
Discovery of Kinase and Carbonic Anhydrase Dual Inhibitors by Machine Learning Classification and Experiments


Abstract
:1. Introduction
2. Results
2.1. The Pair of ECFP4 Fingerprint and Logistic Regression Classifier Was Selected for Screening
2.2. Screening Known Protein-Specific Modulators by ML Classified the Candidate Inhibitors for CA
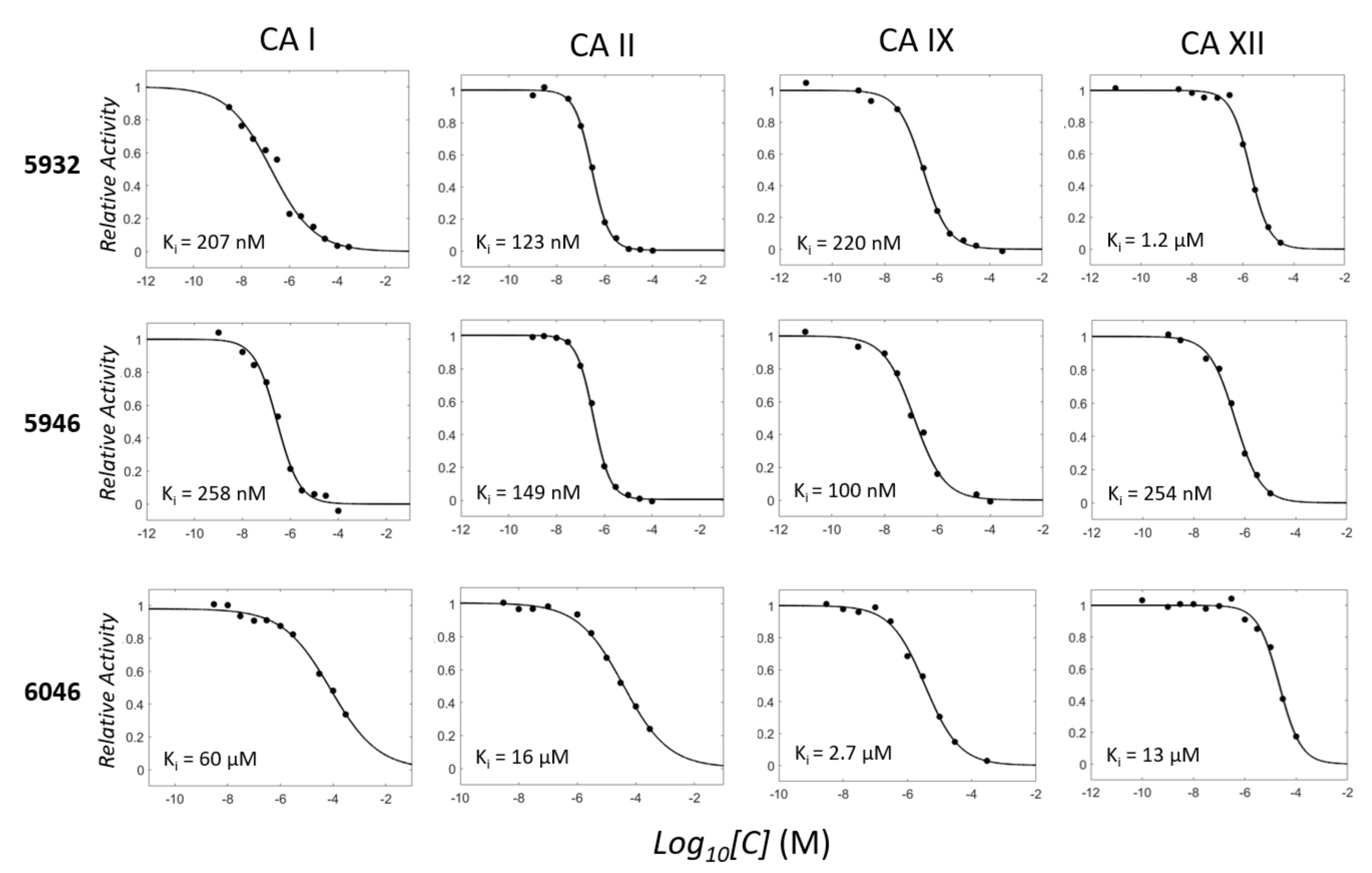
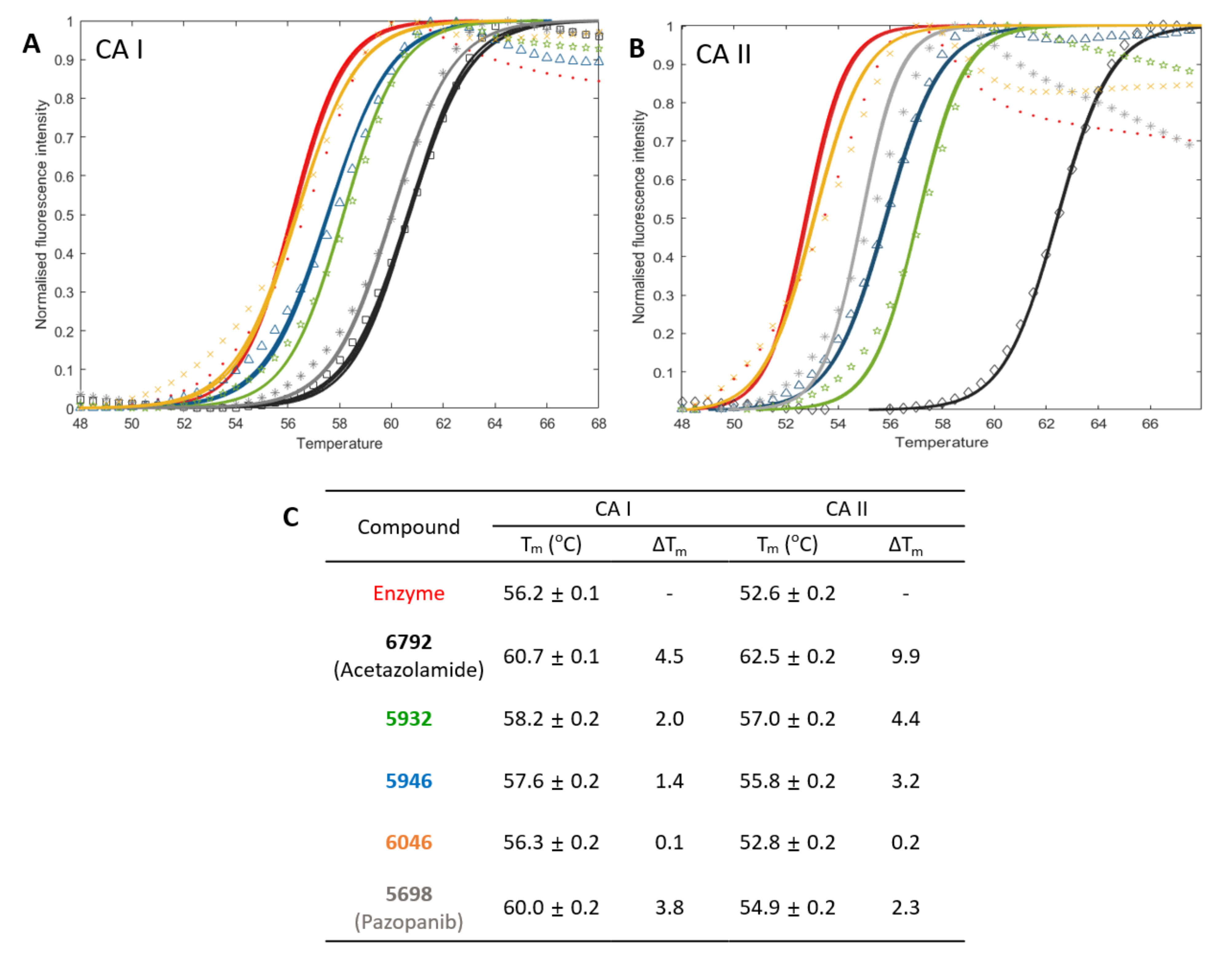
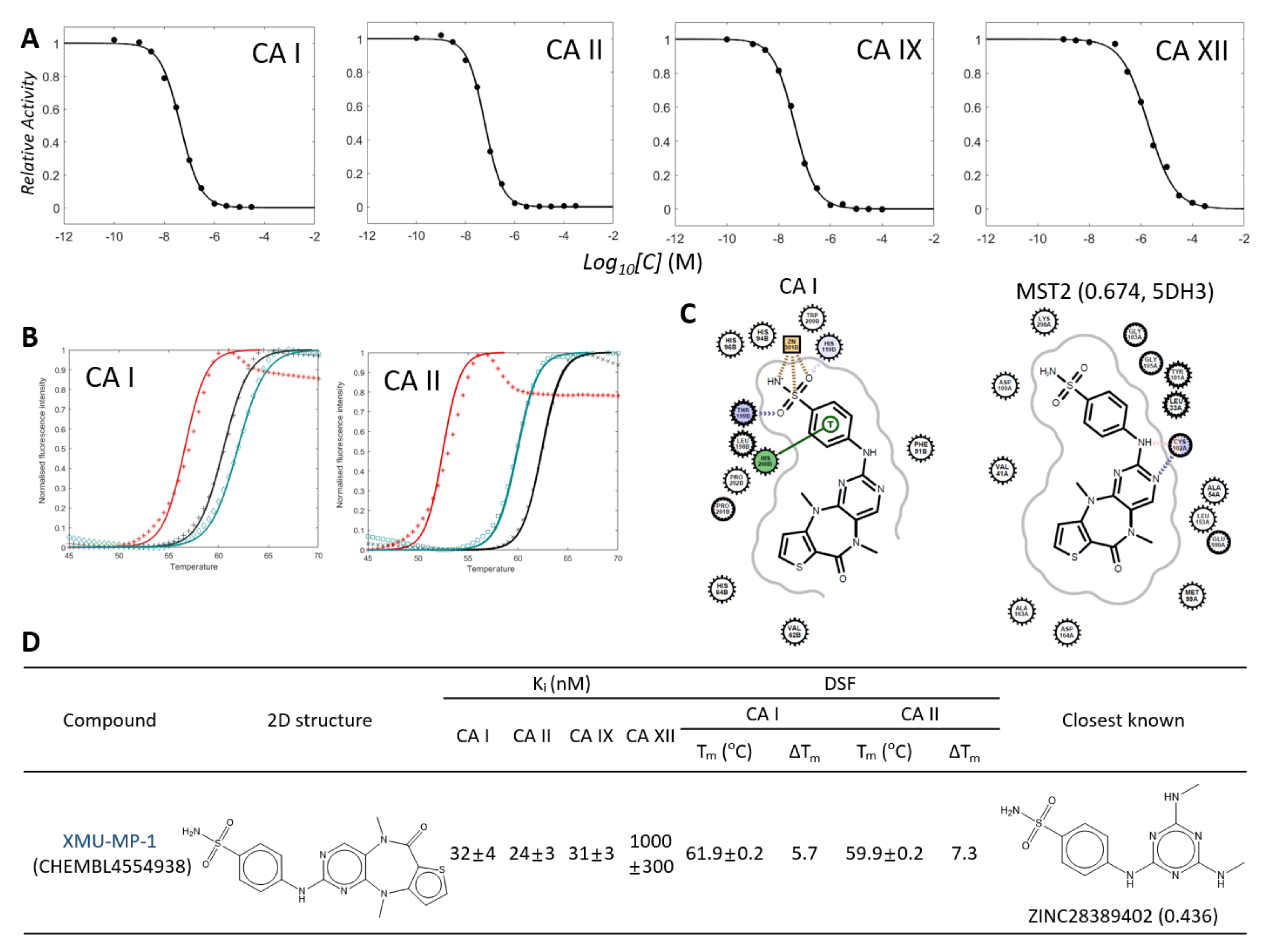
2.3. Two Candidates, 5932 and 5946, Were Highly Inhibitory for CAs
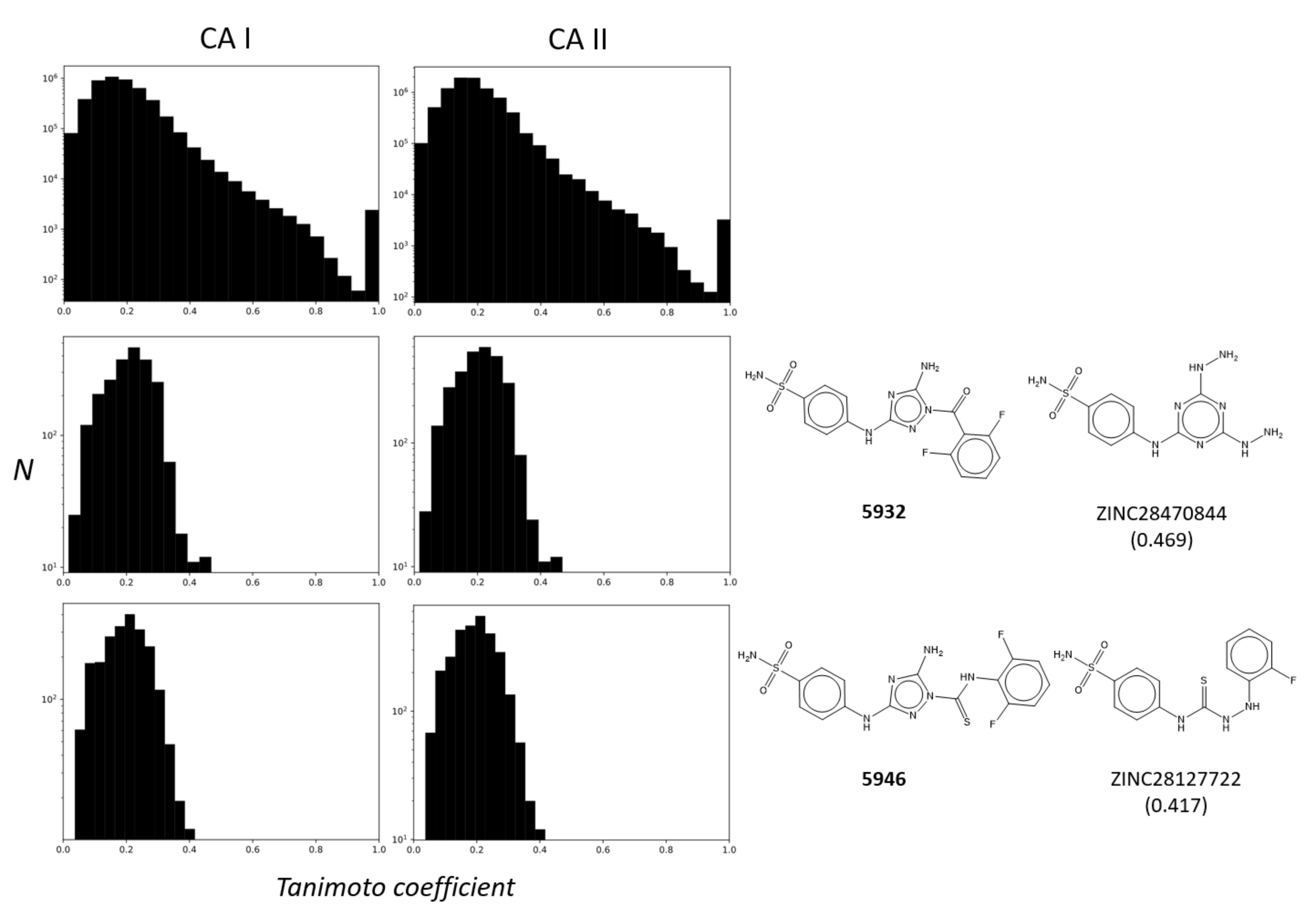
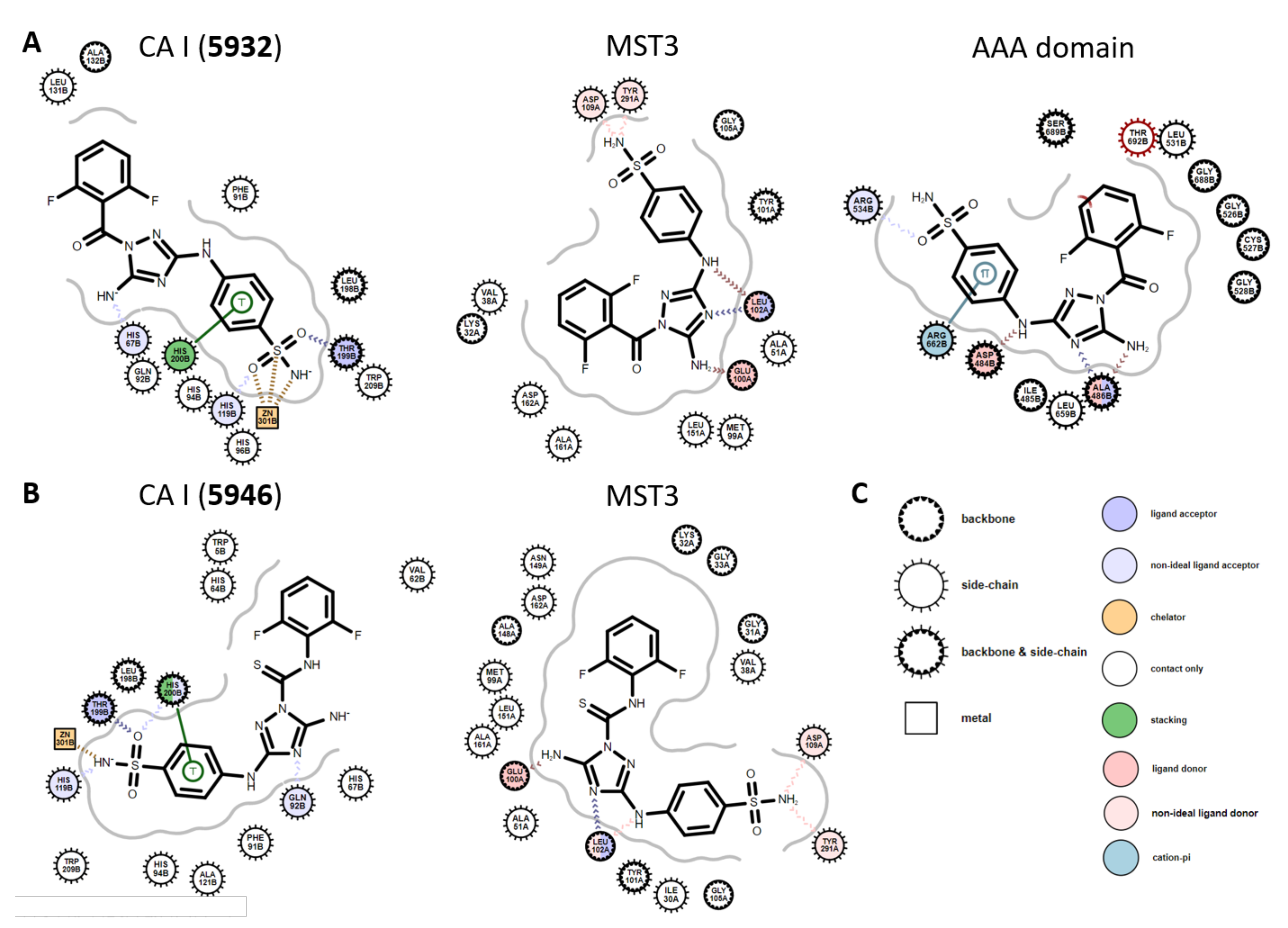
2.4. Cheminformatics Demonstrated Unique Features Compared to Other CA Inhibitors
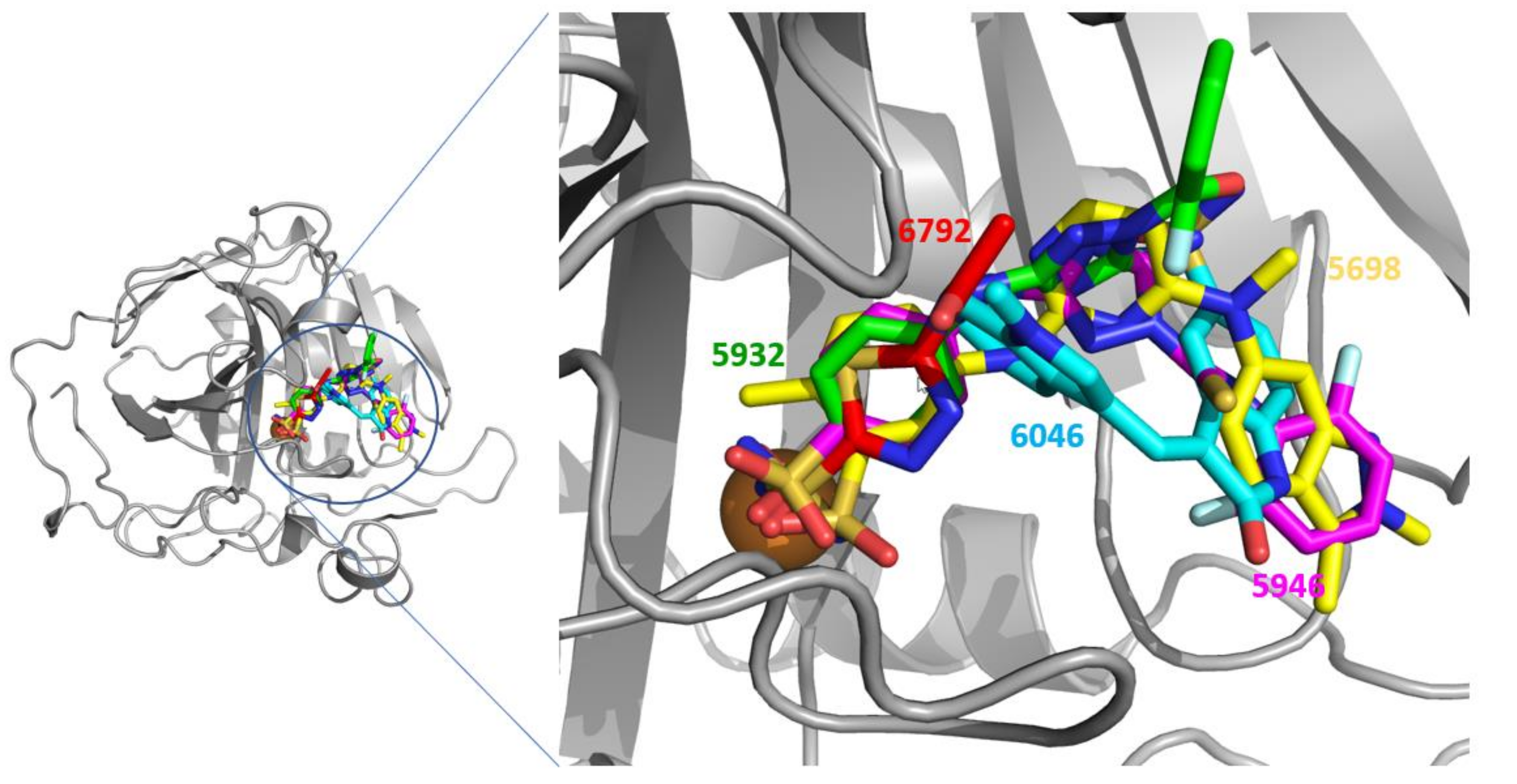
2.5. Docking Simulation Predicted the Binding Modes of the Dual Inhibitors
2.6. ML Screening of the Known Kinase Inhibitors Led to Discovering a New Potent CA Inhibitor, XMU-MP-1
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Brotz-Oesterhelt, H.; Brunner, N.A. How many modes of action should an antibiotic have? Curr. Opin. Pharmacol. 2008, 8, 564–573. [Google Scholar] [CrossRef]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2018, 62, 420–444. [Google Scholar] [CrossRef]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2021, 165, 105463. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Zatovicova, M.; Pastorek, J. Cancer-associated carbonic anhydrases and their inhibition. Curr. Pharm. Des. 2008, 14, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Georgey, H.H.; Manhi, F.M.; Mahmoud, W.R.; Mohamed, N.A.; Berrino, E.; Supuran, C.T. 1,2,4-Trisubstituted imidazolinones with dual carbonic anhydrase and p38 mitogen-activated protein kinase inhibitory activity. Bioorg. Chem. 2019, 82, 109–116. [Google Scholar] [CrossRef]
- Olesen, S.H.; Zhu, J.Y.; Martin, M.P.; Schonbrunn, E. Discovery of Diverse Small-Molecule Inhibitors of Mammalian Sterile20-like Kinase 3 (MST3). ChemMedChem 2016, 11, 1137–1144. [Google Scholar] [CrossRef]
- Winum, J.Y.; Maresca, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. Polypharmacology of sulfonamides: Pazopanib, a multitargeted receptor tyrosine kinase inhibitor in clinical use, potently inhibits several mammalian carbonic anhydrases. Chem. Commun. 2012, 48, 8177–8179. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Pottel, J.; Armstrong, D.; Zou, L.; Fekete, A.; Huang, X.P.; Torosyan, H.; Bednarczyk, D.; Whitebread, S.; Bhhatarai, B.; Liang, G.; et al. The activities of drug inactive ingredients on biological targets. Science 2020, 369, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Cote, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large scale comparison of QSAR and conformal prediction methods and their applications in drug discovery. J. Cheminform. 2019, 11, 4. [Google Scholar] [CrossRef]
- Mathai, N.; Kirchmair, J. Similarity-Based Methods and Machine Learning Approaches for Target Prediction in Early Drug Discovery: Performance and Scope. Int. J. Mol. Sci. 2020, 21, 3585. [Google Scholar] [CrossRef]
- Paricharak, S.; Cortes-Ciriano, I.; AP, I.J.; Malliavin, T.E.; Bender, A. Proteochemometric modelling coupled to in silico target prediction: An integrated approach for the simultaneous prediction of polypharmacology and binding affinity/potency of small molecules. J. Cheminform. 2015, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [PubMed]
- Awale, M.; Reymond, J.L. Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J. Chem. Inf. Modeling 2019, 59, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tao, B.; Chen, C.; Jia, W.; Sun, S.; Zhang, T.; Wang, X. Machine Learning Models Based on Molecular Fingerprints and an Extreme Gradient Boosting Method Lead to the Discovery of JAK2 Inhibitors. J. Chem. Inf. Modeling 2019, 59, 5002–5012. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, W.; Yang, Y.; Hua, Y.; Xing, G.; Liang, L.; Deng, C.; Wang, Y.; Fan, Y.; Liu, H.; et al. Discovery of Dual FGFR4 and EGFR Inhibitors by Machine Learning and Biological Evaluation. J. Chem. Inf. Modeling 2020, 60, 4640–4652. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Modeling 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Gaskins, G.; Sterling, T.; Mysinger, M.M.; Keiser, M.J. Predicted Biological Activity of Purchasable Chemical Space. J. Chem. Inf. Modeling 2018, 58, 148–164. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Bulusu, G.; Roy, A. Accelerating De Novo Drug Design against Novel Proteins Using Deep Learning. J. Chem. Inf. Modeling 2021, 61, 621–630. [Google Scholar] [CrossRef]
- Sun, G.; Fan, T.; Sun, X.; Hao, Y.; Cui, X.; Zhao, L.; Ren, T.; Zhou, Y.; Zhong, R.; Peng, Y. In Silico Prediction of O(6)-Methylguanine-DNA Methyltransferase Inhibitory Potency of Base Analogs with QSAR and Machine Learning Methods. Molecules 2018, 23, 2892. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; Sharman, J.L.; Campo, B.; Cavanagh, D.R.; Alexander, S.P.H.; Davenport, A.P.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 2020, 48, D1006–D1021. [Google Scholar] [CrossRef]
- Carta, F.; Supuran, C.T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Connolly, P.J.; Huang, S.; Wetter, S.K.; Lu, Y.; Murray, W.V.; Emanuel, S.L.; Gruninger, R.H.; Fuentes-Pesquera, A.R.; Rugg, C.A.; et al. 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: Synthesis and evaluation of biological activities. J. Med. Chem. 2005, 48, 4208–4211. [Google Scholar] [CrossRef]
- Bamborough, P.; Angell, R.M.; Bhamra, I.; Brown, D.; Bull, J.; Christopher, J.A.; Cooper, A.W.; Fazal, L.H.; Giordano, I.; Hind, L.; et al. N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of Lck: Indazoles as phenol isosteres with improved pharmacokinetics. Bioorg. Med. Chem. Lett. 2007, 17, 4363–4368. [Google Scholar] [CrossRef]
- Lai, J.Y.; Cox, P.J.; Patel, R.; Sadiq, S.; Aldous, D.J.; Thurairatnam, S.; Smith, K.; Wheeler, D.; Jagpal, S.; Parveen, S.; et al. Potent small molecule inhibitors of spleen tyrosine kinase (Syk). Bioorg. Med. Chem. Lett. 2003, 13, 3111–3114. [Google Scholar] [CrossRef]
- Duan, D.; Torosyan, H.; Elnatan, D.; McLaughlin, C.K.; Logie, J.; Shoichet, M.S.; Agard, D.A.; Shoichet, B.K. Internal Structure and Preferential Protein Binding of Colloidal Aggregates. ACS Chem. Biol. 2017, 12, 282–290. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, C.K.; Duan, D.; Ganesh, A.N.; Torosyan, H.; Shoichet, B.K.; Shoichet, M.S. Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem. Biol. 2016, 11, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Shoichet, M.S.; Shoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1429–1435. [Google Scholar] [CrossRef]
- Coan, K.E.; Shoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Zubriene, A.; Smirnoviene, J.; Smirnov, A.; Morkunaite, V.; Michailoviene, V.; Jachno, J.; Juozapaitiene, V.; Norvaisas, P.; Manakova, E.; Grazulis, S.; et al. Intrinsic thermodynamics of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamide binding to carbonic anhydrases by isothermal titration calorimetry. Biophys. Chem. 2015, 205, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Modeling 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Modeling 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Modi, V.; Dunbrack, R.L., Jr. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; He, Z.; Kong, L.L.; Chen, Q.; Yuan, Q.; Zhang, S.; Ye, J.; Liu, H.; Sun, X.; Geng, J.; et al. Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med. 2016, 8, 352ra108. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef]
- Okuyama, M.; Jiang, W.; Yang, L.; Subramanian, V. Mst1/2 Kinases Inhibitor, XMU-MP-1, Attenuates Angiotensin II-Induced Ascending Aortic Expansion in Hypercholesterolemic Mice. Circ. Rep. 2021, 3, 259–266. [Google Scholar] [CrossRef]
- Tian, Q.; Fan, X.; Ma, J.; Han, Y.; Li, D.; Jiang, S.; Zhang, F.; Guang, H.; Shan, X.; Chen, R.; et al. Resveratrol ameliorates lipopolysaccharide-induced anxiety-like behavior by attenuating YAP-mediated neuro-inflammation and promoting hippocampal autophagy in mice. Toxicol. Appl. Pharmacol. 2020, 408, 115261. [Google Scholar] [CrossRef]
- Seeneevassen, L.; Giraud, J.; Molina-Castro, S.; Sifre, E.; Tiffon, C.; Beauvoit, C.; Staedel, C.; Megraud, F.; Lehours, P.; Martin, O.C.B.; et al. Leukaemia Inhibitory Factor (LIF) Inhibits Cancer Stem Cells Tumorigenic Properties through Hippo Kinases Activation in Gastric Cancer. Cancers 2020, 12, 2011. [Google Scholar] [CrossRef]
- Mitchell, E.; Mellor, C.E.L.; Purba, T.S. XMU-MP-1 induces growth arrest in a model human mini-organ and antagonises cell cycle-dependent paclitaxel cytotoxicity. Cell Div. 2020, 15, 11. [Google Scholar] [CrossRef]
- Liu, L.Y.; Shan, X.Q.; Zhang, F.K.; Fan, X.F.; Fan, J.M.; Wang, Y.Y.; Liu, S.F.; Mao, S.Z.; Gong, Y.S. YAP activity protects against endotoxemic acute lung injury by activating multiple mechanisms. Int. J. Mol. Med. 2020, 46, 2235–2250. [Google Scholar] [CrossRef] [PubMed]
- Faizah, Z.; Amanda, B.; Ashari, F.Y.; Triastuti, E.; Oxtoby, R.; Rahaju, A.S.; Aziz, M.A.; Lusida, M.I.; Oceandy, D. Treatment with Mammalian Ste-20-like Kinase 1/2 (MST1/2) Inhibitor XMU-MP-1 Improves Glucose Tolerance in Streptozotocin-Induced Diabetes Mice. Molecules 2020, 25, 4381. [Google Scholar] [CrossRef] [PubMed]
- Triastuti, E.; Nugroho, A.B.; Zi, M.; Prehar, S.; Kohar, Y.S.; Bui, T.A.; Stafford, N.; Cartwright, E.J.; Abraham, S.; Oceandy, D. Pharmacological inhibition of Hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br. J. Pharmacol. 2019, 176, 3956–3971. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Dai, M.; Wang, D.; Xiong, W.; Zeng, Z.; Guo, C. The regulatory networks of the Hippo signaling pathway in cancer development. J. Cancer 2021, 12, 6216–6230. [Google Scholar] [CrossRef]
- Barelier, S.; Sterling, T.; O’Meara, M.J.; Shoichet, B.K. The Recognition of Identical Ligands by Unrelated Proteins. ACS Chem. Biol. 2015, 10, 2772–2784. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, Y.-M.; Jee, J.-G. Thiopurine drugs repositioned as tyrosinase inhibitors. Int. J. Mol. Sci. 2018, 19, 77. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kim, H.H.; Hyun, J.S.; Choi, J.; Choi, K.E.; Jee, J.G.; Park, S.J. Structural ensemble-based docking simulation and biophysical studies discovered new inhibitors of Hsp90 N-terminal domain. Sci. Rep. 2018, 8, 368. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Choi, K.E.; Park, S.J.; Kim, S.Y.; Jee, J.G. Ensemble-Based Virtual Screening Led to the Discovery of New Classes of Potent Tyrosinase Inhibitors. J. Chem. Inf. Modeling 2016, 56, 354–367. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CA I | ECFP4 | ECFP6 | MACCS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ACC | AUC | MCC | ACC | AUC | MCC | ACC | AUC | MCC | |
| KNN | 0.970 | 0.950 | 0.632 | 0.952 | 0.932 | 0.532 | 0.984 | 0.916 | 0.720 |
| NB | 0.979 | 0.697 | 0.483 | 0.964 | 0.660 | 0.291 | 0.812 | 0.868 | 0.284 |
| Logit | 0.994 | 0.994 | 0.863 | 0.993 | 0.994 | 0.849 | 0.989 | 0.988 | 0.771 |
| DT | 0.988 | 0.889 | 0.757 | 0.985 | 0.858 | 0.703 | 0.985 | 0.864 | 0.703 |
| RF | 0.992 | 0.988 | 0.826 | 0.990 | 0.985 | 0.782 | 0.990 | 0.988 | 0.771 |
| MLP | 0.995 | 0.993 | 0.883 | 0.993 | 0.990 | 0.843 | 0.990 | 0.991 | 0.794 |
| XGBoost | 0.992 | 0.977 | 0.832 | 0.990 | 0.985 | 0.797 | 0.991 | 0.994 | 0.815 |
| CA II | ECFP4 | ECFP6 | MACCS | ||||||
| ACC | AUC | MCC | ACC | AUC | MCC | ACC | AUC | MCC | |
| KNN | 0.981 | 0.973 | 0.734 | 0.972 | 0.965 | 0.659 | 0.984 | 0.929 | 0.739 |
| NB | 0.932 | 0.779 | 0.331 | 0.872 | 0.779 | 0.268 | 0.840 | 0.900 | 0.321 |
| Logit | 0.994 | 0.998 | 0.878 | 0.994 | 0.998 | 0.869 | 0.990 | 0.993 | 0.793 |
| DT | 0.989 | 0.899 | 0.777 | 0.988 | 0.868 | 0.747 | 0.986 | 0.862 | 0.716 |
| RF | 0.992 | 0.997 | 0.831 | 0.991 | 0.996 | 0.801 | 0.992 | 0.995 | 0.815 |
| MLP | 0.994 | 0.996 | 0.872 | 0.993 | 0.995 | 0.848 | 0.991 | 0.996 | 0.812 |
| XGBoost | 0.992 | 0.994 | 0.836 | 0.992 | 0.995 | 0.839 | 0.990 | 0.995 | 0.797 |
| Protein | Classifier | TP1 | TP2 | FP | Sum |
|---|---|---|---|---|---|
| CA I | Logit | 7 | 2 | 9 | |
| MLP | 7 | 1 | 4 | 12 | |
| CA II | Logit | 6 | 5 | 11 | |
| MLP | 6 | 8 | 14 |
| CA I (19) | CA I and CA II (23) | CA II (26) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Prob | Max TC | ZINC ID | ID | Prob | Max TC | ZINC ID | ID | Prob | Max TC | ZINC ID |
| 3463 | 0.982 | 0.394 | 13800463 | 6920 | 0.999 | 0.737 | 28526472 | 9753 | 0.935 | 0.475 | 13780724 |
| 10070 | 0.935 | 0.274 | 28526472 | 6814 | 0.995 | 1.000 | 56721 | 3962 | 0.931 | 0.290 | 13612907 |
| 4840 | 0.900 | 0.288 | 13650488 | 6828 | 0.994 | 1.000 | 8415468 | 3986 | 0.928 | 0.464 | 64527862 |
| 7047 | 0.838 | 0.283 | 58569258 | 7046 | 0.992 | 0.422 | 184018 | 10483 | 0.910 | 0.264 | 34717999 |
| 1376 | 0.834 | 0.342 | 1099 | 4839 | 0.990 | 0.404 | 13800448 | 10664 | 0.903 | 0.353 | 87722919 |
| 10175 | 0.801 | 0.426 | 131170 | 6792 | 0.979 | 1.000 | 3813042 | 4836 | 0.897 | 0.700 | 34799864 |
| 8821 | 0.762 | 0.227 | 13650488 | 10017 | 0.978 | 0.270 | 28388514 | 4742 | 0.850 | 0.328 | 27638369 |
| 980 | 0.751 | 0.328 | 22198192 | 5946 | 0.959 | 0.407 | 28389402 | 10242 | 0.785 | 0.474 | 95586265 |
| 9092 | 0.740 | 0.192 | 2101 | 10149 | 0.948 | 0.639 | 84759371 | 11205 | 0.776 | 0.241 | 64526424 |
| 932 | 0.734 | 0.283 | 595377 | 8146 | 0.912 | 0.413 | 13800465 | 7289 | 0.757 | 0.340 | 13800448 |
| 4702 | 0.724 | 0.464 | 1099 | 2894 | 0.909 | 0.435 | 58569258 | 8316 | 0.714 | 0.375 | 40917210 |
| 960 | 0.685 | 0.324 | 95591272 | 6807 | 0.905 | 0.405 | 13800446 | 4055 | 0.680 | 0.341 | 26387397 |
| 958 | 0.685 | 0.324 | 95591272 | 6810 | 0.884 | 1.000 | 1530622 | 11201 | 0.641 | 0.227 | 64526424 |
| 5501 | 0.685 | 0.324 | 95591272 | 6849 | 0.838 | 0.245 | 34717916 | 4357 | 0.623 | 0.367 | 27636999 |
| 10321 | 0.589 | 0.391 | 84670597 | 9513 | 0.830 | 0.361 | 84670374 | 6574 | 0.599 | 0.195 | 13472881 |
| 8378 | 0.548 | 0.244 | 1099 | 7028 | 0.817 | 0.380 | 84652324 | 4835 | 0.585 | 0.360 | 34799864 |
| 5932 | 0.546 | 0.460 | 28389402 | 4837 | 0.800 | 0.317 | 84670374 | 9129 | 0.583 | 0.243 | 64526424 |
| 7409 | 0.535 | 0.408 | 16525334 | 6797 | 0.722 | 0.542 | 1530622 | 11174 | 0.580 | 0.404 | 27635960 |
| 1394 | 0.509 | 0.239 | 5159179 | 10433 | 0.694 | 0.541 | 13829485 | 10693 | 0.578 | 0.406 | 27741075 |
| 4635 | 0.661 | 0.212 | 13800446 | 7893 | 0.572 | 0.471 | 16525334 | ||||
| 4084 | 0.661 | 0.212 | 13800446 | 2892 | 0.571 | 0.509 | 95586265 | ||||
| 11220 | 0.601 | 0.250 | 26387397 | 6514 | 0.570 | 0.357 | 84669523 | ||||
| 7125 | 0.584 | 0.318 | 28349861 | 6046 | 0.565 | 0.270 | 13804313 | ||||
| 7870 | 0.562 | 0.425 | 131170 | ||||||||
| 7197 | 0.509 | 0.690 | 34799864 | ||||||||
| 6648 | 0.507 | 0.273 | 27644927 | ||||||||
| Compound | 2D Structure | CA I | CA II | CA IX | CA XII |
|---|---|---|---|---|---|
| 6792 (Acetazolamide) |  | 197 ± 18 | 3 ± 1 | 22 ± 5 | 23 ± 10 |
| 5932 |  | 207 ± 66 | 123 ± 16 | 220 ± 32 | 1.2 ± 0.2 (µM) |
| 5946 |  | 258 ± 58 | 149 ± 21 | 100 ± 28 | 254 ± 40 |
| 6046 |  | 60 ± 9 (µM) | 16 ± 5 (µM) | 2.7 ± 0.3 (µM) | 13 ± 4 (µM) |
| 5698 (Pazopanib) |  | 67 ± 10 | 242 ± 32 | 1.7 ± 0.5 (µM) | 0.8 ± 0.3 (µM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-J.; Pandit, S.; Jee, J.-G. Discovery of Kinase and Carbonic Anhydrase Dual Inhibitors by Machine Learning Classification and Experiments. Pharmaceuticals 2022, 15, 236. https://doi.org/10.3390/ph15020236
Kim M-J, Pandit S, Jee J-G. Discovery of Kinase and Carbonic Anhydrase Dual Inhibitors by Machine Learning Classification and Experiments. Pharmaceuticals. 2022; 15(2):236. https://doi.org/10.3390/ph15020236
Chicago/Turabian StyleKim, Min-Jeong, Sarita Pandit, and Jun-Goo Jee. 2022. "Discovery of Kinase and Carbonic Anhydrase Dual Inhibitors by Machine Learning Classification and Experiments" Pharmaceuticals 15, no. 2: 236. https://doi.org/10.3390/ph15020236