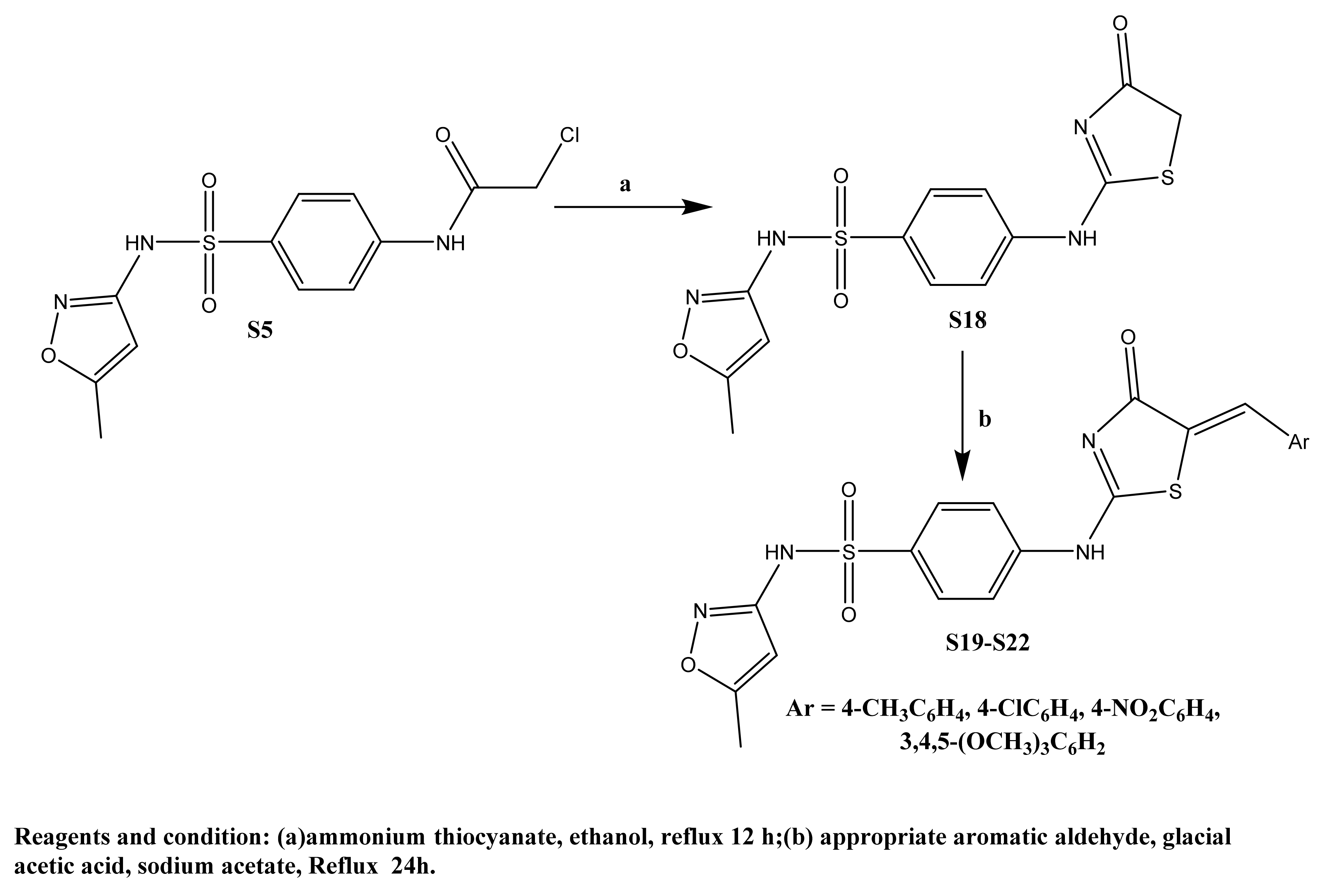
2.1. Chemistry
2.2. Biological Evaluation
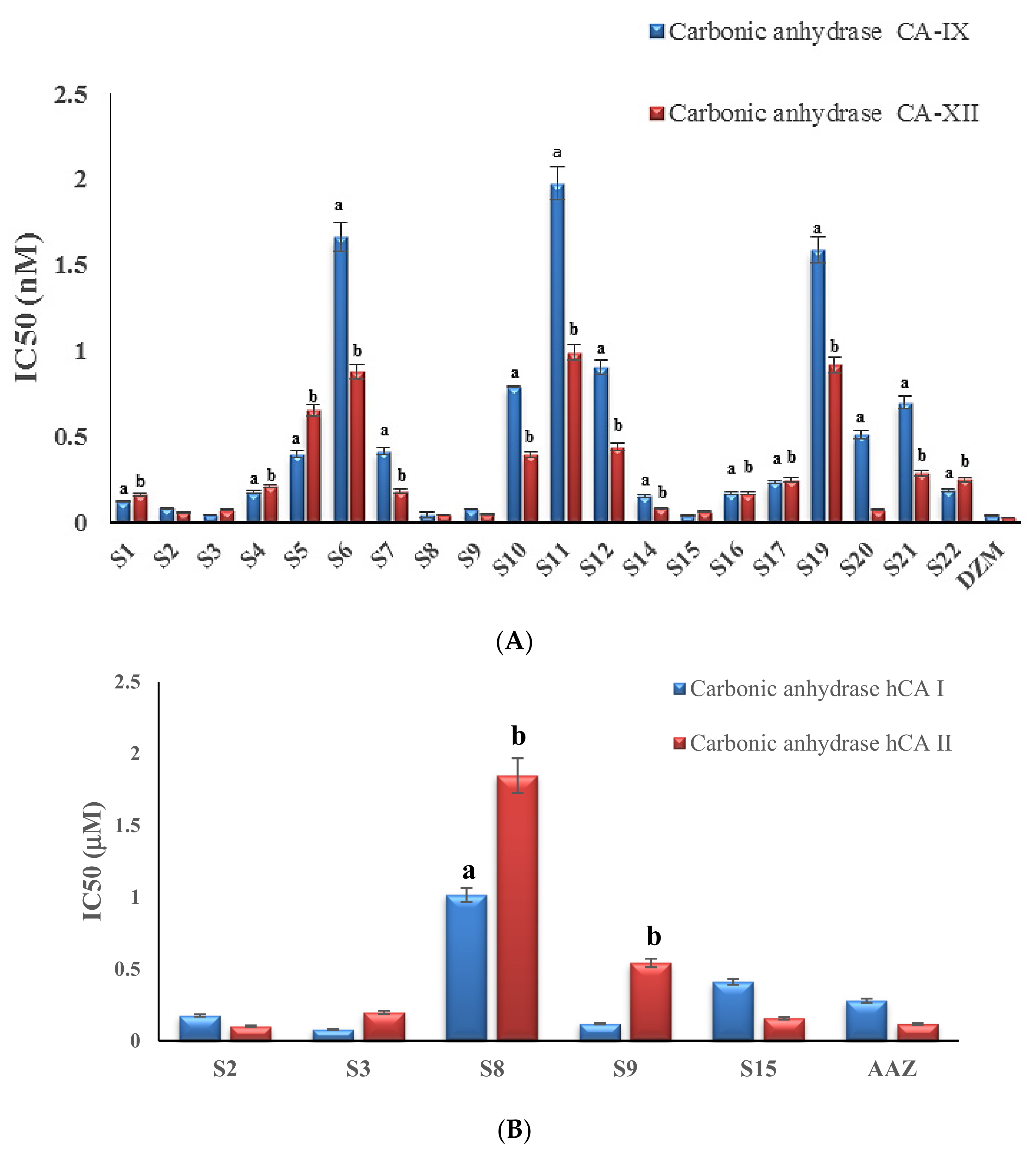
2.2.1. hCA IX and XII Inhibiting Effect
2.2.2. hCA I and II Inhibiting Effect
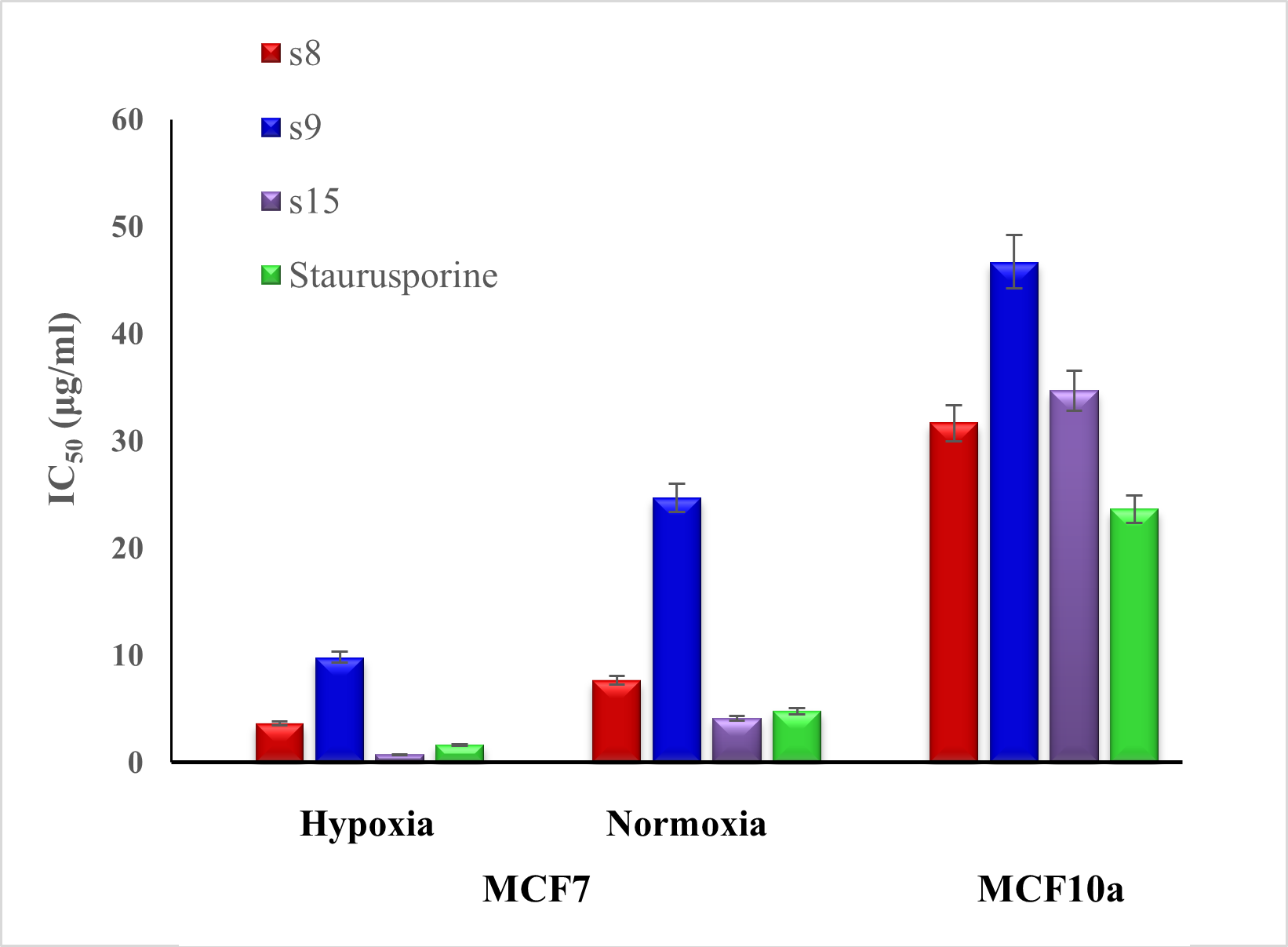
2.2.3. Cytotoxic Activity against MCF7 and MCF10a Cell Lines
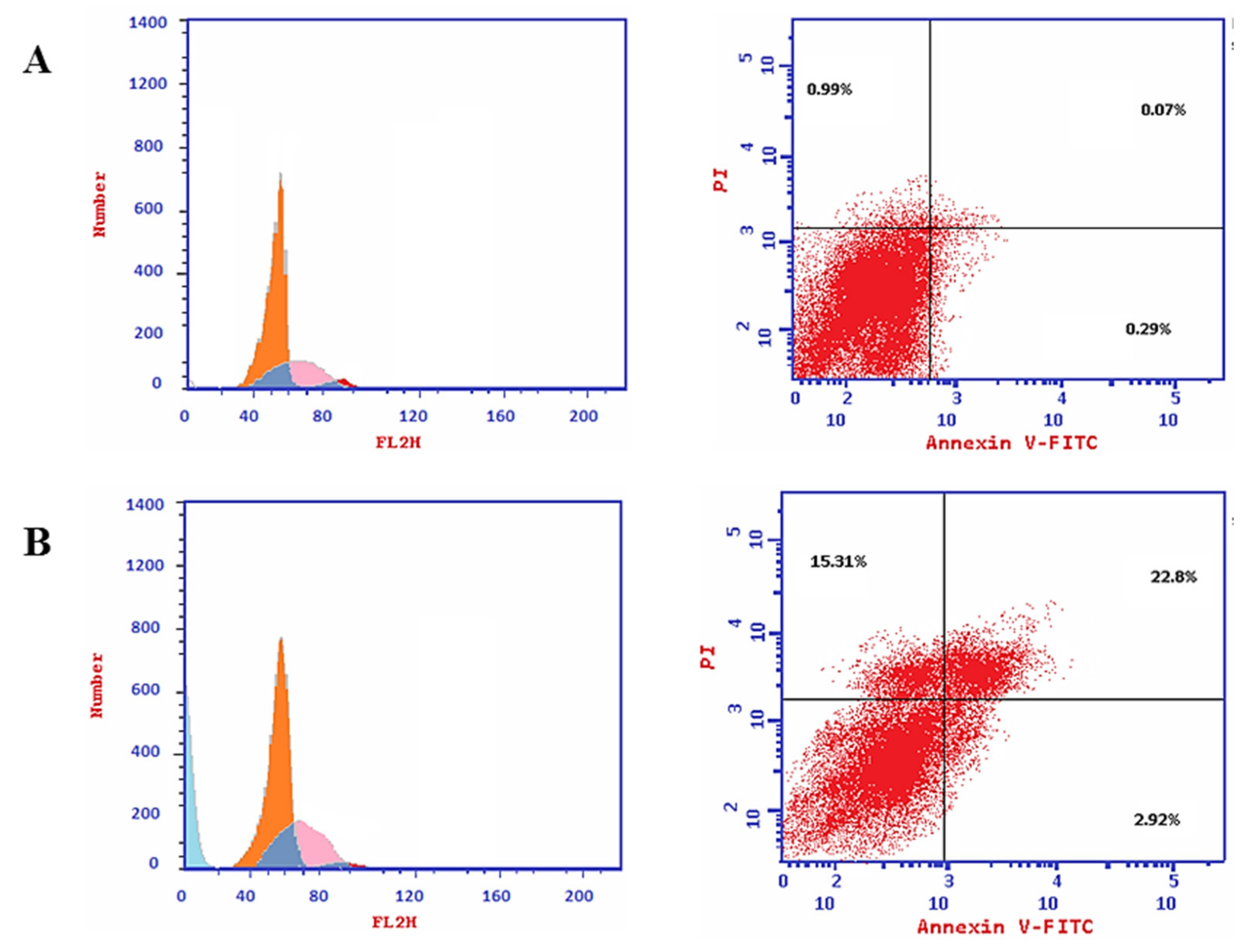
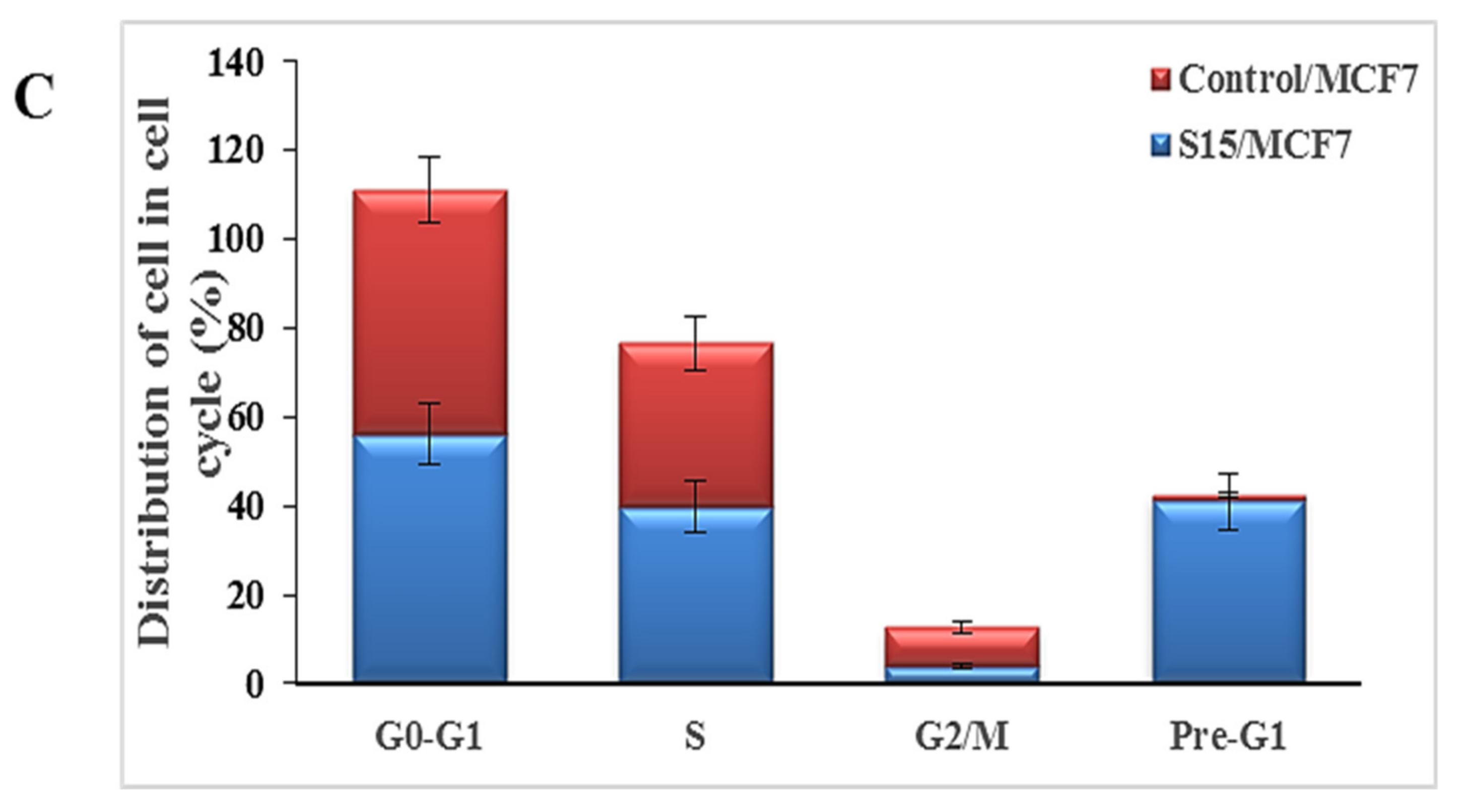
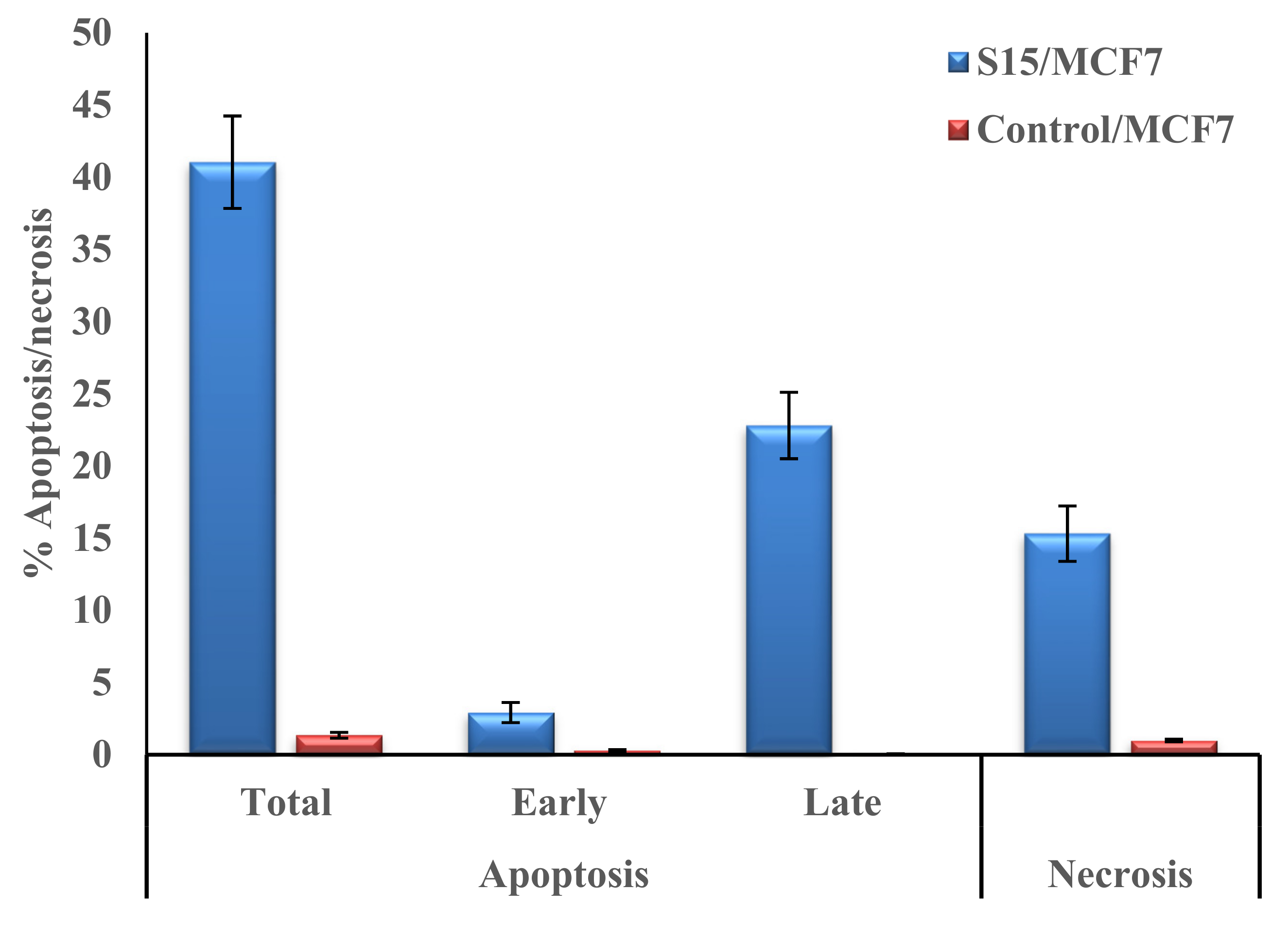
2.2.4. Cell Cycle Analysis and Apoptosis of Compound S15
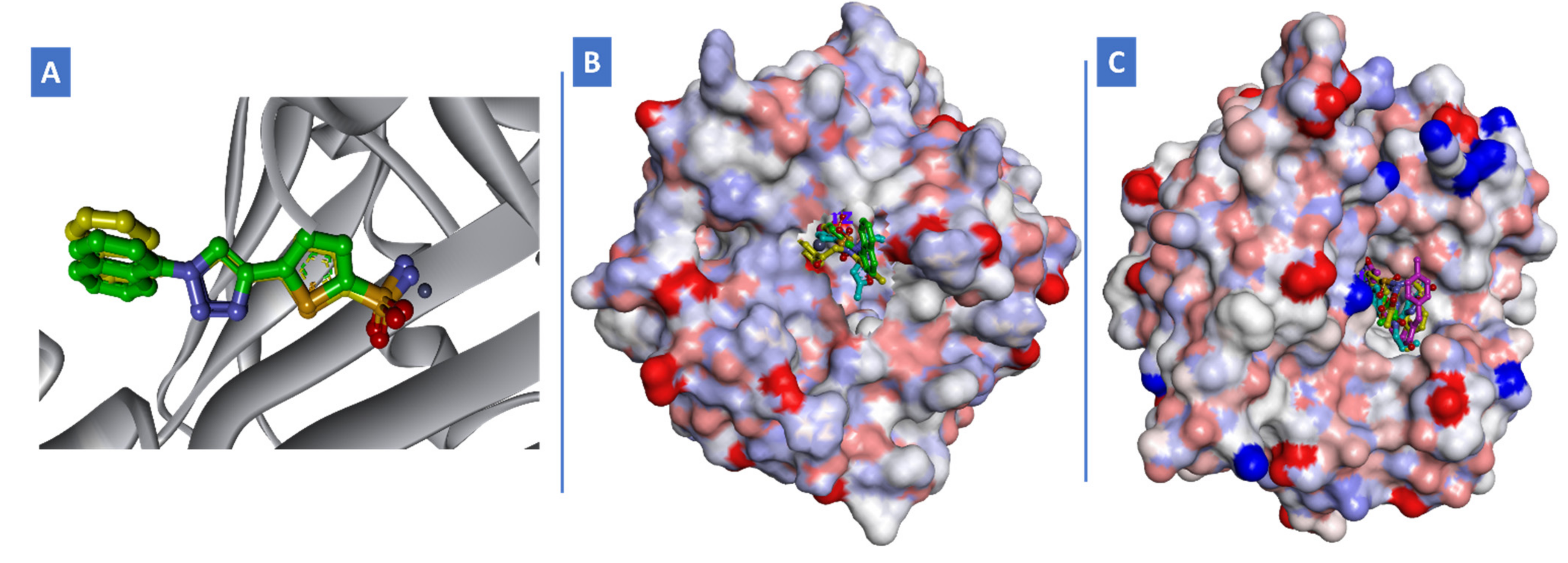
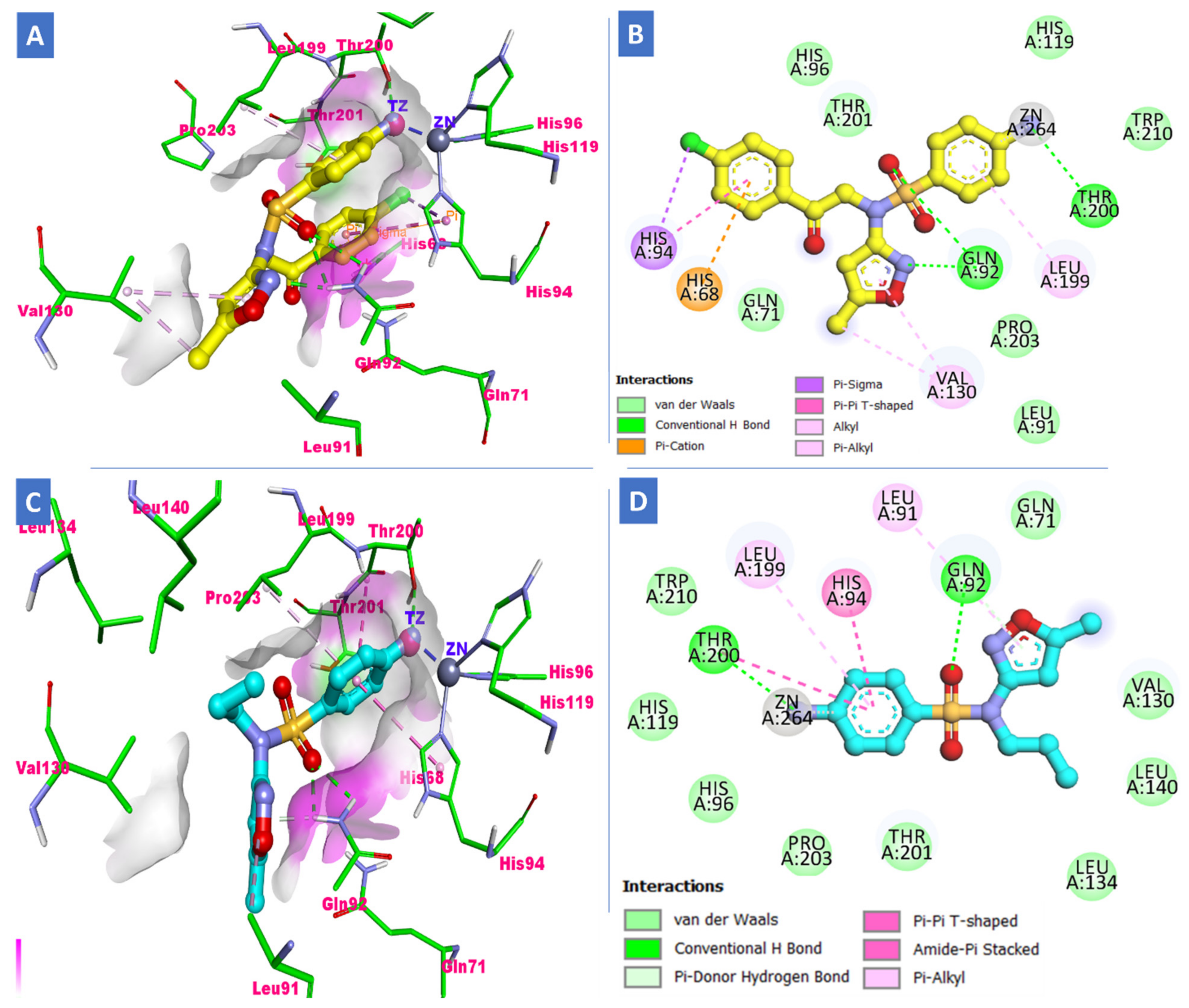
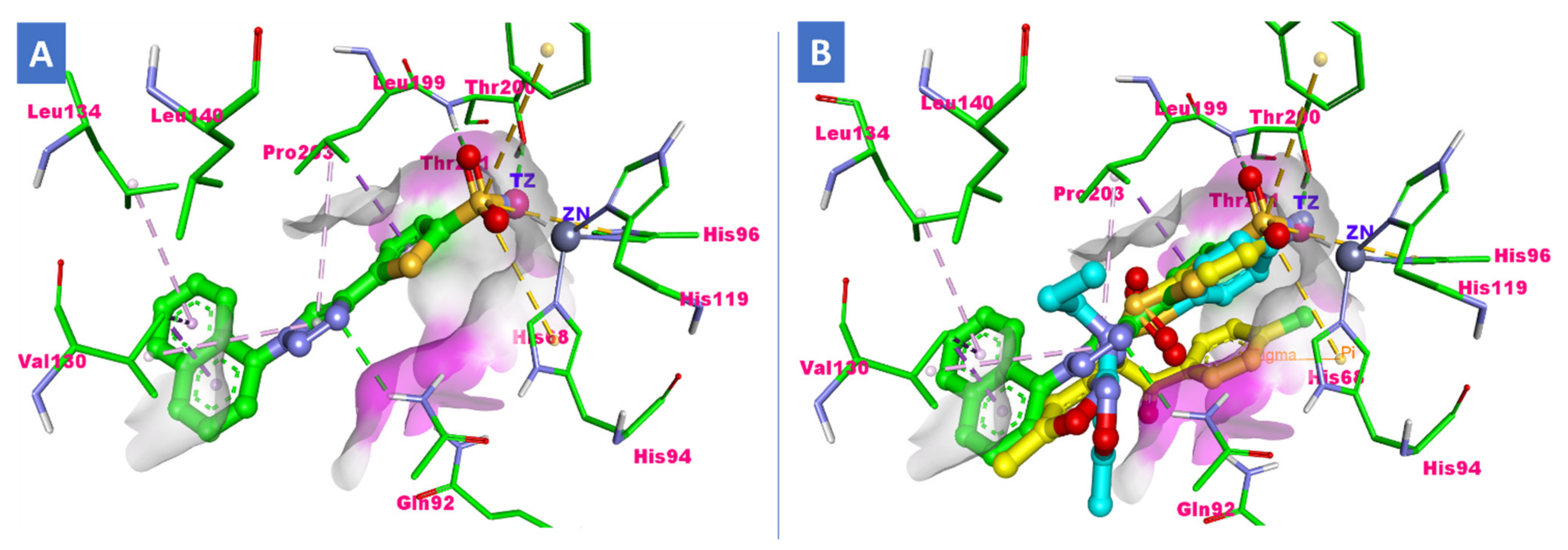
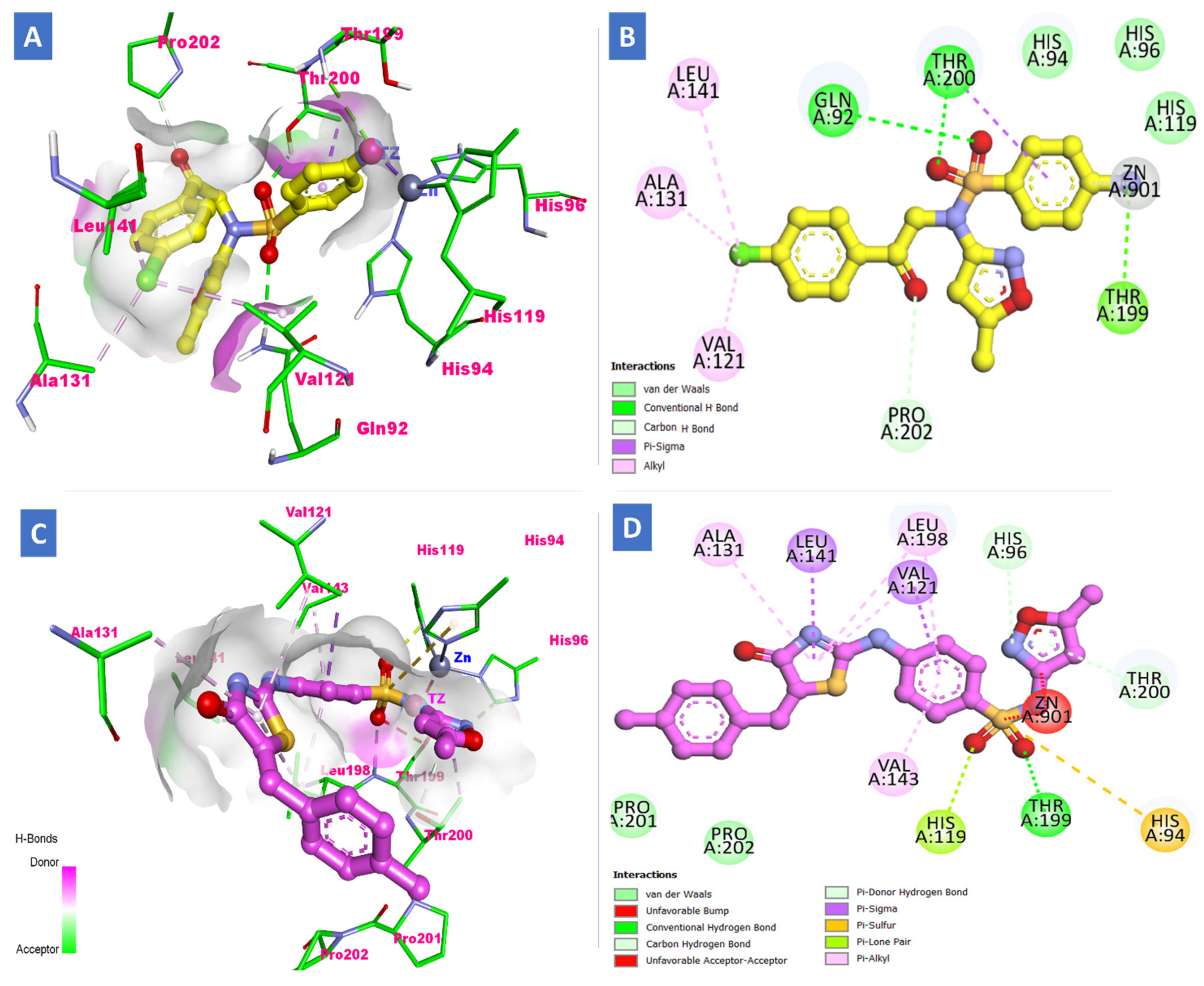
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Method for Preparation of Sulfamethoxazole S1–S4
 (11.27%), 268.41 (100%). Anal. Calcd. for C12H15N3O3S: C, 51.23; H, 5.37; N, 14.94; Found; C, 51.61; H, 5.02; N, 14.86. (15.73%), 166.27 (100%). Anal. Calcd. for C13H15N3O3S: C, 53.23; H, 5.15; N, 14.32; Found; C, 53.11; H, 5.22; N, 14.06. (21.00%), 63.17 (100%). Anal. Calcd. for C13H17N3O3S: C, 52.87; H, 5.80; N, 14.23; Found; C, 52.91; H, 6.02; N, 14.15. (12.93%), 189.82 (100%). Anal. Calcd. for C14H19N3O3S: C, 54.35; H, 6.19; N, 13.58; Found; C, 54.23; H, 6.07; N, 13.66.
(11.27%), 268.41 (100%). Anal. Calcd. for C12H15N3O3S: C, 51.23; H, 5.37; N, 14.94; Found; C, 51.61; H, 5.02; N, 14.86. (15.73%), 166.27 (100%). Anal. Calcd. for C13H15N3O3S: C, 53.23; H, 5.15; N, 14.32; Found; C, 53.11; H, 5.22; N, 14.06. (21.00%), 63.17 (100%). Anal. Calcd. for C13H17N3O3S: C, 52.87; H, 5.80; N, 14.23; Found; C, 52.91; H, 6.02; N, 14.15. (12.93%), 189.82 (100%). Anal. Calcd. for C14H19N3O3S: C, 54.35; H, 6.19; N, 13.58; Found; C, 54.23; H, 6.07; N, 13.66.3.1.2. General Method for Preparation of Compounds S6–S11
(26.11%), 309.58 (100%). Anal. Calcd. for C13H14ClN3O4S: C, 45.42; H, 4.10; N, 12.22; Found; C, 45.71; H, 4.18; N, 12.37. (26.08%), 87.68 (100%). Anal. Calcd. for C14H16ClN3O4S: C, 47.00; H, 4.51; N, 11.74; Found; C, 47.23; H, 4.22; N, 11.67. (11.27%), 285.69 (100%). Anal. Calcd. for C18H17N3O4S: C, 58.21; H, 4.61; N, 11.31; Found; C, 58.40; H, 4.88; N, 11.16. (61.57%), 60.02 (100%). Anal. Calcd. for C17H14N4O6S: C, 50.74; H, 3.51; N, 13.92; Found; C, 51.01; H, 3.32; N, 13.64. (66.30%), 243.77 (100%). Anal. Calcd. for C17H14ClN3O4S: C, 52.11; H, 3.60; N, 10.72; Found; C, 52.03; H, 3.92; N, 10.56. (39.39%), 402.65 (100%). Anal. Calcd. for C20H21N3O7S: C, 53.69; H, 4.73; N, 9.39; Found; C, 53.41; H, 4.85; N, 9.57.3.1.3. Preparation Method of Compounds S12 and S14–S17
(33.65%), 204.82 (100%). Anal. Calcd. for C19H19N3O4S: C, 59.21; H, 4.97; N, 10.90; Found; C, 59.33; H, 4.87; N, 10.76. (26.30%), 211.22 (100%). Anal. Calcd. for C19H20N4O4S: C, 56.99; H, 5.03; N, 13.99; Found; C, 56.41; H, 4.89; N, 14.16. (97.40%), 402.69 (100%). Anal. Calcd. for C18H17ClN4O4S: C, 51.37; H, 4.07; N, 13.31; Found; C, 51.50; H, 4.19; N, 13.38. (25.98%), 231.96 (100%). Anal. Calcd. for C18H17N5O6S: C, 50.11; H, 3.97; N, 16.23; Found; C, 50.23; H, 4.09; N, 16.48. (15.15%), 384.64 (100%). Anal. Calcd. for C21H24N4O7S: C, 52.93; H, 5.08; N, 11.76; Found; C, 52.64; H, 4.99; N, 11.56.3.1.4. Method for Preparation of Benzenesulfonamides S19–S22
(7.45%), 250.90 (100%). Anal. Calcd. for C21H18N4O4S2: C, 55.49; H, 3.99; N, 12.33; Found; C, 55.36; H, 3.61; N, 12.66. (20.45%), 344.74 (100%). Anal. Calcd. for C20H15ClN4O4S2: C, 50.58; H, 3.18; N, 11.80; Found; C, 50.66; H, 3.44; N, 12.06. (24.56%), 253.28 (100%). Anal. Calcd. for C20H15N5O6S2: C, 49.48; H, 3.11; N, 14.43; Found; C, 49.83; H, 3.53; N, 14.76. (29.89%), 49.79 (100%). Anal. Calcd. for C23H22N4O7S2: C, 52.07; H, 4.18; N, 10.56; Found; C, 51.86; H, 4.41; N, 10.79.3.2. Biological Activities
3.2.1. Materials and Methods
Chemicals and Kits
3.2.2. CA IX Inhibitory Assay
3.2.3. hCA XII Inhibitory Assay
3.2.4. CA1 Inhibitory Assay
3.2.5. CAII Inhibitory Assay
3.2.6. Cell Culture Protocol
3.2.7. MTT–Cytotoxicity Assay Protocol
3.2.8. Annexin V-FITC Assay for Apoptosis
3.3. Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wachholtz, A.B.; Fitch, C.E.; Makowski, S.; Tjia, J. A Comprehensive Approach to the Patient at End of Life: Assessment of Multidimensional Suffering. South. Med. J. 2016, 109, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzym. Inhib. Med. Chem. 2015, 31, 345–360. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Angeli, A.; Carta, F.; Nocentini, A.; Winum, J.-Y.; Zalubovskis, R.; Akdemir, A.; Onnis, V.; Eldehna, W.; Capasso, C.; Simone, G.; et al. Carbonic Anhydrase Inhibitors Targeting Metabolism and Tumor Microenvironment. Metabolites 2020, 10, 412. [Google Scholar] [CrossRef]
- Tanpure, R.P.; Ren, B.; Peat, T.S.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Carbonic Anhydrase Inhibitors with Dual-Tail Moieties to Match the Hydrophobic and Hydrophilic Halves of the Carbonic Anhydrase Active Site. J. Med. Chem. 2015, 58, 1494–1501. [Google Scholar] [CrossRef]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Keller, U.A.D.; Leung, S.; Huntsman, D.; et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, M.; Güttler, A.; Funtan, A.; Keßler, J.; Emmerich, D.; Paschke, R.; Vordermark, D.; Bache, M. Combined 3-O-acetylbetulin treatment and carbonic anhydrase IX inhibition results in additive effects on human breast cancer cells. Chem. Interactions 2020, 333, 109326. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Peat, T.S.; Mannelli, L.D.C.; Bartolucci, G.; Capperucci, A.; Ghelardini, C.; Supuran, C.T.; Carta, F. Discovery of New Selenoureido Analogues of 4-(4-Fluorophenylureido)benzenesulfonamide as Carbonic Anhydrase Inhibitors. ACS Med. Chem. Lett. 2017, 8, 963–968. [Google Scholar] [CrossRef]
- Lolak, N.; Akocak, S.; Bua, S.; Koca, M.; Supuran, C.T. Design and synthesis of novel 1,3-diaryltriazene-substituted sulfonamides as potent and selective carbonic anhydrase II inhibitors. Bioorg. Chem. 2018, 77, 542–547. [Google Scholar] [CrossRef]
- Akocak, S.; Lolak, N.; Bua, S.; Turel, I.; Supuran, C.T. Synthesis and biological evaluation of novel N, N′-diaryl cyanoguanidines acting as potent and selective carbonic anhydrase II inhibitors. Bioorg. Chem. 2018, 77, 245–251. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Berrino, E.; Vullo, D.; Ghabbour, H.A.; Al-Rashood, S.T.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Alharbi, A.; et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg. Chem. 2018, 83, 549–558. [Google Scholar] [CrossRef]
- Buemi, M.R.; Di Fiore, A.; De Luca, L.; Angeli, A.; Mancuso, F.; Ferro, S.; Monti, S.M.; Buonanno, M.; Russo, E.; De Sarro, G.; et al. Exploring structural properties of potent human carbonic anhydrase inhibitors bearing a 4-(cycloalkylamino-1-carbonyl)benzenesulfonamide moiety. Eur. J. Med. Chem. 2018, 163, 443–452. [Google Scholar] [CrossRef]
- Murtaza, S.; Altaf, A.A.; Hamayun, M.; Iftikhar, K.; Tahir, M.N.; Tariq, J.; Faiz, K. Synthesis, antibacterial activity and docking studies of chloroacetamide derivatives. Eur. J. Chem. 2019, 10, 358–366. [Google Scholar] [CrossRef]
- Khanusiya, M.; Gadhawala, Z. Design, Synthesis and Biological Evaluation of Some Novel Chalcones-sulphonamide Hybrids. J. Korean Chem. Soc. 2018, 62, 377–385. [Google Scholar]
- Roaiah, H.M.; Ghannam, I.A.; Ali, I.H.; El Kerdawy, A.M.; Ali, M.M.; Abbas, S.E.S.; El-Nakkady, S.S. Design, synthesis, and molecular docking of novel indole scaffold-based VEGFR-2 inhibitors as targeted anticancer agents. Arch. Pharm. 2018, 351, 1700299. [Google Scholar] [CrossRef]
- Ma, L.; Li, S.; Zheng, H.; Chen, J.; Lin, L.; Ye, X.; Chen, Z.; Xu, Q.; Chen, T.; Yang, J.; et al. Synthesis and biological activity of novel barbituric and thiobarbituric acid derivatives against non-alcoholic fatty liver disease. Eur. J. Med. Chem. 2011, 46, 2003–2010. [Google Scholar] [CrossRef]
- Braga, S.F.P.; Martins, L.C.; da Silva, E.B.; Júnior, P.A.S.; Murta, S.M.F.; Romanha, A.J.; de Oliveira, R.B.; Soh, W.T.; Brandstetter, H.; Ferreira, R.S.; et al. Synthesis and biological evaluation of potential inhibitors of the cysteine proteases cruzain and rhodesain designed by molecular simplification. Bioorg. Med. Chem. 2017, 25, 1889–1900. [Google Scholar] [CrossRef]
- Elgaher, W.A.M.; Sharma, K.K.; Haupenthal, J.; Saladini, F.; Pires, M.; Real, E.; Mely, Y.; Hartmann, R.W. Discovery and structure-based optimization of 2-Ureidothiophene-3-carboxylic acids as dual bacterial RNA polymerase and viral reverse transcriptase inhibitors. J. Med. Chem. 2016, 59, 7212–7222. [Google Scholar] [CrossRef]
- Huang, R.-Z.; Hua, S.-X.; Liao, Z.-X.; Huang, X.-C.; Wang, H.-S. Side chain-functionalized aniline-derived ursolic acid derivatives as multidrug resistance reversers that block the nuclear factor-kappa B (NF-κB) pathway and cell proliferation. Medchemcomm 2017, 8, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Awadallah, F.M.; El-Waei, T.A.; Hanna, M.M.; Abbas, S.E.; Ceruso, M.; Oz, B.E.; Guler, O.O.; Supuran, C.T. Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivatives. Eur. J. Med. Chem. 2015, 96, 425–435. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 2020, 43, 484. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic anhydrases: Role in pH control and cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef]
- Hsieh, M.-J.; Chen, K.-S.; Chiou, H.-L.; Hsieh, Y.-S. Carbonic anhydrase XII promotes invasion and migration ability of MDA-MB-231 breast cancer cells through the p38 MAPK signaling pathway. Eur. J. Cell Biol. 2010, 89, 598–606. [Google Scholar] [CrossRef]
- Zheng, G.; Peng, C.; Jia, X.; Gu, Y.; Zhang, Z.; Deng, Y.; Wang, C.; Li, N.; Yin, J.; Liu, X.; et al. ZEB1 transcriptionally regulated carbonic anhydrase 9 mediates the chemoresistance of tongue cancer via maintaining intracellular pH. Mol. Cancer 2015, 14, 84. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Liu, L.-C.; Xu, W.-T.; Wu, X.; Zhao, P.; Lv, Y.-L.; Chen, L. Overexpression of carbonic anhydrase II and Ki-67 proteins in prognosis of gastrointestinal stromal tumors. World J. Gastroenterol. WJG 2013, 19, 2473. [Google Scholar] [CrossRef]
- Favaro, E.; Lord, S.; Harris, A.L.; Buffa, F.M. Gene expression and hypoxia in breast cancer. Genome Med. 2011, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Harris, A.L. Targeting tumour hypoxia in breast cancer. Eur. J. Cancer 2008, 44, 2766–2773. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Innocenti, A.; Kallio, H.; Hilvo, M.; Scozzafava, A.; Supuran, C.T. The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian α-carbonic anhydrase isoforms. Bioorg. Med. Chem. Lett. 2009, 19, 4102–4106. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sławiński, J.; Sączewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Modeling 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Uda, N.R.; Seibert, V.; Stenner-Liewen, F.; Müller, P.; Herzig, P.; Gondi, G.; Zeidler, R.; Van Dijk, M.; Zippelius, A.; Renner, C. Esterase activity of carbonic anhydrases serves as surrogate for selecting antibodies blocking hydratase activity. J. Enzyme Inhib. Med. Chem. 2015, 30, 955–960. [Google Scholar] [CrossRef]
- Liao, S.-Y.; Ivanov, S.; Ivanova, A.; Ghosh, S.; A Cote, M.; Keefe, K.; Coca-Prados, M.; Stanbridge, E.J.; I Lerman, M. Expression of cell surface transmembrane carbonic anhydrase genes CA9 and CA12 in the human eye: Overexpression of CA12 (CAXII) in glaucoma. J. Med Genet. 2003, 40, 257–261. [Google Scholar] [CrossRef]
- Şentürk, M.; Gülçin, I.; Beydemir, Ş.; Küfrevioğlu, Ö.I.; Supuran, C.T. In Vitro Inhibition of Human Carbonic Anhydrase I and II Isozymes with Natural Phenolic Compounds. Chem. Biol. Drug Des. 2011, 77, 494–499. [Google Scholar] [CrossRef]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Biovia, D.S. Discovery Studio Visualizer; San Diego, CA, USA, Volume 936. Available online: https://www.computabio.com/applications.htm (accessed on 18 July 2022).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Carbonic Anhydrase (IC50, nM) | |
|---|---|---|
| Compound | CA-IX | CA-XII |
| S1 | 0.119 ± 0.006 | 0.162 ± 0.008 |
| S2 | 0.083 ± 0.004 | 0.056 ± 0.003 |
| S3 | 0.042 ± 0.002 | 0.07 ± 0.003 |
| S4 | 0.172 ± 0.008 | 0.208 ± 0.01 |
| S5 | 0.398 ± 0.019 | 0.651 ± 0.032 |
| S6 | 1.664 ± 0.081 | 0.877 ± 0.043 |
| S7 | 0.413 ± 0.02 | 0.18 ± 0.009 |
| S8 | 0.042 ± 0.002 | 0.04 ± 0.002 |
| S9 | 0.074 ± 0.004 | 0.047 ± 0.002 |
| S10 | 0.789 ± 0.039 | 0.392 ± 0.019 |
| S11 | 1.977 ± 0.097 | 0.991 ± 0.048 |
| S12 | 0.902 ± 0.044 | 0.437 ± 0.021 |
| S14 | 0.152 ± 0.007 | 0.081 ± 0.004 |
| S15 | 0.037 ± 0.002 | 0.061 ± 0.003 |
| S16 | 0.165 ± 0.008 | 0.164 ± 0.008 |
| S17 | 0.235 ± 0.011 | 0.247 ± 0.012 |
| S19 | 1.587 ± 0.078 | 0.917 ± 0.045 |
| S20 | 0.51 ± 0.025 | 0.069 ± 0.003 |
| S21 | 0.698 ± 0.034 | 0.284 ± 0.014 |
| S22 | 0.186 ± 0.009 | 0.249 ± 0.012 |
| Dorzolamide HCL | 0.036 ± 0.002 | 0.024 ± 0.001 |
| Compound | Carbonic Anhydrase IC50 (uM) | ||
|---|---|---|---|
| Code | MW g/mol | hCA I | hCA II |
| S2 | 293 | 0.177 ± 0.008 | 0.102 ± 0.006 |
| s3 | 295 | 0.08 ± 0.003 | 0.199 ± 0.011 |
| S8 | 471 | 1.017 ± 0.049 a | 1.848 ± 0.12 b |
| S9 | 391 | 0.121 ± 0.006 | 0.543 ± 0.03 b |
| S15 | 420 | 0.411 ± 0.02 | 0.158 ± 0.009 |
| AAZ | 222.245 | 0.281 ± 0.014 | 0.117 ± 0.007 |
| Sample | Cytotoxicity (IC50 µg/mL) | |||
|---|---|---|---|---|
| Compound | MW | MCF7 | MCF10a | |
| Hypoxia | Normoxia | |||
| S8 | 471 | 3.64 ± 0.2 | 7.68 ± 0.41 | 31.65 ± 1.71 |
| S9 | 391 | 9.81 ± 0.53 | 24.69 ± 1.33 | 46.72 ± 2.52 |
| S15 | 420 | 0.73 ± 0.04 | 4.15 ± 0.22 | 34.71 ± 1.87 |
| Staurosporine | 466.5 | 1.63 ± 0.09 | 4.78 ± 0.26 | 23.65 ± 1.27 |
| Apoptosis | Necrosis | |||
|---|---|---|---|---|
| Total | Early | Late | ||
| S15/MCF7 | 41.03 | 2.92 | 22.8 | 15.31 |
| Control/MCF7 | 1.35 | 0.29 | 0.07 | 0.99 |
| Ligand (Isoenzyme) | ΔG (kcal/mol) | H-Bonding Interactions | Hydrophobic Interactions | π-π Stacking | |
|---|---|---|---|---|---|
| Chemical Group | Amino Acids Involved | ||||
| S3 (h-CA-IX) | −29.86 | Amino and sulfonamide | Thr200, Gln92 | Leu91, Val130 and Leu140 | p-chlorophenyl ring with His94 |
| S15 (h-CA-IX) | −28.98 | Amino, isoxazole and sulfonamide | Thr200, Gln92 | Leu91, Val130 and Leu140 | p-chlorophenyl ring with His94 |
| S15 (h-CA-XII) | −27.83 | Amino and sulfonamide | Thr199, Gln92 and Thr200 | Val121, Ala131, Leu141 and Leu198 | -- |
| S19 (h-CA-XII) | −25.21 | sulfonamide | Thr199 | Val121, Ala131, Leu141 and Val143 | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelgawad, M.A.; Bukhari, S.N.A.; Musa, A.; Elmowafy, M.; Elkomy, M.H.; Nayl, A.A.; El-Ghorab, A.H.; Alsohaimi, I.H.; Abdel-Bakky, M.S.; Althobaiti, I.O.; et al. New Sulfamethoxazole Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors: Design, Synthesis, Cytotoxic Activity and Molecular Modeling. Pharmaceuticals 2022, 15, 1134. https://doi.org/10.3390/ph15091134
Abdelgawad MA, Bukhari SNA, Musa A, Elmowafy M, Elkomy MH, Nayl AA, El-Ghorab AH, Alsohaimi IH, Abdel-Bakky MS, Althobaiti IO, et al. New Sulfamethoxazole Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors: Design, Synthesis, Cytotoxic Activity and Molecular Modeling. Pharmaceuticals. 2022; 15(9):1134. https://doi.org/10.3390/ph15091134
Chicago/Turabian StyleAbdelgawad, Mohamed A., Syed N. A. Bukhari, Arafa Musa, Mohammed Elmowafy, Mohammed H. Elkomy, AbdElAziz. A. Nayl, Ahmed H. El-Ghorab, Ibrahim Hotan Alsohaimi, Mohamed Sadek Abdel-Bakky, Ibrahim O. Althobaiti, and et al. 2022. "New Sulfamethoxazole Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors: Design, Synthesis, Cytotoxic Activity and Molecular Modeling" Pharmaceuticals 15, no. 9: 1134. https://doi.org/10.3390/ph15091134