
Optimization of the Pharmacokinetic Profile of [99mTc]Tc-N4-Bombesin Derivatives by Modification of the Pharmacophoric Gln-Trp Sequence
 , , , and
, , , and
Abstract
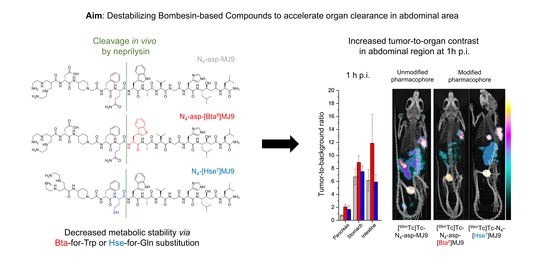
:
1. Introduction
2. Results
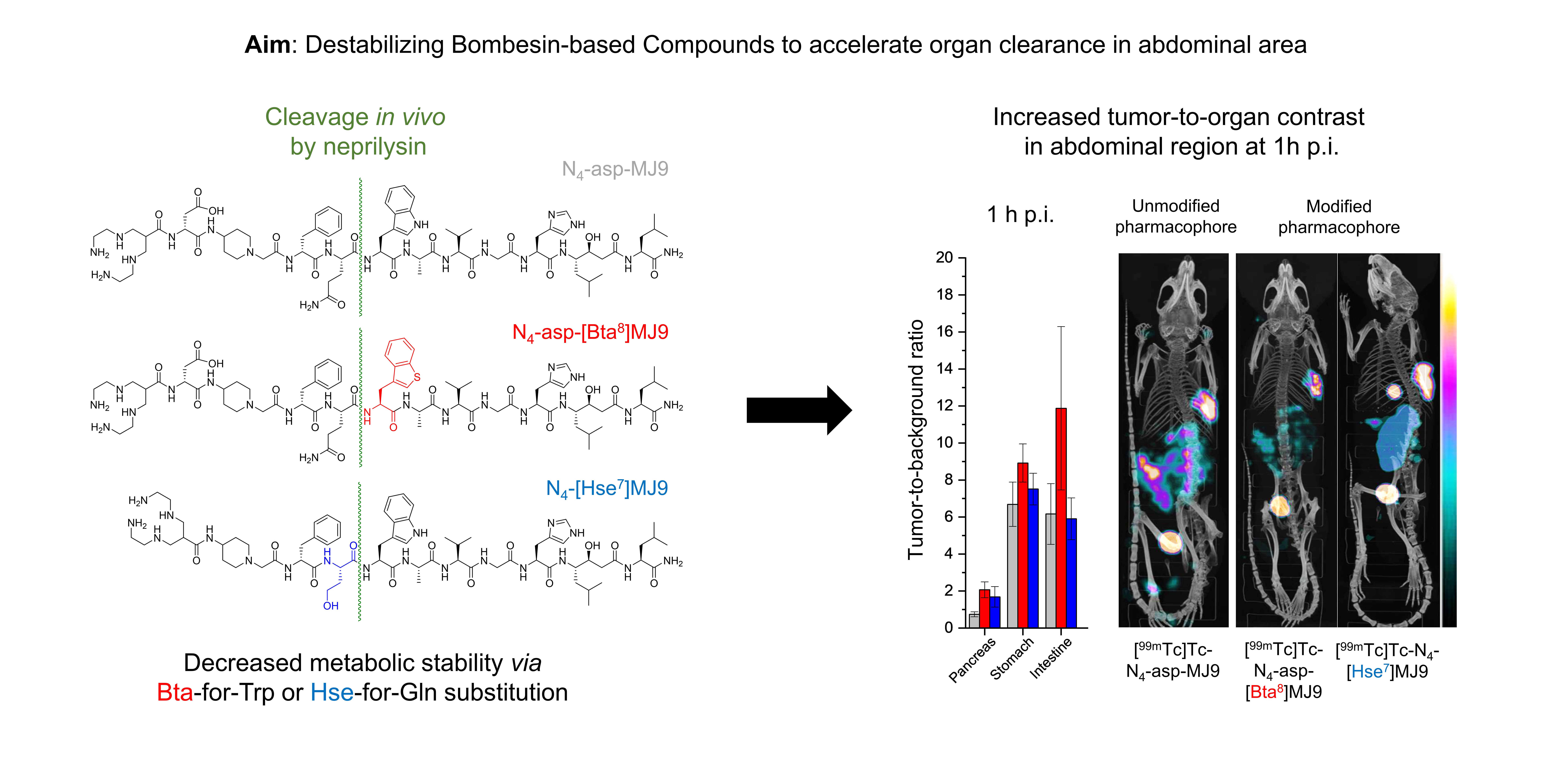
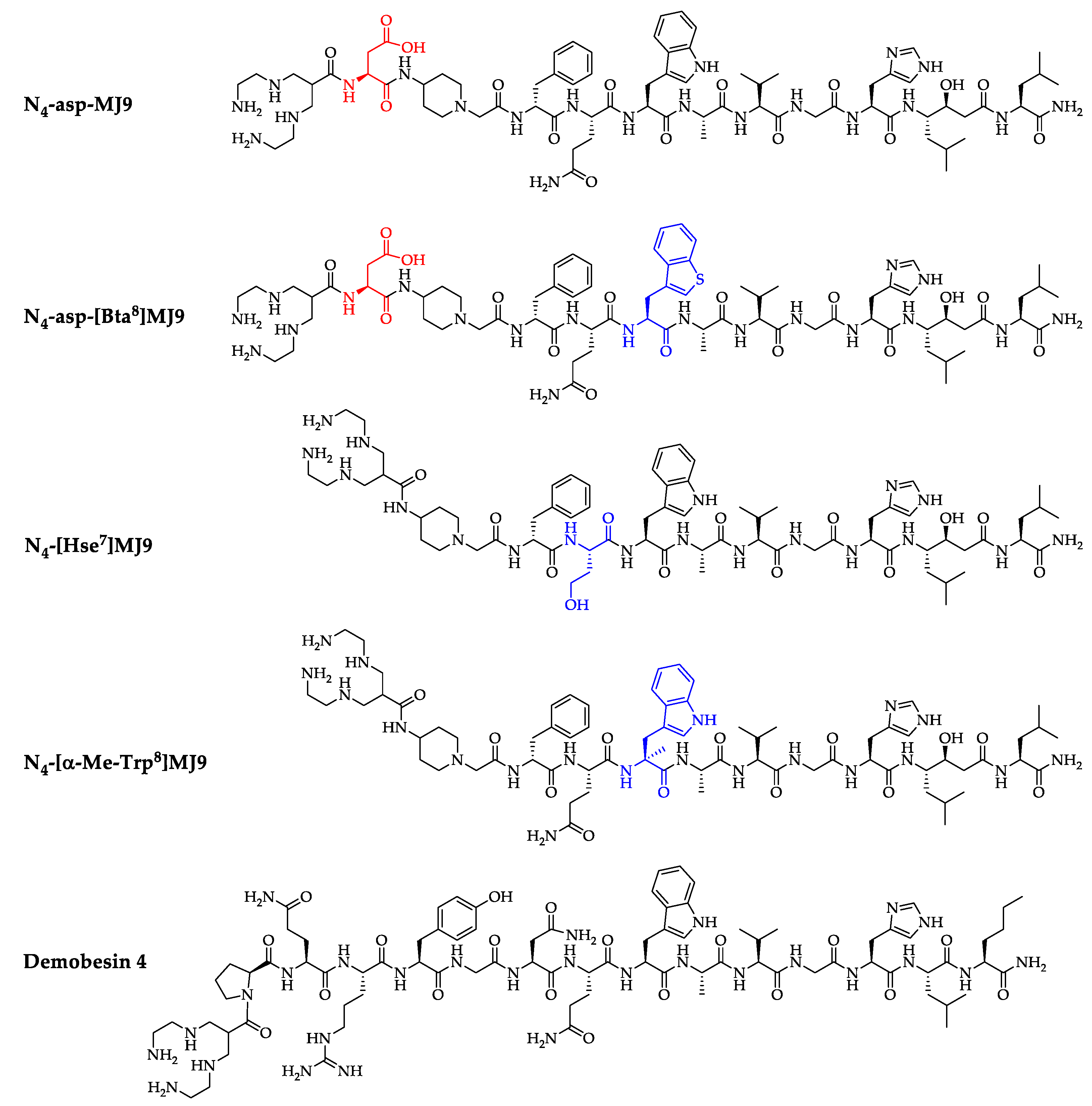
2.1. Synthesis and Radiolabeling
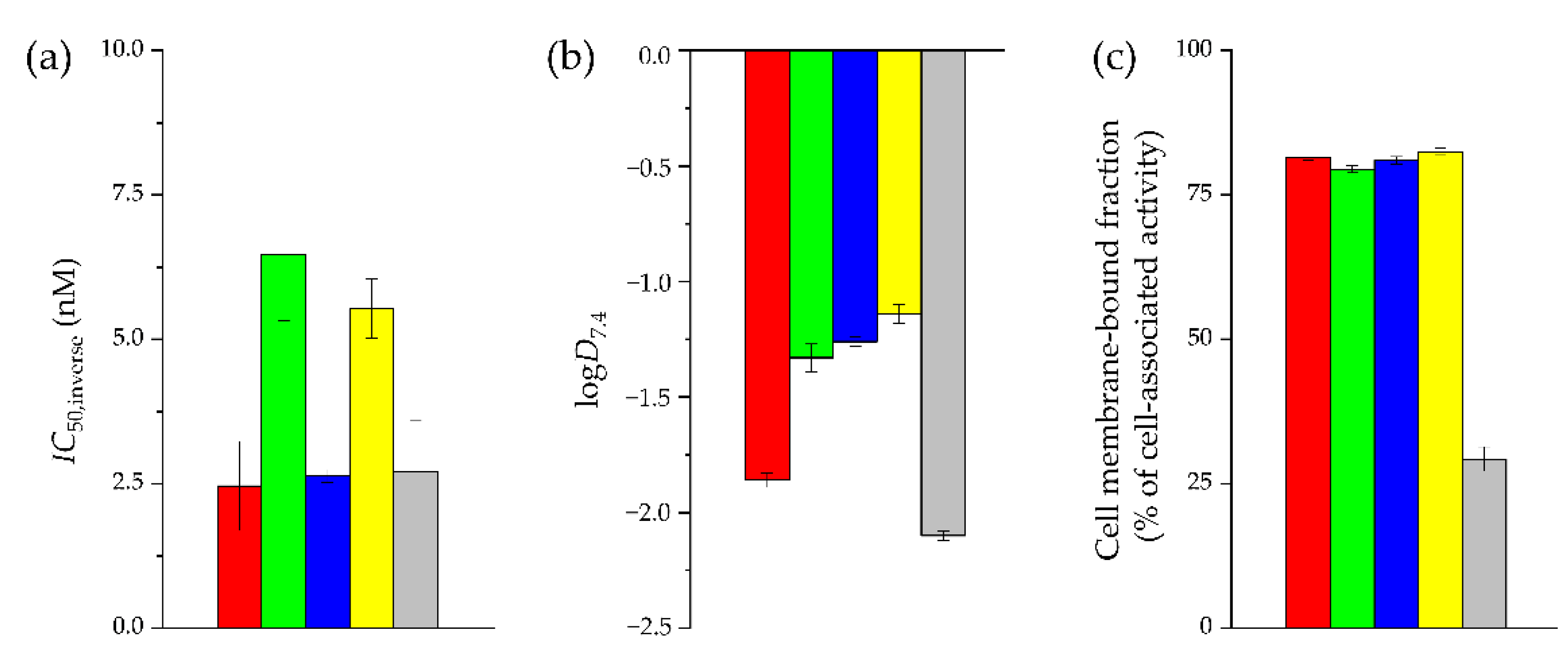
2.2. In Vitro Characterization
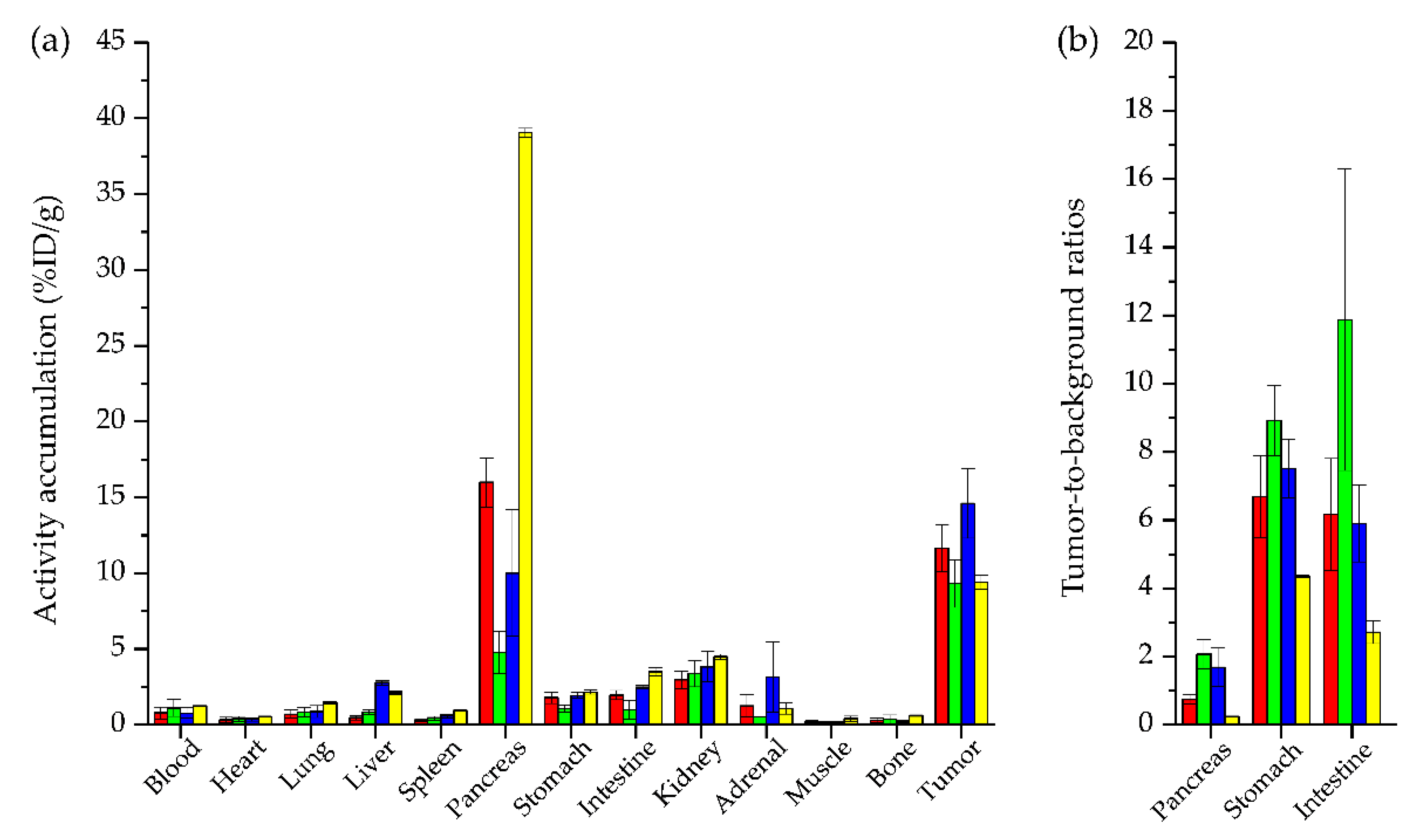
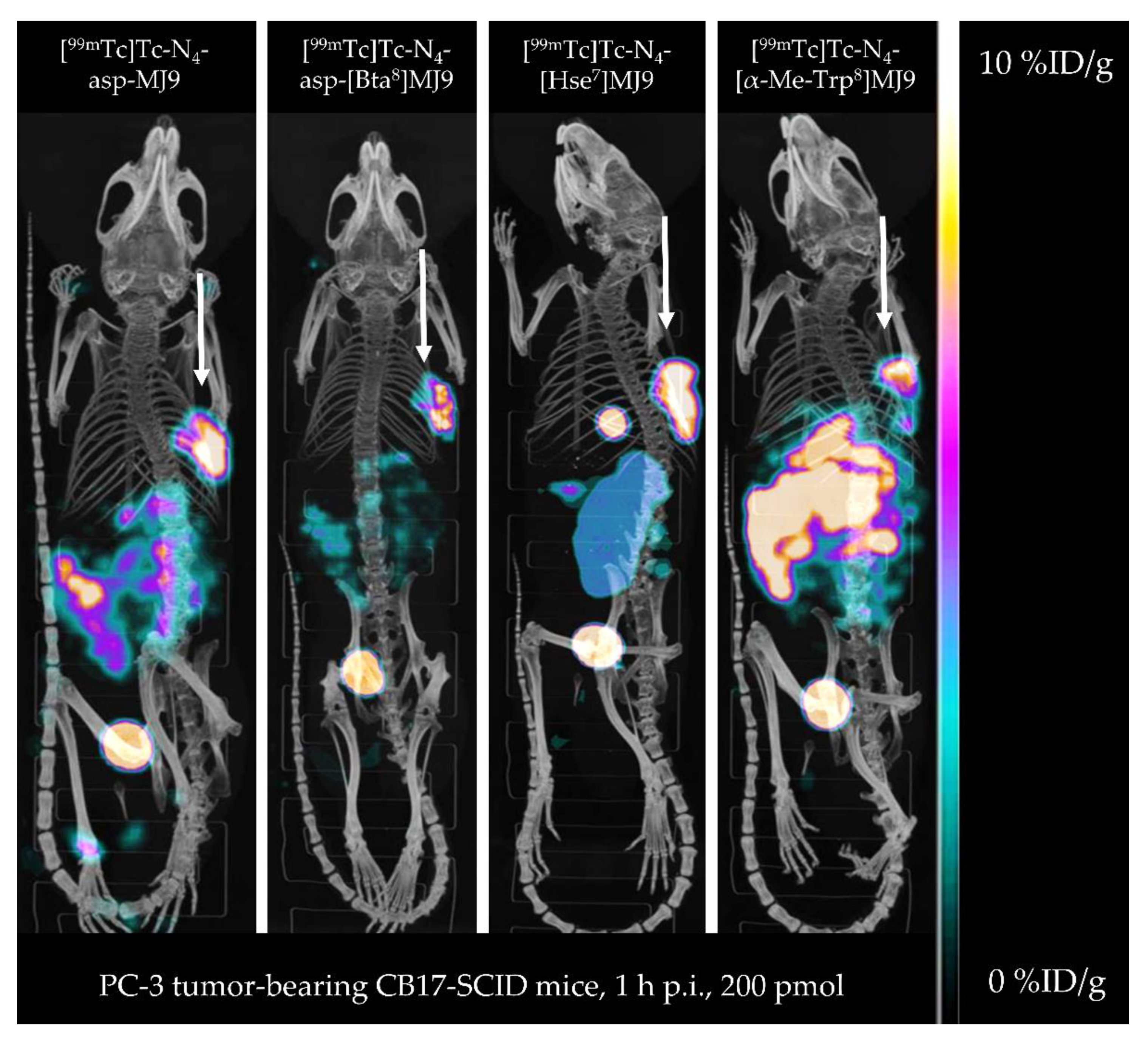
2.3. In Vivo Characterization
3. Discussion
4. Materials and Methods
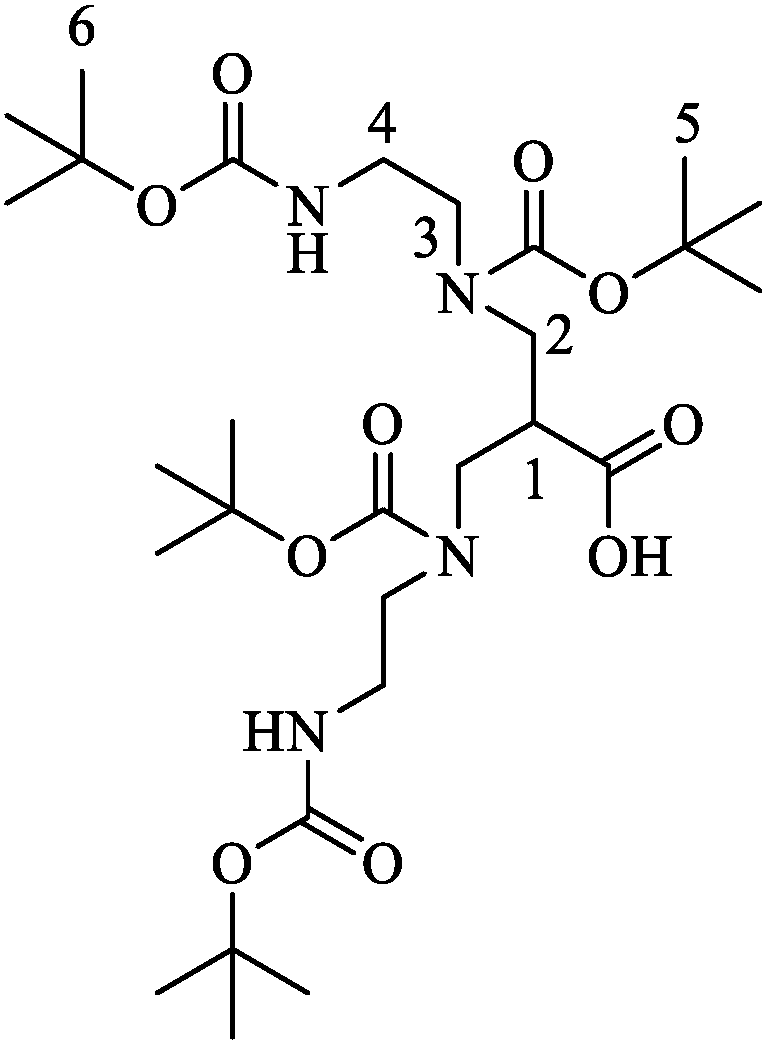
4.1. Synthesis of N,N′,N″,N‴-Tetrakis(tert-butyloxycarbonyl)-6-carboxy-1,4,8,11-tetraazaundecane ((tBu)4N4)
4.2. Compound Synthesis and Labeling Procedures
4.3. In Vitro Experiments
4.4. In Vivo Experiments
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weber, W.A.; Czernin, J.; Anderson, C.J.; Badawi, R.D.; Barthel, H.; Bengel, F.; Bodei, L.; Buvat, I.; DiCarli, M.; Graham, M.M.; et al. The Future of Nuclear Medicine, Molecular Imaging, and Theranostics. J. Nucl. Med. 2020, 61, 263S–272S. [Google Scholar] [CrossRef] [PubMed]
- World Nuclear Association. Radioisotopes in Medicine. Available online: https://world-nuclear.org/information-library/non-power-nuclear-applications/radioisotopes-research/radioisotopes-in-medicine.aspx (accessed on 26 June 2022).
- The Institute of Cancer Research. The Year 2035: What Is the Future of Cancer Research and Treatment? Available online: https://www.icr.ac.uk/blogs/science-talk/page-details/the-year-2035-what-is-the-future-of-cancer-research-and-treatment (accessed on 26 June 2022).
- Rahmim, A.; Zaidi, H. PET versus SPECT: Strengths, limitations and challenges. Nucl. Med. Commun. 2008, 29, 193–207. [Google Scholar] [CrossRef]
- Shahzad, K.; Abdul Majid, A.S.; Khan, M.; Iqbal, M.A.; Ali, A. Recent advances in the synthesis of (99mTechnetium) based radio-pharmaceuticals. Rev. Inorg. Chem. 2021, 41, 151–198. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Correia, J.D.; Paulo, A.; Raposinho, P.D.; Santos, I. Radiometallated peptides for molecular imaging and targeted therapy. Dalton Trans. 2011, 40, 6144–6167. [Google Scholar] [CrossRef] [PubMed]
- Duatti, A. Review on 99mTc radiopharmaceuticals with emphasis on new advancements. Nucl. Med. Biol. 2021, 92, 202–216. [Google Scholar] [CrossRef]
- Nock, B.; Maina, T. Tetraamine-coupled peptides and resulting (99m)Tc-radioligands: An effective route for receptor-targeted diagnostic imaging of human tumors. Curr. Top. Med. Chem. 2012, 12, 2655–2667. [Google Scholar] [CrossRef]
- Hillier, S.M.; Maresca, K.P.; Lu, G.; Merkin, R.D.; Marquis, J.C.; Zimmerman, C.N.; Eckelman, W.C.; Joyal, J.L.; Babich, J.W. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen for molecular imaging of prostate cancer. J. Nucl. Med. 2013, 54, 1369–1376. [Google Scholar] [CrossRef]
- Schmidkonz, C.; Hollweg, C.; Beck, M.; Reinfelder, J.; Goetz, T.I.; Sanders, J.C.; Schmidt, D.; Prante, O.; Bäuerle, T.; Cavallaro, A.; et al. 99mTc-MIP-1404-SPECT/CT for the detection of PSMA-positive lesions in 225 patients with biochemical recurrence of prostate cancer. Prostate 2018, 78, 54–63. [Google Scholar] [CrossRef]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.C.; Gugger, M. Bombesin receptor subtypes in human cancers: Detection with the universal radioligand 125I-[D-TYR6, beta-ALA11, PHE13, NLE14] bombesin(6–14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar]
- Reubi, C.; Gugger, M.; Waser, B. Co-expressed peptide receptors in breast cancer as a molecular basis for In Vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Korner, M.; Waser, B.; Mazzucchelli, L.; Guillou, L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Gourni, E.; Mansi, R.; Jamous, M.; Waser, B.; Smerling, C.; Burian, A.; Buchegger, F.; Reubi, J.C.; Maecke, H.R. N-terminal modifications improve the receptor affinity and pharmacokinetics of radiolabeled peptidic gastrin-releasing peptide receptor antagonists: Examples of 68Ga- and 64Cu-labeled peptides for PET imaging. J. Nucl. Med. 2014, 55, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Popp, I.; Del Pozzo, L.; Waser, B.; Reubi, J.C.; Meyer, P.T.; Maecke, H.R.; Gourni, E. Approaches to improve metabolic stability of a statine-based GRP receptor antagonist. Nucl. Med. Biol. 2017, 45, 22–29. [Google Scholar] [CrossRef]
- Mansi, R.; Wang, X.; Forrer, F.; Waser, B.; Cescato, R.; Graham, K.; Borkowski, S.; Reubi, J.C.; Maecke, H.R. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 97–107. [Google Scholar] [CrossRef]
- Nock, B.A.; Kaloudi, A.; Kanellopoulos, P.; Janota, B.; Brominska, B.; Izycki, D.; Mikolajczak, R.; Czepczynski, R.; Maina, T. [99mTc]Tc-DB15 in GRPR-Targeted Tumor Imaging with SPECT: From Preclinical Evaluation to the First Clinical Outcomes. Cancers 2021, 13, 5093. [Google Scholar] [CrossRef]
- Kaloudi, A.; Lymperis, E.; Giarika, A.; Dalm, S.; Orlandi, F.; Barbato, D.; Tedesco, M.; Maina, T.; de Jong, M.; Nock, B.A. NeoBOMB1, a GRPR-Antagonist for Breast Cancer Theragnostics: First Results of a Preclinical Study with [67Ga]NeoBOMB1 in T-47D Cells and Tumor-Bearing Mice. Molecules 2017, 22, 1950. [Google Scholar] [CrossRef]
- Kanellopoulos, P.; Kaloudi, A.; Rouchota, M.; Loudos, G.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. One Step Closer to Clinical Translation: Enhanced Tumor Targeting of [99mTc]Tc-DB4 and [111In]In-SG4 in Mice Treated with Entresto. Pharmaceutics 2020, 12, 1145. [Google Scholar] [CrossRef]
- Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Comparative evaluation of the new GRPR-antagonist (111) In-SB9 and (111) In-AMBA in prostate cancer models: Implications of in vivo stability. J. Label. Compd. Radiopharm. 2019, 62, 646–655. [Google Scholar] [CrossRef]
- Nock, B.A.; Charalambidis, D.; Sallegger, W.; Waser, B.; Mansi, R.; Nicolas, G.P.; Ketani, E.; Nikolopoulou, A.; Fani, M.; Reubi, J.C.; et al. New Gastrin Releasing Peptide Receptor-Directed [99mTc]Demobesin 1 Mimics: Synthesis and Comparative Evaluation. J. Med. Chem. 2018, 61, 3138–3150. [Google Scholar] [CrossRef]
- Xiao, D.; Wang, J.; Hampton, L.L.; Weber, H.C. The human gastrin-releasing peptide receptor gene structure, its tissue expression and promoter. Gene 2001, 264, 95–103. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Bakker, I.L.; van Tiel, S.T.; Haeck, J.; Doeswijk, G.N.; de Blois, E.; Segbers, M.; Maina, T.; Nock, B.A.; de Jong, M.; Dalm, S.U. In Vivo Stabilized SB3, an Attractive GRPR Antagonist, for Pre- and Intra-Operative Imaging for Prostate Cancer. Mol. Imaging Biol. 2018, 20, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Merkens, H.; Zeisler, J.; Zhang, C.; Roxin, A.; Tan, R.; Benard, F.; Lin, K.S. 68Ga-Labeled [Leu13ψThz14]Bombesin(7–14) Derivatives: Promising GRPR-Targeting PET Tracers with Low Pancreas Uptake. Molecules 2022, 27, 3777. [Google Scholar] [CrossRef]
- Maina, T.; Kaloudi, A.; Valverde, I.E.; Mindt, T.L.; Nock, B.A. Amide-to-triazole switch vs. in vivo NEP-inhibition approaches to promote radiopeptide targeting of GRPR-positive tumors. Nucl. Med. Biol. 2017, 52, 57–62. [Google Scholar] [CrossRef]
- Chatalic, K.L.; Konijnenberg, M.; Nonnekens, J.; de Blois, E.; Hoeben, S.; de Ridder, C.; Brunel, L.; Fehrentz, J.A.; Martinez, J.; van Gent, D.C.; et al. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016, 6, 104–117. [Google Scholar] [CrossRef]
- Guenther, T.; Deiser, S.; Felber, V.; Beck, R.; Wester, H.J. Substitution of L-Trp by α-methyl-L-Trp in 177Lu-RM2 results in 177Lu-AMTG, a high affinity GRPR ligand with improved In Vivo stability. J. Nucl. Med. 2022, 63, 1364–1370. [Google Scholar] [CrossRef]
- Guenther, T.; Wester, H.J. Modified grpr Antagonist Peptides for Imaging and Therapy of Cancer. Patent Application No. WO2021121734A1, 19 December 2019. [Google Scholar]
- Mather, S.J.; Nock, B.A.; Maina, T.; Gibson, V.; Ellison, D.; Murray, I.; Sobnack, R.; Colebrook, S.; Wan, S.; Halberrt, G.; et al. GRP Receptor Imaging of Prostate Cancer Using [99mTc]Demobesin 4: A First-in-Man Study. Mol. Imaging Biol. 2014, 16, 888–895. [Google Scholar] [CrossRef]
- Nock, B.A.; Nikolopoulou, A.; Galanis, A.; Cordopatis, P.; Waser, B.; Reubi, J.C.; Maina, T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: A preclinical study. J. Med. Chem. 2005, 48, 100–110. [Google Scholar] [CrossRef]
- Dalm, S.U.; Bakker, I.L.; de Blois, E.; Doeswijk, G.N.; Konijnenberg, M.W.; Orlandi, F.; Barbato, D.; Tedesco, M.; Maina, T.; Nock, B.A.; et al. 68Ga/177Lu-NeoBOMB1, a Novel Radiolabeled GRPR Antagonist for Theranostic Use in Oncology. J. Nucl. Med. 2017, 58, 293–299. [Google Scholar] [CrossRef]
- Abiraj, K.; Ursillo, S.; Tamma, M.L.; Rylova, S.N.; Waser, B.; Constable, E.C.; Fani, M.; Nicolas, G.P.; Reubi, J.C.; Maecke, H.R. The tetraamine chelator outperforms HYNIC in a new technetium-99m-labelled somatostatin receptor 2 antagonist. EJNMMI Res. 2018, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouzayed, A.; Rinne, S.S.; Sabahnoo, H.; Sorensen, J.; Chernov, V.; Tolmachev, V.; Orlova, A. Preclinical Evaluation of 99mTc-Labeled GRPR Antagonists maSSS/SES-PEG2-RM26 for Imaging of Prostate Cancer. Pharmaceutics 2021, 13, 182. [Google Scholar] [CrossRef] [PubMed]
- Guenther, T.; Fischer, S.; Beck, R.; Wester, H. Preclinical results of novel GRPR-targeted antagonists with modified binding sequences. J. Nucl. Med. 2020, 61, 1054. [Google Scholar]
- Lau, J.; Rousseau, E.; Zhang, Z.; Uribe, C.F.; Kuo, H.T.; Zeisler, J.; Zhang, C.; Kwon, D.; Lin, K.S.; Benard, F. Positron Emission Tomography Imaging of the Gastrin-Releasing Peptide Receptor with a Novel Bombesin Analogue. ACS Omega 2019, 4, 1470–1478. [Google Scholar] [CrossRef]
- Lymperis, E.; Kaloudi, A.; Sallegger, W.; Bakker, I.L.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Radiometal-Dependent Biological Profile of the Radiolabeled Gastrin-Releasing Peptide Receptor Antagonist SB3 in Cancer Theranostics: Metabolic and Biodistribution Patterns Defined by Neprilysin. Bioconjug. Chem. 2018, 29, 1774–1784. [Google Scholar] [CrossRef]
- Kaloudi, A.; Lymperis, E.; Kanellopoulos, P.; Waser, B.; de Jong, M.; Krenning, E.P.; Reubi, J.C.; Nock, B.A.; Maina, T. Localization of 99mTc-GRP Analogs in GRPR-Expressing Tumors: Effects of Peptide Length and Neprilysin Inhibition on Biological Responses. Pharmaceuticals 2019, 12, 42. [Google Scholar] [CrossRef]
- Gainkam, L.O.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Xavier, C.; Boerman, O.; Muyldermans, S.; Bossuyt, A.; Lahoutte, T. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol. Imaging 2011, 6, 85–92. [Google Scholar] [CrossRef]
- Makris, G.; Bandari, R.P.; Kuchuk, M.; Jurisson, S.S.; Smith, C.J.; Hennkens, H.M. Development and Preclinical Evaluation of 99mTc- and 186Re-Labeled NOTA and NODAGA Bioconjugates Demonstrating Matched Pair Targeting of GRPR-Expressing Tumors. Mol. Imaging Biol. 2020, 23, 52–61. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A.; Zhang, H.; Nikolopoulou, A.; Waser, B.; Reubi, J.C.; Maecke, H.R. Species differences of bombesin analog interactions with GRP-R define the choice of animal models in the development of GRP-R-targeting drugs. J. Nucl. Med. 2005, 46, 823–830. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organs | [99mTc]Tc-N4-asp-MJ9 | [99mTc]Tc-N4-asp-[Bta8]MJ9 | [99mTc]Tc-N4-[Hse7]MJ9 | [99mTc]Tc-N4-[α-Me-Trp8]MJ9 | [99mTc]Tc-Demobesin 4 * |
|---|---|---|---|---|---|
| Blood | 0.79 ± 0.39 | 1.11 ± 0.55 | 0.77 ± 0.34 | 1.24 ± 0.01 | ~1.3 ± 0.5 |
| Heart | 0.32 ± 0.18 | 0.40 ± 0.16 | 0.36 ± 0.10 | 0.54 ± 0.04 | n.a. |
| Lung | 0.70 ± 0.27 | 0.83 ± 0.30 | 0.88 ± 0.39 | 1.46 ± 0.07 | n.a. |
| Liver | 0.48 ± 0.14 | 0.84 ± 0.16 | 2.78 ± 0.16 | 2.11 ± 0.13 | ~2.5 ± 0.3 |
| Spleen | 0.31 ± 0.10 | 0.43 ± 0.13 | 0.59 ± 0.18 | 0.95 ± 0.02 | n.a. |
| Pancreas | 15.99 ± 1.62 | 4.77 ± 1.38 | 10.00 ± 4.17 | 39.06 ± 0.28 | ~39.0 ± 7.0 |
| Stomach | 1.79 ± 0.38 | 1.07 ± 0.25 | 1.94 ± 0.19 | 2.17 ± 0.13 | ~2.4 ± 0.4 |
| Intestine | 1.96 ± 0.31 | 1.00 ± 0.61 | 2.49 ± 0.13 | 3.50 ± 0.26 | ~7.5 ± 1.5 |
| Kidney | 2.98 ± 0.57 | 3.37 ± 0.87 | 3.84 ± 1.00 | 4.49 ± 0.17 | 20.0 ± 4.0 |
| Adrenal | 1.25 ± 0.74 | 0.53 ± 0.04 | 3.17 ± 2.31 | 1.07 ± 0.36 | n.a. |
| Muscle | 0.17 ± 0.09 | 0.19 ± 0.05 | 0.16 ± 0.04 | 0.40 ± 0.17 | ~0.25 ± 0.05 |
| Bone | 0.30 ± 0.14 | 0.37 ± 0.28 | 0.22 ± 0.08 | 0.61 ± 0.05 | n.a. |
| Tumor | 11.66 ± 1.55 | 9.31 ± 1.56 | 14.59 ± 2.28 | 9.42 ± 0.46 | ~10.5 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Günther, T.; Konrad, M.; Stopper, L.; Kunert, J.-P.; Fischer, S.; Beck, R.; Casini, A.; Wester, H.-J. Optimization of the Pharmacokinetic Profile of [99mTc]Tc-N4-Bombesin Derivatives by Modification of the Pharmacophoric Gln-Trp Sequence. Pharmaceuticals 2022, 15, 1133. https://doi.org/10.3390/ph15091133
Günther T, Konrad M, Stopper L, Kunert J-P, Fischer S, Beck R, Casini A, Wester H-J. Optimization of the Pharmacokinetic Profile of [99mTc]Tc-N4-Bombesin Derivatives by Modification of the Pharmacophoric Gln-Trp Sequence. Pharmaceuticals. 2022; 15(9):1133. https://doi.org/10.3390/ph15091133
Chicago/Turabian StyleGünther, Thomas, Matthias Konrad, León Stopper, Jan-Philip Kunert, Sebastian Fischer, Roswitha Beck, Angela Casini, and Hans-Jürgen Wester. 2022. "Optimization of the Pharmacokinetic Profile of [99mTc]Tc-N4-Bombesin Derivatives by Modification of the Pharmacophoric Gln-Trp Sequence" Pharmaceuticals 15, no. 9: 1133. https://doi.org/10.3390/ph15091133